

Abstract
Mononuclear nonheme iron(V)-oxo complexes have been reported previously. Herein, we report the first example of a mononuclear nonheme iron(V)-imido complex bearing a tetraamido macrocyclic ligand (TAML), [(TAML)FeV(NTs)]− (1). The spectroscopic characterization of 1 revealed an S = 1/2 Fe(V) oxidation state, an Fe—N bond length of 1.65(4) Å, and an Fe—N vibration at 817 cm−1. The reactivity of 1 was demonstrated in C—H bond functionalization and nitrene transfer reactions.
High-valent iron-imido (Fen+═NR) species, which are iron-oxo (Fen+═O) analogs,1 have been proposed as reactive intermediates in aziridination and amination reactions.2,3 A number of synthetic iron-imido complexes have been isolated and characterized structurally in heme and nonheme iron systems.4 Recently, a mononuclear nonheme iron(IV)-imido complex bearing a pentadentate N4Py ligand, [(N4Py)-FeIV(NTs)]2+ (N4Py = (N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)-methylamine; NTs = tosylimido), which is an iron(IV)-oxo analog (i.e., [(N4Py)FeIV(O)]2+),5 was successfully synthesized and investigated in the C—H bond activation and nitrene transfer reactions.6 However, to the best of our knowledge, high-valent iron(V)-imido complexes have not been reported in nonheme iron models. Encouraged by the previous report of the iron(V)-oxo complex bearing a tetraamido macrocyclic ligand (TAML), [(TAML)FeV(O)]−,7 we attempted to synthesize an iron(V)-imido complex using the TAML supporting ligand. Herein, we report the first example of a mononuclear nonheme iron(V)-imido complex, [(TAML)-FeV(NTs)]− (1) (Scheme 1).
Scheme 1.
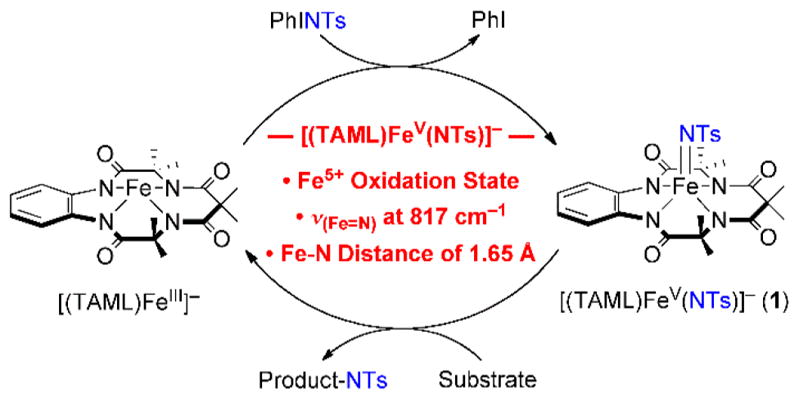
Schematic Representation Showing the Synthesis, Characterization, and Reactivity of [(TAML)FeV(NTs)]−
Addition of solid N-tosyliminophenyliodinane (PhINTs; 3 equiv) to the CH3CN solution of Na[(TAML)FeIII] at 15 °C afforded an immediate color change from orange to dark green (see Experimental Section in Supporting Information (SI) for detailed synthetic procedures). The intermediate, denoted as 1, was relatively stable at 25 °C (t1/2 ~ 12 min), allowing us to characterize it with various spectroscopic methods. The UV–vis spectrum of 1 showed two characteristic absorption bands at λmax = 595 nm (ε = 5200 M−1 cm−1) and 915 nm (ε = 2200 M−1 cm−1) (Figure 1a). Cold-spray ionization time-of-flight mass spectrum (CSI-MS) of 1 in negative mode exhibited a prominent ion peak at m/z of 595.1, whose mass and isotopic distribution patterns correspond to [(TAML)Fe(NTs)]−(calculated m/z of 595.1) (Figure 1a, inset; Figure S1). A 15N-labeling experiment performed with PhI15NTs resulted in one mass unit shift from m/z 595.1 to 596.1 (Figure 1a, inset), indicating that 1 contains an NTs group. X-band EPR spectra of 1 displayed an isotropic signal at g = 2.00 in CH3CN and the anisotropic signals with gxx, = 2.04, gyy = 2.00, and gzz = 1.96 in butyronitrile (Figure S2), indicating a low-spin (S = 1/2) state of an iron(V) ion, as reported in the iron(V)-oxo analog, [(TAML)FeV(O)]−.7
Figure 1.
