

Abstract
Over the last quarter-century, there has been tremendous progress in genetics research that has defined molecular causes for cardiomyopathies. More than a thousand mutations have been identified in many genes with varying ontologies, therein indicating the diverse molecules and pathways that cause hypertrophic, dilated, restrictive, and arrhythmogenic cardiomyopathies. Translation of this research to the clinic via genetic testing can precisely group affected patients according to molecular etiology, and identify individuals without evidence of disease who are at high risk for developing cardiomyopathy. These advances provide insights into the earliest manifestations of cardiomyopathy and help to define the molecular pathophysiological basis for cardiac remodeling. Although these efforts remain incomplete, new genomic technologies and analytic strategies provide unparalleled opportunities to fully explore the genetic architecture of cardiomyopathies. Such data hold the promise that mutation-specific pathophysiology will uncover novel therapeutic targets, and herald the beginning of precision therapy for cardiomyopathy patients.
Keywords: dilated cardiomyopathy, hypertrophic cardiomyopathy, molecular etiology
Cardiomyopathies are disorders with primary abnormalities in the structure and function of the heart. Addition of the historical term idiopathic was used to denote an enigmatic etiology that specifically excluded pre-existing cardiovascular or systemic diseases. These disorders are commonly grouped into morphological subtypes that include hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), and left ventricular noncompaction (LVNC) (1,2). Early clinical investigations recognized familial transmission for many cardiomyopathies, suggesting a critical role for genetics. Research advances over the past 30 years confirmed this hypothesis, and today many cardiomyopathies are recognized as monogenic disorders.
Cardiomyopathy mutations are notable for substantial variation in clinical expression. Both genetic heterogeneity (e.g., distinct genes that cause the same disease) and allelic variation (distinct mutations in the same gene) contribute to variable morphological phenotypes and disease severity. Background genomic variation, lifestyle, and exposures are other contributory factors that account for differences in clinical manifestations in family members with identical mutations. These complexities pose substantial challenges for clinicians faced with interpreting genetic testing results and effectively using this information to improve patient management, as well as for scientists trying to decipher disease mechanisms. Herein, we review the current understanding of cardiomyopathy genetics, and discuss how the rapid evolution of genomic technologies is changing this landscape at both the bench and the bedside.
DEFINING PATHOGENIC VARIANTS
The era of genetic medicine in cardiomyopathy research began with studies that used genome-wide linkage analysis to identify mutations that displayed classic Mendelian inheritance (3). These studies surveyed all chromosomes, and then restricted analyses of genes and variants using familial cosegregation to achieve strong statistical evidence that an identified candidate gene was likely to cause disease. Additional evidence included expression of the candidate gene in the disease tissue, demonstration that the mutation was absent from the general population, and that it altered an amino-acid residue that was highly conserved across species, thus implying functional consequences. Definitive pathogenic confirmation required the identification of additional mutations in the same gene via analyses of unrelated affected patients or demonstration that the human phenotype was recapitulated by expression of the variant in a model system (Table 1). This overarching strategy has now been supplemented with direct sequence analyses using arrays or next-generation sequencing to study the exome (sequences that encode all proteins) and genome.
TABLE 1.
Criteria for Defining a Variant as Pathogenic
| Criterion | Description and Significance |
|---|---|
| Nonsynonymous genetic variant | A genetic variant that alters the amino-acid sequence |
| Expression | Confirmation that the protein product of the gene is expressed in diseased tissue |
| Unbiased genetic analysis | Genome-wide analysis (linkage analysis, genome-wide association study, whole-exome or whole-genome sequencing) reduces the possibility of identifying a coincidental and unrelated mutation or a disease modifying mutation |
| Conservation | Conservation of the affected amino acid in different species through evolution suggests a critical biological role of that particular residue |
| Control population | Absence from a large number of unaffected controls, or confirmation of very low minor allele frequency in a population of interest demonstrates that the mutation is unlikely to be a polymorphism unrelated to the disease |
| Cosegregation | Demonstrates that the mutation is found in those with disease only (note: incomplete and age-dependent penetrance is common); identification of the disease phenotype in those without the mutation proves nonpathogenicity |
| Multiplicity of events | Demonstration that a variant may be causal in ≥2 unrelated probands greatly increases confidence in pathogenicity |
| Animal models | Recapitulation of the disease phenotype by the variant in animals genetically engineered to express the mutation strongly supports causality |
Core sets of pathogenic cardiomyopathy genes have been successfully identified, but knowledge of the full genetic architecture remains incomplete. Novel disease genes have been reported in association with a cardiomyopathy, but with less definitive proof of pathogenicity. This can reflect small family size that limits statistical power, incomplete segregation analyses due to sample access, and the considerable effort and expense required for comprehensive study of all family members, and/or reliance on predicted pathogenicity on the basis of variant characteristics. Variants that cause major structural changes in the encoded protein (truncating, nonsense, and canonical splice-site mutations) are usually considered more likely pathogenic than missense mutations that alter 1 amino acid, but neither assumption is uniformly true. The development of in silico algorithms has improved the prediction of pathogenicity on the basis of expected protein structural changes, evolutionary conservation of amino acids across protein orthologs, knowledge of protein function, and altered messenger RNA (mRNA) transcript splicing. But even with these tools, the specificity is low and overall accuracy is only ~65% to 80% for accurately predicting pathogenicity (4).
A recently developed and publically available resource, the Exome Aggregation Consortium (ExAC)(5) has further improved variant classification. ExAC data summarizes the individual frequencies of all variants identified in exome sequences (which encode all proteins in the genome) from over 60,000 unrelated individuals. Notably, the frequency of specific variants differs among peoples of different ancestries. On the basis of the assumption that pathogenic variants will be very rare or absent in a general population with shared ancestry, the ExAC frequency provides a benchmark for whether or not a variant is likely to cause cardiomyopathy. Already this dataset has provided supportive evidence that prioritized certain variants for further testing, while also casting doubt on the pathogenicity of some cardiomyopathy-associated variants that were identified by candidate gene analyses (6). Further expansion of this database to include many more distinct populations will continue to improve variant interpretation.
The genetic diversity that underlies the cardiomyopathies remains a complex issue. Next-generation sequence analyses of disease and control cohorts have demonstrated that 60% to 90% of rare cardiomyopathy mutations are “private” and found only in single families (7,8). In addition, haplotype analyses of identical mutations in separate families have confirmed that most are recent and independent mutational events (9). Consequently, clinical genetic testing results often contain a nuanced interpretation, rather than definitively labeling a variant as pathogenic or benign (Table 2).
TABLE 2.
Variant Classification
| Designation | Definition |
|---|---|
| Pathogenic | Variants with very low population frequency (i.e., MAF <0.02%) and with strong evidence for cosegregation with disease phenotype, conservation across species, and/or functional evidence |
| Likely Pathogenic | Variants with very low frequency population (i.e., MAF <0.02%) in the population, but with lower strength for cosegregation and/or limited functional evidence |
| Variant of Uncertain Significance | Variant with restricted evidence for cosegregation, limited or contradictory functional evidence, and variable conservation across species |
| Likely Benign | Variant identified at low frequency in the general population (i.e., MAF >0.3%) and not conserved across species |
| Benign | Variant identified at moderate levels in the general population (i.e., MAF >1%) |
Adapted from Richards et al. (4). MAF = minor allele frequency.
GENETIC CARDIOMYOPATHIES
HYPERTROPHIC CARDIOMYOPATHY
HCM is a primary disorder of the heart that is unaccompanied by pathology in another organ (10). The pathological hallmark of HCM is unexplained left ventricular hypertrophy (LVH). Diagnosis requires exclusion of secondary causes of LVH, such as hypertension and aortic stenosis, or physiological hypertrophy, as seen in highly trained athletes. Classically, HCM results in asymmetric septal hypertrophy, although almost any pattern of LVH can be associated with the disease (11,12).