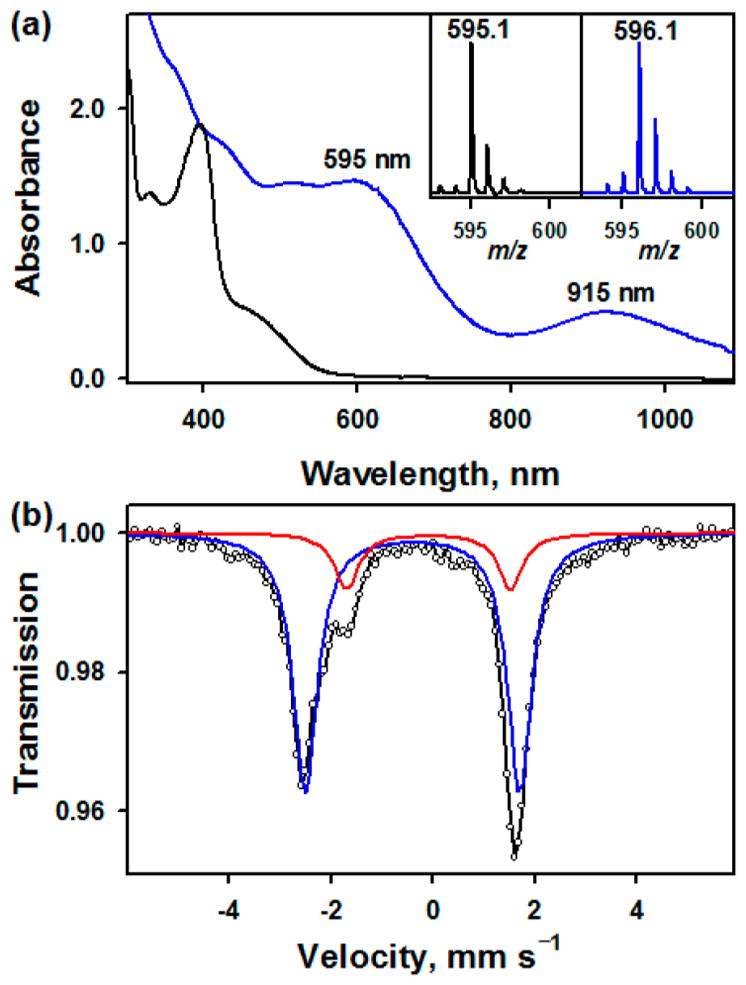
(a) UV–vis spectra of [FeIII(TAML)]− (0.25 mM; black line) and 1 (blue line) produced by reacting [FeIII(TAML)]− (0.25 mM) with solid PhINTs (3 equiv) in CH3CN at 15 °C. Insets show CSI-MS spectra with the isotopic distribution patterns of the peaks at m/z of 595.1 for 1—14NTs (left panel) and m/z of 596.1 for 1—15NTs (right panel). (b) Mössbauer spectrum (black dotted line) for 1 recorded at 6 K and 0 T. The black solid line is a superposition of the simulated 1 (85%, blue line) and the decay product of 1 (15%, red line) (see Figures S4 and S5).
The resonance Raman (rR) spectrum of 1, upon 441.6 nm-excitation in CH3CN at −40 °C, exhibited an isotopically sensitive doublet feature centered at 817 cm−1 (Figure S3), which shifted to a new doublet centered at 796 cm−1 upon 15N-labeling of 1 (Figure S3; see the detailed discussion in Supplementary Results and Analysis in SI). This isotopic shift of 21 cm−1 is in a good agreement with the calculated value of 22 cm−1 for a diatomic Fe—N oscillator estimated by Hooke’s Law and the DFT-calculated value of 20 cm−1 (Table S1). The rR data indicate that the iron and nitrogen bond in 1 possesses double bond character (vide infra), as shown in mononuclear nonheme iron(IV)-oxo complexes with ν(Fe═O) vibrations in the rage of ~810–840 cm−1.8 It is worth noting that iron-imido complexes with a pseudotetrahedral geometry and Cs symmetry show Fe—N stretching vibrations at >1000 cm−1 with short Fe—N bond distances (~1.63 Å) and nearly linear Fe—N—R angles (vide infra), though it should be noted that these geometric parameters are dependent on the electronic structure and spin state of the complexes in addition to their coordination number.4,9,10
Mössbauer data were collected on three 57Fe samples of 1 at 6 K, as shown in Figures 1b and S4. Although there was a degree of variability to these samples due to instability, the three data sets could be simultaneously fit to variable amounts of the same two species with parameters δ = −0.40 mm/s and ΔEQ = 3.83 mm/s and δ = −0.09 mm/s and ΔEQ = 3.23 mm/s, respectively; those are consistent with the XAS data on parallel samples (vide infra). The parameters for the latter component, which comprised 15–30% of the Mössbauer spectrum, are identical to those measured for the decay product of 1 (Figure S5). The parameters for the former component, accounting for 70–85% of the spectrum, are very similar to those previously reported for [(TAML)FeV(O)]− (e.g., δ = −0.42 mm/s and ΔEQ = 4.25 mm/s),7a leading us to assign the formal oxidation state of +5 for the iron ion in 1.
X-ray absorption spectroscopy (XAS) data were also collected on these three different 57Fe samples having different amounts of 1 and its decay product (Figures 2, S6–S10, and Table S2). The normalized X-ray absorption near-edge structure (XANES) at the Fe K-edge of 1 displayed an edge at a higher energy than the [FeIII(TAML)]− reference edge and comparable to the [(TAML)FeV(O)]− edge (Figure 2a),7a indicating a formal +5 oxidation state of the iron ion as determined from Mössbauer. The pre-edge energy of 1 (7113.2 eV) is also higher than the pre-edge energy of the [FeIII(TAML)]− reference (7112.1 eV) and the FeIV decay product (7112.9 eV) and similar to the pre-edge energy of [(TAML)FeV(O)]− (7113.3 eV),7a consistent with an FeV (Figure 2a, inset). Fits to the pre-edge feature at 7113.2 eV of 1 found a total pre-edge intensity of 51 units, which is normalized to 52 units to account for the 15–30% decay product in the sample, similar to [(TAML)FeV(O)]− (48 units, normalized to 60 units after accounting for 30% decay; see Figure S7).7a The normalization procedure is explained in the SI. The lower pre-edge intensity for 1 vs [(TAML)FeV(O)]− reflects its weaker axial bonding (vide infra).
Figure 2.