HCM is a monogenic disorder caused by mutations in genes that encode the protein components of the cardiac sarcomere (Figure 1, Online Table 1). Clinical studies define the population prevalence for unexplained LVH as 1:500 (13,14), which is far higher than the prevalence of pathogenic mutations. However, next-generation sequence analyses of sarcomere genes have identified considerable numbers of rare variants in population cohorts, including among individuals with some clinical features of cardiomyopathy. Together, these data imply that variants in sarcomere genes may individually or collectively affect cardiac morphology and function, even without causing overt HCM (8,15–17).
FIGURE 1. A Schematic of Definitive (Bolded) and Posited HCM Genes With the Subcellular Localization of the Encoded Proteins.
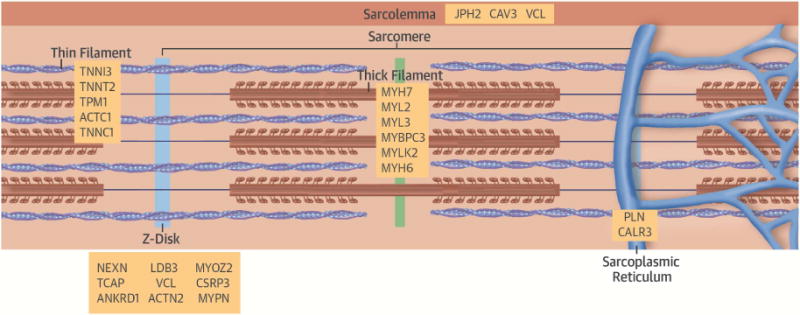
All pathogenic genes encode sarcomere proteins. Putative HCM genes encode these and sarcomere-associated molecules. HCM = hypertrophic cardiomyopathy.
HCM is transmitted as a dominant trait; hence, first-degree relatives of affected individuals have a 50% risk of developing disease. However, penetrance is incomplete and lowest at very young ages, which often delays clinical diagnosis until adolescence or adulthood. Identical HCM mutations can produce distinct LVH morphologies, varying amounts of myocardial fibrosis, and differing susceptibility to arrhythmias. Genetic modifiers, epigenetic differences, and distinct environmental factors are likely to influence these variables.
HCM Genes
Multiple independent studies have demonstrated pathogenic mutations causing HCM in 8 genes: β-myosin heavy chain (MYH7) (18,19), α-tropomyosin (TPM1), cardiac troponin T (TNNT2) (20,21), cardiac myosin binding protein-C (MYBPC3) (22,23), myosin regulatory light chain (MYL2), myosin essential light chain (MYL3) (24), cardiac troponin I (TNNI3) (25), and cardiac α-actin (ACTC1) (26,27). Combined, MYBPC3 and MYH7 account for up to 50% of all clinically recognized cases of HCM, and constitute at least 75% of probands where a mutation is identified, whereas other HCM genes account for <10% of cases (28–33). Disease-causing mutations are identified in ~30% to 60% of probands in studies that include these 8 pathogenic HCM genes, (28,29,34,35), with the highest detection rate among probands with: 1) early-onset disease; 2) greater severity of LVH; 3) asymmetric septal hypertrophy; and 4) family history of HCM (34,36,37).
Mutations in other genes have been associated with HCM. Unbiased genome-wide studies on 2 separate families have identified mutations in myozenin 2 (MYOZ2) (38) and α-actinin 2 (ACTN2)(39), molecules that interact with the sarcomere, but are not directly involved in force generation. Although mutations in these genes are likely pathogenic, identification of additional families or evidence that animal models carrying these mutations cause disease is needed to support their pathogenicity in HCM. In contrast, mutations in TTN, encoding the giant protein titin, are infrequently reported in HCM patients (40), and the pathogenicity of these mutations has been contested by multiple subsequent studies (29,41,42). Similarly, identical mutations in CSRP3, encoding the Z-disc molecule cardiac LIM protein, have been associated with both HCM and DCM through candidate gene analyses in separate cohorts (43–45).
Functional consequences of HCM mutations
Although the genetic basis for a large proportion of HCM is well established, the biochemical and biophysical mechanisms by which sarcomere gene mutations cause disease remains incompletely understood. To inform these processes, HCM mutations have been engineered into mice to permit detailed biochemical and molecular analyses. Although informative, interpretation of these models is complicated by the predominant expression of similar, but nonidentical myosin heavy chain isoforms (human: β-myosin; mouse: α-myosin), and differences in calcium hemostasis and cardiac physiology (such as the 10-fold difference in human and mouse heart rates). Despite these complexities, mouse models of HCM reveal unexpected consequences of how dominant mutant proteins that are incorporated into myofilaments perturb sarcomere function.
The myosin head region is the domain that generates force, hydrolyzes adenosine triphosphate (ATP) and interacts with regulatory light chain, essential light chain, troponin T, and actin. Mutations in this domain exhibit biophysical properties that enhance contraction, but impair relaxation. Studies of isolated myofibrillar tissue preparations from these models and assessment of the chemo-mechanical cycle in ex vivo preparations of sarcomere proteins with HCM mutations confirm these observations (46). Consistent with this mechanism, MYK-461, a small molecule that inhibits the myosin adenosine triphosphatase (ATPase) and reduces sarcomere power, was shown to attenuate the development of HCM in mice that carry pathogenic myosin mutations. (47)
Far less is known about how MYBPC3 mutations cause HCM. Myosin binding protein-C does not directly participate in force generation, but modulates contractile performance through interaction with myosin and titin. HCM mutations in other genes encode missense mutations that alter only 1 amino acid. In contrast, many MYBPC3 mutations produce truncated proteins (48). If incorporated into the sarcomere, these mutant proteins could cause disease. Alternatively, if mutant transcripts or proteins are cleared by cell surveillance mechanisms, disease would result from haploinsufficiency, an inadequate amount of functional myosin binding protein-C for normal sarcomere performance (49–51).
In addition to the biophysical consequences on sarcomere performance, HCM mutations increase the energetic cost of contraction. Cardiac tissues from humans and model organisms show a reduced phosphorylated creatinine to ATP ratio, altered ATPase activity, and an overall increased energetic cost of cross-bridge cycling (52–56). These data suggest that perturbation of myocardial energetics is a common underlying molecular feature of HCM, which may account for the similar phenotypes derived from these different mutations (53,56).
Genotype-phenotype correlation
The large numbers of pathogenic mutations in different sarcomere protein genes, combined with modifying genetic, epigenetic, and environmental factors, accounts for why genotype alone cannot predict patient-specific outcomes in HCM. Despite this, clinical data from large families with specific mutations have provided some noteworthy insights into how genotype can impact phenotype.
Adverse clinical outcomes have been identified in several recurrent MYH7 mutations, each of which causes substantial cardiac remodeling (Arg403Gln, associated with increased risk for sudden cardiac death; Arg719Trp and Arg453Cys, associated with increased risk for end-stage heart failure [HF]), whereas other MYH7 mutations produce milder phenotypes and have a good prognosis (57–59). HCM caused by MYBPC3 mutations generally presents later in life, and displays less morbidity and lower penetrance (60,61). Supporting this, HCM founder mutations occur almost exclusively in MYBPC3 (62). Two such mutations are particularly illustrative. Among individuals with southeast Asian ancestry, ~4% of the population carries a MYBPC3 mutation that is estimated to have arisen ~30,000 years ago (63). This mutation is associated with a 7-fold increased risk of HF late in life. Another MYBPC3 founder mutation dating to the 15th century today accounts for nearly 60% of HCM in Iceland, and is associated with adverse outcomes later in life (64). Maintenance of such variants through hundreds to thousands of years and generations would only occur via neutral selection with no impact during childbearing years; only recently, with increased life expectancy, has disease become apparent.
STORAGE AND METABOLIC CARDIOMYOPATHIES
Mutations in genes that encode proteins that function in cardiac metabolism or clearance of cellular byproducts also cause LVH (Online Table 2). Although these are often labeled as “HCM phenocopies” on the basis of cardiac imaging findings that mimic HCM, histopathology demonstrates the absence of cardiomyocyte disarray and the presence of cytoplasmic vacuoles that contain lipid (GLA mutations), lysosomal remnants (LAMP2 mutations), and glycogen (PRKAG2 and GAA mutations). These cardiomyopathies have distinct modes of inheritance, including autosomal-dominant (PRKAG2 mutations), X-linked (GLA and LAMP2 mutations) and autosomal-recessive patterns (GAA mutations).
Gene-based diagnosis, unlike cardiac imaging, can readily distinguish these disorders from HCM, which is important for appropriate management of patients and their families. Diagnosis of PRKAG2 cardiomyopathy necessitates electrophysiological monitoring due to the high incidence of conduction defects. LAMP2 mutations cause childhood-onset cardiomyopathy with prevalent arrhythmias, often in the context of Danon disease (neurocognitive deficits and hepatic dysfunction), which has a very poor prognosis (65) and requires early triage to heart transplantation. GLA mutations cause Fabry disease (LVH with renal, eye, and skin manifestations), accounting for at least 1% of all cases of unexplained LVH and an even higher percentage of patients who present after age 40 (66–68). Recognition of Fabry cardiomyopathy has critical therapeutic implications, as early treatment with enzyme replacement therapy limits myocardial remodeling and maintains cardiac function (69–71).
RESTRICTIVE CARDIOMYOPATHY
Primary RCM is a very rare heart muscle disease that is diagnosed by functional, rather than anatomic findings. RCM causes increased myocardial stiffness that results in a rapid rise in ventricular pressure, despite only small increases in ventricular volumes (1,2). This functional definition complicates disease classification because restrictive physiology also occurs with HCM and DCM. Among 1,226 patients with familial HCM (688 families), 1.5% had phenotypes diagnostic of RCM (72). Half of the probands with RCM had pathogenic mutations in either MYH7 or TNNI3; all patients with an identified mutation either had marked myofibrillar disarray on biopsy or had relatives with an “unequivocal” diagnosis of HCM. Notably, restrictive physiology was associated with a higher symptom burden, lower exercise tolerance, and increased complications (72).
Additional mutations in TNNI3 (71), as well as mutations in other sarcomere genes (73–75), and an unidentified gene at locus 10q23.3 (76) are also associated with RCM. Affected family members had HCM, DCM, or desmin-associated myopathy. Mutations in MYPN, which encodes the sarcomere-associated protein myopalladin, can also cause myofibrillar disarray and restrictive physiology (77). However the resulting clinical diagnoses associated with MYPN mutations include HCM, DCM, or RCM (77–80).
Two mutations associated with RCM have been modeled in mice. Mutant animals carrying a TNNI3 missense mutation that was associated with human RCM and HCM, developed early diastolic dysfunction, small chamber size and reduced LV systolic function, atypical manifestations of HCM (81). A MYPN mutation produced subtle manifestations of increased cardiac fibrosis, reduced LV chamber size, and minimal diastolic dysfunction (82).
In aggregate, these data illuminate the variable physiological and anatomic responses to sarcomere mutations, and implicate factors beyond a specific mutation in clinical manifestation. RCM does not appear to constitute a distinct genetic cardiomyopathy, but rather represents part of a phenotypic spectrum of HCM that occurs with limited hypertrophy and restrictive physiology.
DILATED CARDIOMYOPATHY
DCM is characterized by ventricular chamber enlargement and systolic dysfunction (1). LV mass is often greatly increased in DCM, but in contrast to HCM, LV wall thickness is normal. The histopathology of DCM is typically unrevealing of the underlying cause: myocytes are enlarged, with characteristics of cell demise, and myocardial fibrosis is increased, which are abnormalities that promote arrhythmias and HF. DCM is the commonest cause for heart transplantation. Approximately 50% of nonischemic DCM has no identifiable etiology (83), and in both familial and sporadic cases, genetic causes are increasingly identified.
Early population data using echocardiography estimated the prevalence of unexplained DCM at 1:2,500 (84). However, extrapolation of epidemiological data for DCM and HF suggests a much higher prevalence, possibly as high as 1:250 (85), an estimate that is aligned with the prevalence of likely pathogenic variants identified by next-generation sequencing (7,17).
DCM genes encode a heterogeneous group of molecules that participate in force generation, force transmission, sarcomere integrity, cytoskeletal and nuclear architecture, electrolyte homeostasis, mitochondrial function, and transcription (Figure 2, Online Table 3). Although most mutations are transmitted as dominant traits, a few exhibit recessive, X-linked, and matrilineal inheritance. Several issues contribute to the limited clinical recognition of familial DCM. First, the nonspecific manifestations of genetic DCM are frequently misattributed to other common and prevalent cardiovascular diseases. Secondly, DCM mutations exhibit age-dependent penetrance, and clinical expression may be delayed until after the fifth or sixth decade. Even with overt familial transmission, the penetrance of DCM mutations can be incomplete, an observation that indicates the potential for compensatory mechanisms, and modifying genetic and epigenetic factors. Finally, the substantial genetic heterogeneity of DCM has made comprehensive genetic testing technically difficult and costly. Until recently, most DCM diagnosis panels failed to screen all potential genes, resulting in disappointingly low sensitivity for detection of pathogenic mutations. Next-generation sequencing that can fully interrogate all DCM genes should substantially increase mutation detection in both familial and sporadic cases.
FIGURE 2. A Schematic of Definitive and Posited DCM Genes With the subcellular Localization of the Encoded Proteins.
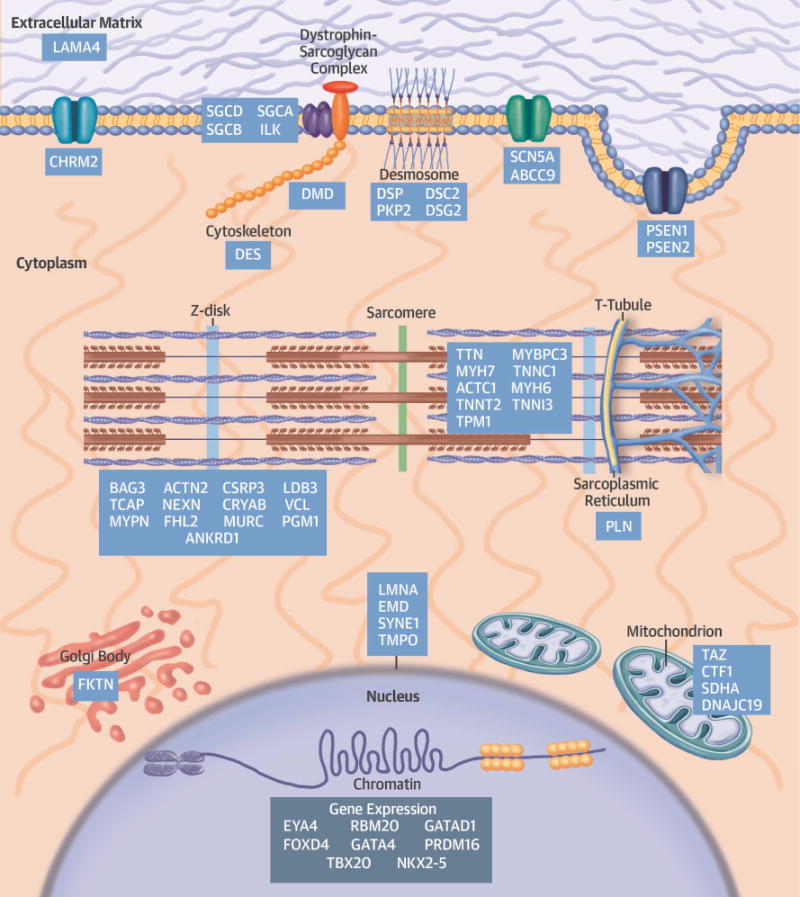
Pathogenic genes encode proteins that participate in many diverse biological processes of cardiomyocytes. DCM = dilated cardiomyopathy.