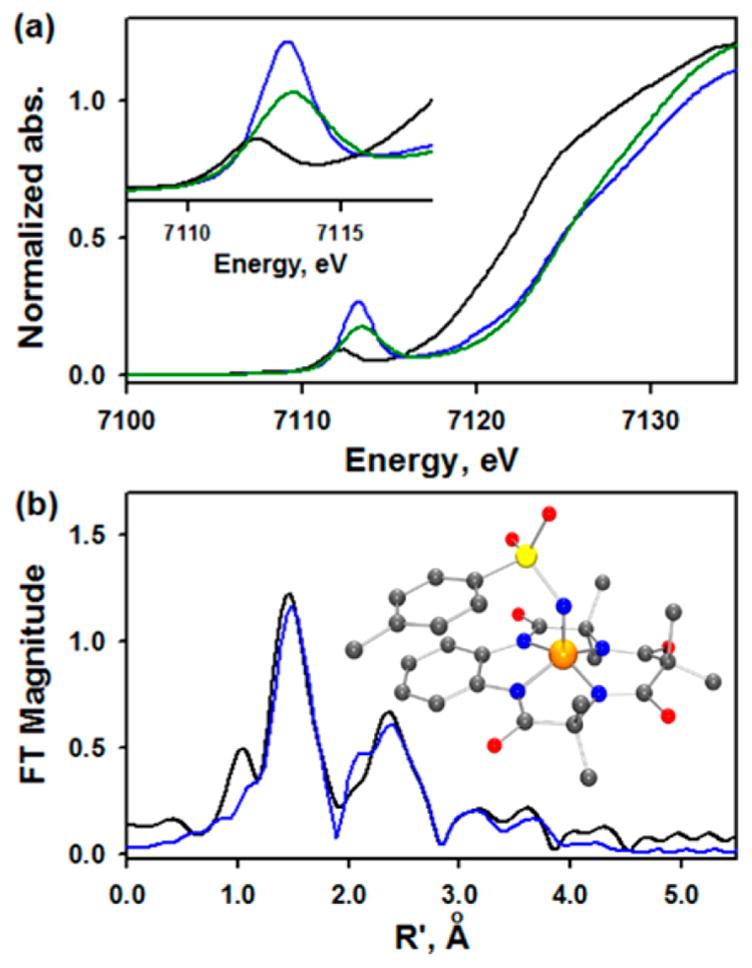
(a) XAS data of 1 (pre-edge = 51 units) (blue line), [FeIII(TAML)]− (black line), and 70% [(TAML)FeV(O)]− (48 units, fit of this pre-edge is given in Figure S7) (green line). Inset shows the expansion of pre-edge region. (b) Fourier transform of 1 (black line) with fits (blue line). Raw EXAFS data are in Figure S10. The EXAFS data were fit with 30% FeIV decay, consistent with parallel Mössbauer data. Inset shows the DFT-optimized structure of 1.
Direct evidence for the iron-tosylimido unit is found in the analysis of the extended X-ray absorption fine structure (EXAFS) region (Figures 2b and S10). Varying the first shell at 0.70–0.85 N scatterer (determined from Mössbauer), the fits across all data sets give an Fe—N bond length of 1.65 ± 0.04 Å (Table S3). This is longer than the Fe—O bond length of 1.58 Å found for [(TAML)FeV(O)]−,7a which leads to less Fe 4p mixing in the unoccupied orbitals of 1 and decreased pre-edge intensity. Two other shells showed four N/O scatterers at 1.87 Å and eight C scatterers at 2.84 Å, consistent with low Z atoms from the TAML ligand (Table S3) and similar to the fit for [(TAML)FeV(O)]−. The fit also found an S scatterer at 2.95 Å, consistent with the tosylimido group binding to the iron. Full fit parameters for the different samples are given in Table S4.
Density functional theory (DFT) calculations were used to correlate to the above data and further define the electronic structure of 1. 1 and [(TAML)FeV(O)]− for calibration purposes were geometrically optimized on the S = 1/2 surface using the BP86 and B3LYP functionals in combination with the 6-311G+(3df) basis set on Fe and the tosylimido N and the 6-311G basis set on all other atoms.11 Further computational details can be found in the SI. Table S1 summarizes the results of these calculations and the comparison with the relevant experimental data. The BP86 calculation gives the best agreement with experiment. The electronic structure (Figure S13) associated with this calculation is a [(TAML)FeV(NTs)]−(see the Supplementary Results and Analysis in the SI for a full discussion of the electronic structure of 1 and a description of its Fe—N bonding). On the basis of the spectroscopic characterization and DFT calculations, we conclude that an iron(V)-imido complex, [(TAML)FeV(NTs)]− (1), was successfully synthesized in the reaction of [FeIII(TAML)]− and PhINTs (Scheme 1). Note that an equivalent DFT calculation using the B3LYP functional gave an FeIV-tosylimido radical with spectral features that do not agree with experiment (see SI).