Selected DCM genes and functional consequences
Mutations affecting sarcomere function
Mutations in TTN, which encodes titin, a massive sarcomere protein, are the most prevalent genetic cause of DCM, accounting for ~15% to 20% of all DCM cases, including those with ambulatory and end-stage disease (42,86–88). Mutations that truncate titin (nonsense, frameshift, or splice-site and copy-number variants) cosegregate with familial DCM, providing overwhelming statistical evidence for pathogenicity (combined logarithm of odds [LOD] score >11), and display nearly complete penetrance after 40 years of age (42). As clinical outcomes, including arrhythmic risk and progressive functional deficits, are worse in DCM patients with TTN truncating mutations than in patients with unknown etiologies, genetic diagnosis is expected to improve risk stratification and interventions (88).
Titin contains 4 major protein domains (Z-disk, I-band, A-band, M-band) that span half the length of the sarcomere (Figure 3A). There is considerable variation in the usage of exons that encode the I-band domains, whereas most other domains incorporate all exons (89). As a consequence, truncating mutations within I-band exons, if excluded from the mature transcript, will not cause disease, thereby accounting for the fact that ~3% of the general population with an I-band mutation do not have DCM (Figures 3B and 3C) (88). Genetic testing laboratories need to interpret TTN variants in light of exon usage (90) to appropriately discriminate between pathogenic and benign variants.
FIGURE 3. Titin Mutations in DCM.
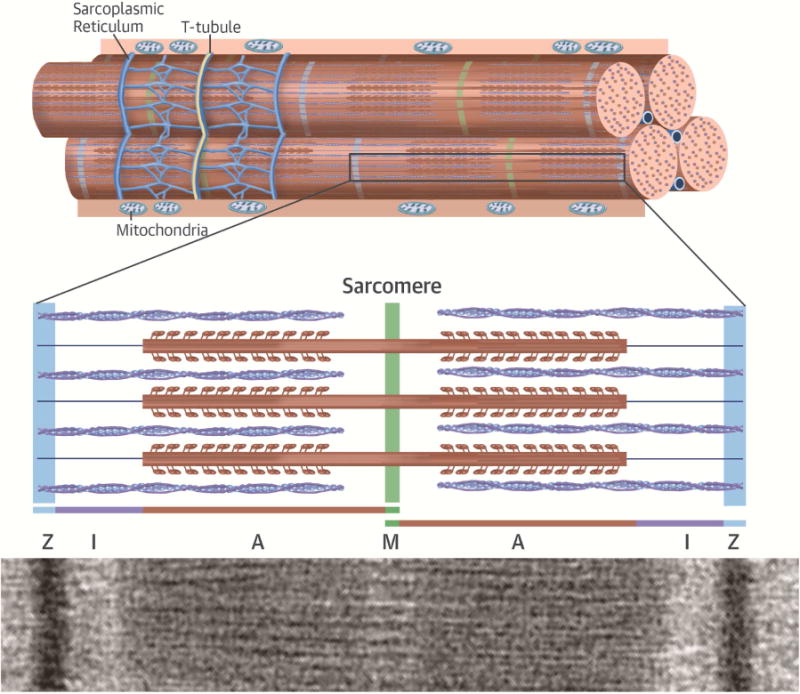

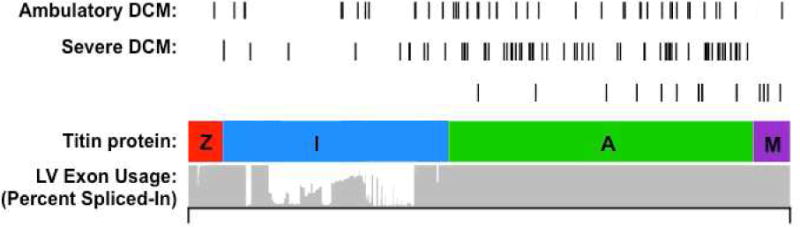
(A) Schematic representation and electron micrograph demonstrating the location of titin protein in the sarcomere. Titin molecules are composed of 4 protein domains, including the Z-disc (Z), I-band, A-band, and M-band, that span half the length of the sarcomere. (B) Bar graph demonstrating the percentage of identified TTN truncating variants in cohorts with a range of cardiovascular physiology. (C) Schematic demonstrating the location of TTN truncating variants with respect to protein domain. The percent spliced-in is a measure of exon usage in TTN messenger RNA from different cardiac tissue samples and demonstrates reduced exon usage in the I-band, which would exclude the expression of most mutations located in this domain. In contrast, mutations causing DCM reside in fully expressed exons. Adapted from Roberts et al. (88). DCM = dilated cardiomyopathy; OR = odds ratio.
TTN sequence analyses have also revealed innumerable missense variants. Indeed, the Exome Sequencing Project identified on average 23 TTN missense variants per individual without cardiomyopathy (87), including approximately 1 rare TTN missense mutation (42). Functional analyses of cardiomyocytes derived from induced pluripotent human stem cells with a TTN missense mutation have demonstrated that at least some missense mutations can cause contractile dysfunction (89). Although the clinical significance of TTN missense variants remains unclear at present, this is an area of active investigation, and we expect at least some such mutations will be shown to cause disease, thereby further improving the clinical interpretation of these genetic variants.
RBM20 encodes RNA binding motif 20 protein, a chaperone protein that binds mRNA and regulates exon splicing to produce different gene isoforms. Among several cardiac targets, RBM20 regulates splicing of titin (91). An RBM20 deletion in rats alters titin splicing and produces a DCM phenotype (92), providing strong evidence that both RBM20 mutations and TTN abnormalities can cause cardiomyopathy. Multiple human RBM20 mutations have now been identified, including a mutational hot spot in exon 9 (93–96). Recurrent missense mutations in this exon alter an arginine-serine-rich region, which disrupts binding with other splicing factors and alters transcript processing, causing highly penetrant DCM (91).
Mutations affecting electrolyte homeostasis
Phospholamban mutations cause DCM by altering calcium homeostasis. Encoded by PLN, phospholamban regulates calcium uptake by the sarcoplasmic/endoplasmic reticulum calcium transporting ATPase (SERCA2a), functioning as a molecular brake on calcium cycling. Multiple distinct human mutations have been identified and confirmed with animal models (97–99). The Arg9Cys mutation in PLN is particularly severe, having been consistently identified with progressive HF requiring heart transplantation in early adult life in unrelated families (97,100,101). This mutation blunts β-adrenergic control of calcium cycling secondary to the reduced ability of protein kinase A to phosphorylate PLN (97,102,103). In turn, altered calcium kinetics leads to depressed contractility and DCM.
A single amino acid deletion in phospholamban (Arg14del) has been reported in association with mild and severe DCM phenotypes, perhaps indicating this as a genetic modifier rather than a pathogenic mutation (99,104). Consistent with this concept, PLN Arg14del has also been identified as a founder mutation that is >500 years of age, and which constitutes 15% of index DCM cases in the Netherlands (105). Interestingly, this mutation also occurs in 12% of desmosome mutation-negative ARVC cases, all of which had a high burden of ventricular arrhythmias (105). The substantial clinical and pathological overlap observed in these subjects supports the growing finding that at least some forms of ARVC and DCM are not distinct clinical entities, but rather may constitute a condition sometimes labeled arrhythmogenic cardiomyopathy.
Some mutations in SCN5A, which encodes the sarcolemmal transmembrane cardiac voltage-gated sodium channel that functions in developing cardiac action potentials, are implicated in DCM (106–108). SCN5A mutations also cause a high burden of arrhythmias, and constitute another example of arrhythmogenic cardiomyopathy (109). There are also many allelic variants in SCN5A, including those causing Brugada syndrome (110), idiopathic ventricular fibrillation (111), and familial atrial fibrillation (112).
Mutations affecting nonsarcomere structural proteins
Multiple DCM genes cause both heart and skeletal muscle phenotypes (Online Table 3), including LMNA, which encodes lamin A/C, a ubiquitously expressed inner nuclear membrane protein that plays a role in maintenance of proper nuclear structure. Dominant LMNA mutations occur in approximately 6% of DCM cases, and are far more common in DCM with conduction system disease (113). Electrophysiological abnormalities (conduction system block and atrial fibrillation) often precede DCM that relentlessly progresses to HF (114,115). The severity of the associated skeletal myopathy is variable. Most LMNA mutations cause haploinsufficiency, and mouse models of these mutations demonstrate inadequate response to mechanical strain, which may promote premature cardiomyocyte death (116).
DCM genotype-phenotype correlations
The marked genetic heterogeneity of DCM and variable penetrance of mutations has impeded the direct application of most genotypes to clinical management. A notable exception is LMNA mutations, which are highly predictive for progressive conduction disease and risk of sudden cardiac death (114,117,118). As initial manifestations of LMNA mutations are often subtle (e.g., first-degree atrioventricular block), clinical recognition of affected individuals at high risk for sudden death is difficult. This has prompted recommendations to restrict mutation carriers of any age from participation in competitive sports (117). Assessment for prophylactic implantable cardioverter-defibrillator (ICD) placement to treat malignant ventricular arrhythmias should be considered for patients receiving a pacemaker for LMNA-associated conduction system disease, independent of ejection fraction (119,120). Likewise, atrial lead placement should be considered in LMNA+ individuals receiving an ICD who do not otherwise meet criteria for dual-chamber pacemaker implantation.
OTHER GENETIC CARDIOMYOPATHIES
Mutations in additional genes have been identified as causing other cardiomyopathies, including LVNC (Online Table 4) (121,122), ARVC (Online Table 5) (123), mitochondrial cardiomyopathies (124), and the restrictive desminopathies (125). The interested reader is referred to the suggested reviews for further details.
GENETIC TESTING IN CLINICAL PRACTICE
Defining the precise genetic cause of a cardiomyopathy in patients can improve clinical management. The identification of a pathogenic mutation provides diagnostic certainty and eliminates ambiguities associated with phenotypic variation. Genetic data may also help to guide the use of emerging therapies that target the biophysical consequences associated with mutations (47). Genetic diagnosis enables cost-effective screening of first-degree family members and eliminates healthcare expenditures for relatives without pathogenic mutations, resulting in substantial health care cost savings (35).
Cardiomyopathy gene panels continue to evolve, but increasingly include comprehensive analyses of all genes implicated in HCM, DCM, ARVC, or LVNC. Commercially available genetic testing for cardiomyopathies (and other disorders) can be found at the GeneTests website (126). This site details available gene panels and performing laboratories with contact information and costs. Additional genetic testing information is found in the National Institutes of Health Genetic Testing Registry (127). Genetic testing costs vary considerably between commercial and academic laboratories, technology used, and numbers of genes analyzed. In the United States, patients are initially studied using large multigene cardiomyopathy panels that are sequenced by next-generation technologies (at commercial costs from $1,500 to $5,000). If negative, whole-exome or whole-genome sequencing analyses are considered. With the continued fall in technical and analysis costs, these broad-based platforms may become the preferred strategy for genetic testing of cardiomyopathies. The identification of a definitive mutation allows targeted analysis of first-degree relatives at substantially reduced costs (typically only a few hundred dollars).
INTERPRETING GENETIC TESTING
Genetic testing can yield 5 distinct results (Table 2)(4): 1) Identification of a definitively pathogenic mutation. This outcome confirms the diagnosis, establishes etiology, and identifies a target for familial screening; 2) Identification of a probable pathogenic mutation. This potentially useful result supports the clinical diagnosis. But additional evidence, such as familial cosegregation or confirmatory data in unrelated affected patients, is necessary to establish causality; 3). Identification of a variant of unknown significance (VUS). This result does not distinguish whether a variant is causative or represents a rare polymorphism unrelated to the disease. Familial segregation of a VUS can support research aimed at identifying new disease genes or can be used to characterize a newly identified variant as likely pathogenic; 4) Identification of benign variants. This result identifies a polymorphism that is not disease-associated and occurs in the general population; 5) No variant is identified. Scenarios 4 and 5 can indicate that an unknown gene, or a gene not included in the analyses causes the disease, or that the condition is not genetic. In these instances, continued clinical surveillance, rather than genetic testing of first-degree relatives, is necessary.
Increasingly, genetic testing reports provide an assessment of the pathogenicity of all identified variants. A clinician trained in medical genetics or a clinical genetic counselor should always review the interpretation of the results (128). Clinical genetics is a rapidly advancing field, and proper communication of genetic testing results requires knowledge of advances in testing, emerging data on newly identified variants and clinical correlations, and an understanding of the complex allelic heterogeneity that characterizes inherited cardiomyopathies. Complicated scenarios can arise, including the discovery of multiple variants that may influence disease. Furthermore, given the age of onset for many genetic cardiomyopathies, a major point of discussion is reproductive counseling. The limits of cardiovascular disease training, the rapidly advancing field of clinical genetics, and the constraints on physician time often require the involvement of a clinical genetic counselor to fully communicate these results.
The Genotype-Positive Phenotype-Negative Individual
Genetic testing of cardiomyopathies has uncovered a new class of patients: mutation-positive/phenotype-negative individuals. Often these individuals are younger than the typical age at which clinical disease is recognized. This group poses many uncertainties, including the periodicity for serial clinical evaluations, whether participation or exclusion from intensive athletics is warranted, and whether this status and family history of sudden cardiac death warrants prophylactic interventions. The complexities of managing genotype-positive phenotype-negative individuals reflect: 1) a paucity of outcome data; 2) incomplete knowledge about the molecular mechanisms that link gene mutation and disease; and 3) variable disease penetrance, which may indicate uncharacterized compensatory mechanisms that modify disease. Although consensus documents provide opinions from experts in genetic cardiomyopathies, this is an area of active research that may influence recommendations (Central Illustration) (10,129–131).
CENTRAL ILLUSTRATION. Genetics of Inherited Cardiomyopathies.
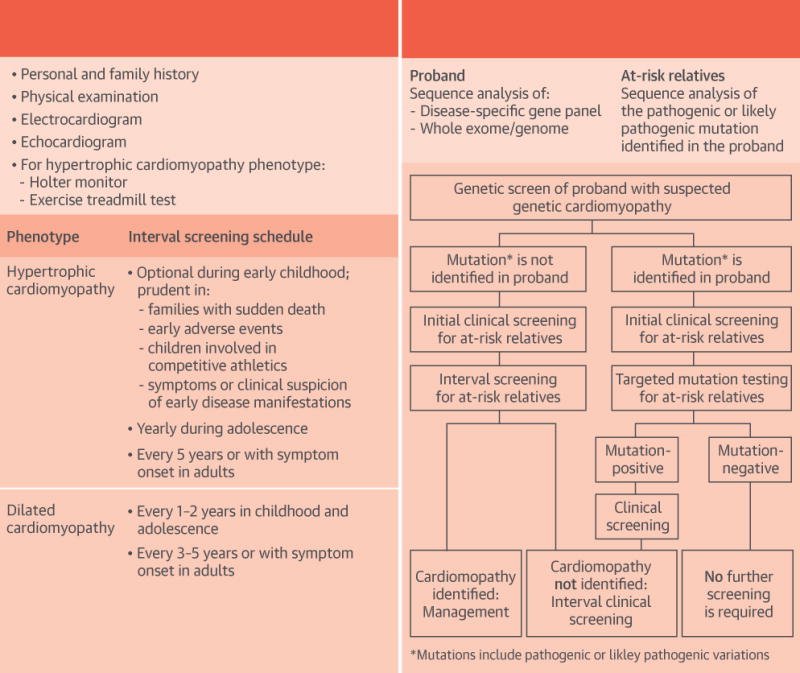
Clinical (A) or cascade genetic (B) screening strategies for familial cardiomyopathies. Initial clinical and genetic screening, as well as interval follow-up, is recommended as per guidelines (10,143,144).