The electronic structures calculated for 1 and its oxo analog7a using BP86 give insight into the nature of the Fe—N bond. The Fe—N bond of 1 is longer than that of the oxo analogue (1.65 vs 1.58 Å), consistent with its lower Mayer bond order (1.686 vs 1.950), due to one N(Ts) orbital being unavailable for bonding with Fe as it is dominantly involved in the N Ts bond (see SI). A previous study described differences in bonding between the S = 1 complexes [(N4Py)FeIV(O)]2+ and [(N4Py)FeIV(NTs)]2+, also attributed to the Ts group.6a Interestingly, in that case, going from the FeIV═O to the FeIV—NTs was found to increase the bond length by a larger amount, from 1.64 to 1.73 Å, and the Fe—N bond was described as consisting primarily of π bonding with only a weak σ bond.6a In addition, Mayer bond orders calculated for the FeIV structures reported6a decrease more in going from oxo to NTs, from 1.931 to 1.528. The six-coordinate [(N4Py)-FeIV(NTs)]2+ complex has a more linear calculated Fe—N—S angle (152°) relative to 1 (126°), due to its additional ligand trans to the NTs (see the SI for details). This trans ligand interacts with the Fe dz2 orbital, making it less available for σ type bonding with the NTs in the bent structure, leading the FeIV complex to adopt a more linear structure that has π rather than σ overlap between the Fe and NTs (Figure S18). Thus, the lack of a trans axial ligand in 1 allows a more bent Fe—N—S unit that replaces an Fe—N π bond with a stronger σ bond relative to the FeIV complex (see the SI for more details). Note that this bending is not a Jahn–Teller (J-T) distortion: in the ~C4v symmetry of 1 with the Fe—N—S angle constrained at near linear (13 kcal/mol higher in energy than the freely optimized structure), the ground state is 2E and a J-T distorting mode would have to have a symmetry that is contained in the symmetric direct product of E × E (= A1 + B1 + B2). The normal mode associated with bending of the Fe—N—S unit has E symmetry and is not J-T active.
The reactivity of 1 was investigated in C—H bond activation and nitrene transfer reactions. Addition of xanthene to a CH3CN solution of 1 resulted in the disappearance of the absorption bands at 595 and 915 nm due to 1 (Figure S19). The first-order rate constants increased linearly with the increase of xanthene concentration (Figure S20), giving a second-order rate constant (k2) of 4.7(4) M−1 s−1 at 15 °C. A kinetic isotope effect (KIE) value of 11(1) was determined in the oxidation of xanthene and xanthene-d2 by 1 (Figure S21); a KIE value of 11(1) was obtained in the oxidation of ethylbenzene by the iron(V)-oxo analog, [(TAML)FeV(O)]−.7d We also determined second-order rate constants in the oxidation of other substrates, such as 9,10-dihydroanthracene (DHA), indene, and fluorene (Figure S20 and Table S5). As shown in Figure 3a, a good linear correlation between the reaction rates and the C—H bond dissociation energies (BDEs) of substrates was obtained. On the basis of the observations of the large KIE value and the good correlation between the reaction rates and the C—H BDEs of substrates, we propose that a hydrogen atom (H atom) abstraction from the C—H bonds of substrates by 1 is the rate-determining step (r.d.s.) in the C—H bond activation reactions, as also observed in the C—H bond activation of hydrocarbons by its iron(V)-oxo analog, [(TAML)FeV(O)]−.7d Product analysis of the fluorene oxidation by 1 revealed that the oxidized product of fluorene was fluorene—N(H)Ts as a major product (~90% yield) and the reduced product of 1 was the starting [FeIII(TAML)]− complex (Figure S22). We therefore conclude that the amination reaction by 1 occurs via a H atom abstraction of substrates to give the aminated product and the starting [FeIII(TAML)]− complex (see Scheme 2, reaction (i)).