Longitudinal study of genotype-positive/phenotype-negative individuals provides the unique opportunity to study the natural history of disease, including phenotype conversion. Recent advances in the development of human cardiomyocytes derived from induced pluripotent stem cells with cardiomyopathy mutations is expected to provide a more comprehensive understanding of critical molecular signals that may not be recapitulated by mouse models (132). The study of human subjects, coupled with animal and cell models, should help elucidate the roles of genetic modifiers and epigenetic changes that likely contribute to the variability in phenotype (133).
Detailed analyses of genotype-positive/phenotype-negative subjects have also identified functional abnormalities that precede cardiac remodeling (134–136). These subtle manifestations raise a critical question: can development of cardiomyopathy be delayed or prevented by treatment of those with preclinical disease? Disruption of key molecular pathways in animal models of HCM before disease onset has been shown to blunt and even reverse hypertrophic remodeling (137,138). Although results from small clinical trials have been promising (139,140), the recently published INHERIT trial failed to show a reduction in LVH with losartan treatment in HCM subjects with established disease (141), suggesting that strategies which capitalize on genetic testing to identify early disease in mutation-positive patients may be warranted. The currently enrolling VANISH trial (NCT01912534) seeks to assess whether treatment with valsartan early in HCM can slow disease progression.
SUMMARY AND RECOMMENDATIONS
The use of clinical genetic testing is rapidly evolving in conjunction with advances in sequencing technology, new discoveries about the genetic basis of disease, and reduced costs, leading to more widespread coverage by medical insurance. As such, the number of cardiomyopathy patients who should have genetic testing is rapidly growing. Although the decision to perform genetic testing should be individualized, when applied appropriately, genetic testing assists with diagnosis and guides the clinical management of the patient and family members (Central Illustration).
Genetic testing can remove the ambiguity of clinical diagnosis (i.e., mild ventricular remodeling in a competitive athlete), and influence clinical screening and management of family members. This is particularly important with children and adolescents who may not yet have manifested clinical features of disease, but who may be at risk for sudden death. Contemporary guidelines recommend targeted genetic testing within families with a definitive pathogenic mutation. Genotype-positive/phenotype-negative individuals require continued clinical screening (Central Illustration), whereas relatives without the familial mutation can be reassured and require no further screening. Although first-degree relatives with overt disease may not directly benefit from genetic testing, such results can inform the clinical interpretation of variants with less certain pathogenicity. For the individual with a definitive clinical diagnosis of cardiomyopathy and no relatives at risk for disease, genetic testing can still contribute to increasing knowledge of how genotype influences phenotype and outcomes.
FUTURE DIRECTIONS
The last quarter-century has provided remarkable progress in cardiomyopathy genetics, but there is much yet to learn. Continued application of technical advances in genomic sequencing is expected to further unlock the genetic basis of these enigmatic disorders. In addition to identifying new mutations that alter protein sequences, these technologies can identify structural genomic changes and variations in regulatory elements that can alter gene dosage without affecting protein sequences. Additional research will improve our understanding of the complex and longitudinal molecular changes that lead from gene mutation to clinical expression.
Future strategies may directly target the mutant allele itself. Preclinical assessment of this approach in an HCM mouse model successfully employed adenoviral constructs to silence the mutant, but not the wild type allele, resulting in complete abrogation of the disease phenotype for many months (142). Further development of similar approaches raises the possibility that someday genetic cardiomyopathies will not only be treated, but also cured.
Supplementary Material
Acknowledgments
This work was supported by: the Leducq Foundation Transatlantic Networks of Excellence (to Mr. Cook and Drs. J.G. and C.E. Seidman; the John S. LaDue Memorial Fellowship at Harvard Medical School, by the National Institutes of Health (Clinical Skills Development Core Training grant NHLBI #U10HL110337 to Dr. Burke; and grants NHLBI HL080494 and HL084553 to Drs. J.G. and C.E. Seidman; U01-HG006500: Seidman); and by the Howard Hughes Medical Institutes to Dr. C.E. Seidman.
ABBREVIATIONS AND ACRONYMS
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- HF
heart failure
- LVH
left ventricular hypertrophy
- LVNC
left ventricular noncompaction
- RCM
restrictive cardiomyopathy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: Drs. J.G. and C.E. Seidman are founders of and own shares in Myokardia, Inc., a startup company developing therapeutics that target the sarcomere. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
References
- 1.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation. 2006;113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 2.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–6. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 3.Jarcho JA, McKenna W, Pare JA, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–8. doi: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 4.Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants : a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lek Monkol, Karczewski Konrad, Minikel Eric, et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh R, Thomson KL, Ware JS, et al. Exome Aggregation Consortium Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2016 Aug 17; doi: 10.1038/gim.2016.90. [E-pub ahead of print], http://dx.doi.org/10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed]
- 7.Norton N, Robertson PD, Rieder MJ, et al. National Heart, Lung and Blood Institute GO Exome Sequencing Project Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet. 2012;5:167–74. doi: 10.1161/CIRCGENETICS.111.961805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan S, Caleshu CA, Dunn KE, et al. Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation. Circ Cardiovasc Genet. 2012;5:602–10. doi: 10.1161/CIRCGENETICS.112.963421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watkins H, Thierfelder L, Anan R, et al. Independent origin of identical beta cardiac myosin heavy-chain mutations in hypertrophic cardiomyopathy. Am J Hum Genet. 1993;53:1180–5. [PMC free article] [PubMed] [Google Scholar]
- 10.Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58:e212–e260. doi: 10.1016/j.jacc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 11.Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation. 1995;92:1680–92. doi: 10.1161/01.cir.92.7.1680. [DOI] [PubMed] [Google Scholar]
- 12.Nagueh SF, Mahmarian JJ. Noninvasive cardiac imaging in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2006;48:2410–22. doi: 10.1016/j.jacc.2006.07.065. [DOI] [PubMed] [Google Scholar]
- 13.Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Circulation. 1995;92:785–9. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 14.Zou Y, Song L, Wang Z, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med. 2004;116:14–8. doi: 10.1016/j.amjmed.2003.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Morita H, Larson MG, Barr SC, et al. Single-gene mutations and increased left ventricular wall thickness in the community: the Framingham Heart Study. Circulation. 2006;113:2697–705. doi: 10.1161/CIRCULATIONAHA.105.593558. [DOI] [PubMed] [Google Scholar]
- 16.Bick AG, Flannick J, Ito K, et al. Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am J Hum Genet. 2012;91:513–9. doi: 10.1016/j.ajhg.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Golbus JR, Puckelwartz MJ, Fahrenbach JP, et al. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–9. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisterfer-Lowrance AAT, Kass S, Tanigawa G, et al. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 19.Tanigawa G, Jarcho JA, Kass S, et al. A molecular basis for familial hypertrophic cardiomyopathy: an α/β cardiac myosin heavy chain hybrid gene. Cell. 1990;62:991–8. doi: 10.1016/0092-8674(90)90273-h. [DOI] [PubMed] [Google Scholar]
- 20.Thierfelder L, Watkins H, MacRae C, et al. α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–12. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 21.Watkins H, McKenna WJ, Thierfelder L, et al. Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–64. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 22.Bonne G, Carrier L, Bercovici J, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–40. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 23.Watkins H, Conner D, Thierfelder L, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–7. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 24.Poetter K, Jiang H, Hassanzadeh S, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–9. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 25.Kimura A, Harada H, Park JE, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–82. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 26.Mogensen J, Klausen IC, Pedersen AK, et al. α-Cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–43. doi: 10.1172/JCI6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olson TM, Doan TP, Kishimoto NY, et al. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–94. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- 28.Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy. Circulation. 2003;107:2227–32. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 29.Andersen PS, Havndrup O, Hougs L, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–70. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 30.Millat G, Bouvagnet P, Chevalier P, et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet. 2010;53:261–7. doi: 10.1016/j.ejmg.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Curila K, Benesova L, Penicka M, et al. Spectrum and clinical manifestations of mutations in genes responsible for hypertrophic cardiomyopathy. Acta Cardiol. 2012;67:23–9. doi: 10.1080/ac.67.1.2146562. [DOI] [PubMed] [Google Scholar]
- 32.Brito D, Miltenberger-Miltenyi G, Vale Pereira S, et al. Sarcomeric hypertrophic cardiomyopathy: genetic profile in a Portuguese population. Rev Port Cardiol. 2012;31:577–87. doi: 10.1016/j.repc.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 33.Liu W, Liu W, Hu D, et al. Mutation spectrum in a large cohort of unrelated Chinese patients with hypertrophic cardiomyopathy. Am J Cardiol. 2013;112:585–9. doi: 10.1016/j.amjcard.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Van Driest SL, Ommen SR, Tajik AJ, et al. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:739–44. doi: 10.1016/S0025-6196(11)61527-9. [DOI] [PubMed] [Google Scholar]
- 35.Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity [Published correction appears in Genet Med 2015;17:319] Genet Med. 2015;17:880–8. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 36.Solomon SD, Wolff S, Watkins H, et al. Left ventricular hypertrophy and morphology in familial hypertrophic cardiomyopathy associated with mutations of the beta-myosin heavy chain gene. J Am Coll Cardiol. 1993;22:498–505. doi: 10.1016/0735-1097(93)90055-6. [DOI] [PubMed] [Google Scholar]
- 37.Binder J, Ommen SR, Gersh BJ, et al. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc. 2006;81:459–67. doi: 10.4065/81.4.459. [DOI] [PubMed] [Google Scholar]
- 38.Osio A, Tan L, Chen SN, et al. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100:766–8. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiu C, Bagnall RD, Ingles J, et al. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. 2010;55:1127–35. doi: 10.1016/j.jacc.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 40.Satoh M, Takahashi M, Sakamoto T, et al. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun. 1999;262:411–7. doi: 10.1006/bbrc.1999.1221. [DOI] [PubMed] [Google Scholar]
- 41.Bos JM, Poley RN, Ny M, et al. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metabl. 2006;88:78–85. doi: 10.1016/j.ymgme.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herman DS, Lam L, Taylor MRG, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knöll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–55. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 44.Geier C, Gehmlich K, Ehler E, et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet. 2008;17:2753–65. doi: 10.1093/hmg/ddn160. [DOI] [PubMed] [Google Scholar]
- 45.Geier C, Perrot A, Ozcelik C, et al. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation. 2003;107:1390–5. doi: 10.1161/01.cir.0000056522.82563.5f. [DOI] [PubMed] [Google Scholar]
- 46.Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–49. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–21. doi: 10.1126/science.aad3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–64. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marston S, Copeland ON, Jacques A, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–22. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 50.van Dijk SJ, Dooijes D, dos Remedios C, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–83. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 51.Barefield D, Kumar M, Gorham J, et al. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J Mol Cell Cardiol. 2015;79:234–43. doi: 10.1016/j.yjmcc.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyska MJ, Hayes E, Giewat M, et al. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res. 2000;86:737–44. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 53.Crilley JG, Boehm EA, Blair E, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–82. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 54.Sommese RF, Sung J, Nag S, et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc Natl Acad Sci USA. 2013;110:12607–12. doi: 10.1073/pnas.1309493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bloemink M, Deacon J, Langer S, et al. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin. J Biol Chem. 2014;289:5158–67. doi: 10.1074/jbc.M113.511204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Witjas-Paalberends ER, Güçlü A, Germans T, et al. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res. 2014;103:248–57. doi: 10.1093/cvr/cvu127. [DOI] [PubMed] [Google Scholar]
- 57.Anan R, Greve G, Thierfelder L, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest. 1994;93:280–5. doi: 10.1172/JCI116957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ko YL, Chen JJ, Tang TK, et al. Malignant familial hypertrophic cardiomyopathy in a family with a 453Arg–>Cys mutation in the beta-myosin heavy chain gene: coexistence of sudden death and end-stage heart failure. Hum Genet. 1996;97:585–90. doi: 10.1007/BF02281865. [DOI] [PubMed] [Google Scholar]
- 59.Tesson F, Richard P, Charron P, et al. Genotype-phenotype analysis in four families with mutations in β-myosin heavy chain gene responsible for familial hypertrophic cardiomyopathy. Hum Mutat. 1998;12:385–92. doi: 10.1002/(SICI)1098-1004(1998)12:6<385::AID-HUMU4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 60.Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–57. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 61.Page SP, Kounas S, Syrris P, et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet. 2012;5:156–66. doi: 10.1161/CIRCGENETICS.111.960831. [DOI] [PubMed] [Google Scholar]
- 62.Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743–50. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhandapany PS, Sadayappan S, Xue Y, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–91. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adalsteinsdottir B, Teekakirikul P, Maron BJ, et al. Nationwide study on hypertrophic cardiomyopathy in Iceland: evidence of a MYBPC3 founder mutation. Circulation. 2014;130:1158–67. doi: 10.1161/CIRCULATIONAHA.114.011207. [DOI] [PubMed] [Google Scholar]
- 65.Maron BJ, Roberts WC, Arad M, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301:1253–9. doi: 10.1001/jama.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sachdev B, Takenaka T, Teraguchi H, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407–11. doi: 10.1161/01.cir.0000012626.81324.38. [DOI] [PubMed] [Google Scholar]
- 67.Chimenti C, Pieroni M, Morgante E, et al. Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation. 2004;110:1047–53. doi: 10.1161/01.CIR.0000139847.74101.03. [DOI] [PubMed] [Google Scholar]
- 68.Monserrat L, Gimeno-Blanes JR, Marín F, et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50:2399–403. doi: 10.1016/j.jacc.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 69.Eng CM, Guffon N, Wilcox WR, et al. International Collaborative Fabry Disease Study Group Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 70.Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 71.Kampmann C, Perrin A, Beck M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years’ treatment. Orphanet J Rare Dis. 2015;10:125. doi: 10.1186/s13023-015-0338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kubo T, Gimeno JR, Bahl A, et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. 2007;49:2419–26. doi: 10.1016/j.jacc.2007.02.061. [DOI] [PubMed] [Google Scholar]
- 73.Kaski JP, Syrris P, Burch M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–84. doi: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- 74.Menon SC, Michels VV, Pellikka PA, et al. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet. 2008;74:445–54. doi: 10.1111/j.1399-0004.2008.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu W, Lu CX, Wang YN, et al. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J Am Heart Assoc. 2015;4:e001879. doi: 10.1161/JAHA.115.001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang J, Kumar A, Stalker HJ, et al. Clinical and molecular studies of a large family with desmin-associated restrictive cardiomyopathy. Clin Genet. 2001;59:248–56. doi: 10.1034/j.1399-0004.2001.590406.x. [DOI] [PubMed] [Google Scholar]
- 77.Purevjav E, Arimura T, Augustin S, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21:2039–53. doi: 10.1093/hmg/dds022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duboscq-Bidot L, Xu P, Charron P, et al. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res. 2008;77:118–25. doi: 10.1093/cvr/cvm015. [DOI] [PubMed] [Google Scholar]
- 79.Bagnall RD, Yeates L, Semsarian C. Analysis of the Z-disc genes PDLIM3 and MYPN in patients with hypertrophic cardiomyopathy. Int J Cardiol. 2010;145:601–2. doi: 10.1016/j.ijcard.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 80.Mogensen J, Kubo T, Duque M, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations [Published correction appears in J Clin Invest 2003;111:925] J Clin Invest. 2003;111:209–16. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du J, Liu J, Feng HZ, et al. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol. 2008;294:H2604–13. doi: 10.1152/ajpheart.91506.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huby AC, Mendsaikhan U, Takagi K, et al. Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J Am Coll Cardiol. 2014;64:2765–76. doi: 10.1016/j.jacc.2014.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kasper EK, Agema WR, Hutchins GM, et al. The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll Cardiol. 1994;23:586–90. doi: 10.1016/0735-1097(94)90740-4. [DOI] [PubMed] [Google Scholar]
- 84.Codd MB, Sugrue DD, Gersh BJ, et al. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80:564–72. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 85.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–47. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 86.Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30:201–4. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 87.Norton N, Li D, Rampersaud E, et al. National Heart, Lung, and Blood Institute GO Exome Sequencing Project and the Exome Sequencing Project Family Studies Project Team Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–53. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hinson JT, Chopra A, Nafissi N, et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349:982–6. doi: 10.1126/science.aaa5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Locus Reference Genomic. 2011 Available at: http://www.lrg-sequence.org. Accessed October 17, 2016.
- 91.Maatz H, Jens M, Liss M, et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest. 2014;124:3419–30. doi: 10.1172/JCI74523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo W, Schafer S, Greaser ML, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–73. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brauch KM, Karst ML, Herron KJ, et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–41. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li D, Morales A, Gonzalez-Quintana J, et al. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3:90–7. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Millat G, Bouvagnet P, Chevalier P, et al. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Genet. 2011;54:e570–5. doi: 10.1016/j.ejmg.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 96.Wells QS, Becker JR, Su YR, et al. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:317–26. doi: 10.1161/CIRCGENETICS.113.000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–3. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 98.Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–76. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haghighi K, Kolokathis F, Gramolini AO, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:1388–93. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Truszkowska GT, Bilińska ZT, Kosińska J, et al. A study in Polish patients with cardiomyopathy emphasizes pathogenicity of phospholamban (PLN) mutations at amino acid position 9 and low penetrance of heterozygous null PLN mutations. BMC Med Genet. 2015;16:21. doi: 10.1186/s12881-015-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fish M, Shaboodien G, Kraus S, et al. Mutation analysis of the phospholamban gene in 315 South Africans with dilated, hypertrophic, peripartum and arrhythmogenic right ventricular cardiomyopathies. Sci Rep. 2016;6:22235. doi: 10.1038/srep22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ha KN, Masterson LR, Hou Z, et al. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc Natl Acad Sci U S A. 2011;108:2735–40. doi: 10.1073/pnas.1013987108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Abrol N, de Tombe PP, Robia SL. Acute inotropic and lusitropic effects of cardiomyopathic R9C mutation of phospholamban. J Biol Chem. 2015;290:7130–40. doi: 10.1074/jbc.M114.630319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DeWitt MM, MacLeod HM, Soliven B, et al. Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1396–8. doi: 10.1016/j.jacc.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 105.van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–207. doi: 10.1093/eurjhf/hfs119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Olson TM, Keating MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest. 1996;97:528–32. doi: 10.1172/JCI118445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McNair WP, Ku L, Taylor MRG, et al. Familial Cardiomyopathy Registry Research Group SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–7. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 108.Olson TM, Michels VV, Ballew JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–54. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McNair WP, Sinagra G, Taylor MRG, et al. Familial Cardiomyopathy Registry Research Group SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011;57:2160–8. doi: 10.1016/j.jacc.2010.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 111.Watanabe H, Nogami A, Ohkubo K, et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4:874–81. doi: 10.1161/CIRCEP.111.963983. [DOI] [PubMed] [Google Scholar]
- 112.Ellinor PT, Nam EG, Shea MA, et al. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 2008;5:99–105. doi: 10.1016/j.hrthm.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 113.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–24. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 115.Taylor MRG, Fain PR, Sinagra G, et al. Familial Dilated Cardiomyopathy Registry Research Group Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–80. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 116.Nikolova V, Leimena C, McMahon AC, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–69. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–60. doi: 10.1016/j.jacc.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 118.van Rijsingen IAW, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol. 2012;59:493–500. doi: 10.1016/j.jacc.2011.08.078. [DOI] [PubMed] [Google Scholar]
- 119.Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–10. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- 120.Anselme F, Moubarak G, Savouré A, et al. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm. 2013;10:1492–8. doi: 10.1016/j.hrthm.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 121.Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol. 2014;64:1840–50. doi: 10.1016/j.jacc.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hussein A, Karimianpour A, Collier P, et al. Isolated noncompaction of the left ventricle in adults. J Am Coll Cardiol. 2015;66:578–85. doi: 10.1016/j.jacc.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 123.Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 2013;61:1945–8. doi: 10.1016/j.jacc.2013.01.073. [DOI] [PubMed] [Google Scholar]
- 124.Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J. 2013;40:385–94. [PMC free article] [PubMed] [Google Scholar]
- 125.Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24:214–20. doi: 10.1097/hco.0b013e32832a1d2e. [DOI] [PubMed] [Google Scholar]
- 126.GeneTests. 2016. Available at: https://www.genetests.org. Accessed October 17, 2016.
- 127.GTR: Genetic Testing Registry. National Institutes of Health. Available at: https://www.ncbi.nlm.nih.gov/gtr/.
- 128.Hershberger RE, Cowan J, Morales A, et al. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2:253–61. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Levine BD, Baggish AL, Kovacs RJ, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 1: Classification of Sports: Dynamic, Static, and Impact: A Scientific Statement From the American Heart Association and American College of Cardiology. J Am Coll Cardiol. 2015;66:2350–5. doi: 10.1016/j.jacc.2015.09.033. [DOI] [PubMed] [Google Scholar]
- 130.Maron BJ, Levine BD, Washington RL, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 2: Preparticipation Screening for Cardiovascular Disease in Competitive Athletes: A Scientific Statement From the American Heart Association and American College of Cardiology. J Am Coll Cardiol. 2015;66:2356–61. doi: 10.1016/j.jacc.2015.09.034. [DOI] [PubMed] [Google Scholar]
- 131.Maron BJ, Udelson JE, Bonow RO, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 3: Hypertrophic Cardiomyopathy, Arrhythmogenic Right Ventricular Cardiomyopathy and Other Cardiomyopathies, and Myocarditis: A Scientific Statement From the American Heart Association and American College of Cardiology. J Am Coll Cardiol. 2015;66:2362–71. doi: 10.1016/j.jacc.2015.09.035. [DOI] [PubMed] [Google Scholar]
- 132.Karakikes I, Termglinchan V, Wu JC. Human-induced pluripotent stem cell models of inherited cardiomyopathies. Curr Opin Cardiol. 2014;29:214–9. doi: 10.1097/HCO.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dorn GW, II, McNally EM. Two strikes and you’re out: gene-gene mutation interactions in HCM. Circ Res. 2014;115:208–10. doi: 10.1161/CIRCRESAHA.114.304383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ho CY, López B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363:552–63. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lakdawala NK, Thune JJ, Colan SD, et al. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ Cardiovasc Genet. 2012;5:503–10. doi: 10.1161/CIRCGENETICS.112.962761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Valente AM, Lakdawala NK, Powell AJ, et al. Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet. 2013;6:230–7. doi: 10.1161/CIRCGENETICS.113.000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lim DS, Lutucuta S, Bachireddy P, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–91. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Teekakirikul P, Eminaga S, Toka O, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J Clin Invest. 2010;120:3520–9. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shimada YJ, Passeri JJ, Baggish AL, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol HF. 2013;1:480–7. doi: 10.1016/j.jchf.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ho CY, Lakdawala NK, Cirino AL, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. J Am Coll Cardiol HF. 2015;3:180–8. doi: 10.1016/j.jchf.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Axelsson A, Iversen K, Vejlstrup N, et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015;3:123–31. doi: 10.1016/S2213-8587(14)70241-4. [DOI] [PubMed] [Google Scholar]
- 142.Jiang J, Wakimoto H, Seidman JG, et al. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–4. doi: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hershberger RE, Lindenfeld J, Mestroni L, et al. Genetic evaluation of cardiomyopathy—A Heart Failure Society of America Practice Guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 144.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace. 2011;13:1077–109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.