Figure 3.
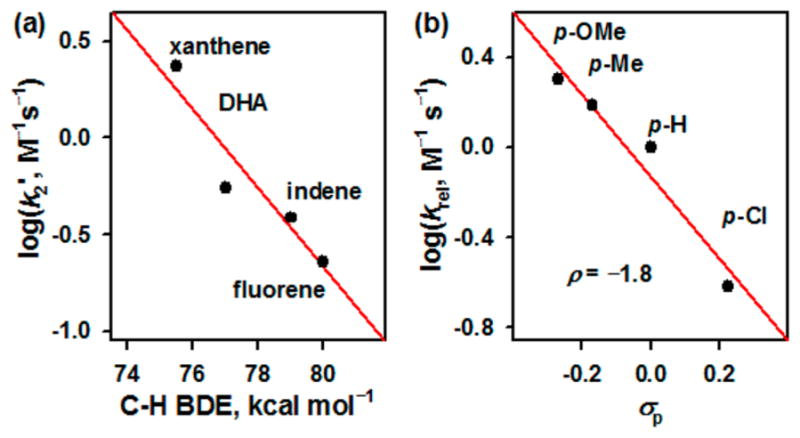
(a) Plot of log(k2′) against C—H BDE of substrates for the reactions of 1 (Table S5). (b) Hammett plot of log(krel) against σp of para-substituted thioanisole derivatives in the nitrene transfer reaction of 1 with thioanisole derivatives in CH3CN at 15 °C (Table S6).
Scheme 2.
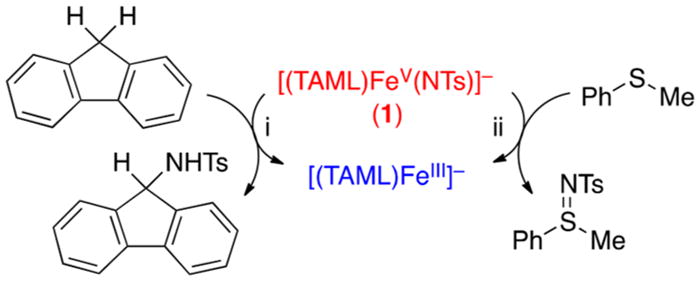
Reactions of 1 in the Amination and Nitrene Transfer Reactions
1 was also shown to transfer its NTs group to thioanisole and para-X-substituted thioanisoles (X = OMe, Me, and Cl). The kobs of the sulfimidation reaction was determined by pseudo-first-order fitting of the decay of 1 monitored at 595 and 915 nm (Figure S23a). The kobs value increased linearly with increasing thioanisole concentration, affording a k2 value of 2.6(2) × 10−2 M−1 s−1 at 15 °C (Figure S23b and Table S6). The product analysis of the thioanisole oxidation by 1 revealed the formation of PhS(═NTs)CH3 (~85% yield). In addition, on the basis of spectroscopic characterization of the decay product of 1 using UV–vis, X-band EPR, and ESI-MS (Figures S23a and S24), we conclude that 1 transfers its NTs group to thioanisole to give the PhS(═NTs)CH3 and [FeIII(TAML)]− products (Scheme 2, reaction (ii). The electrophilic character of the iron(V)-imido complex was also demonstrated by the ρ value of −1.8 in the sulfimidation reaction of para-X-substituted thioanisoles by 1 (Figure 3b). In the reactivity studies, we have shown that the iron(V)-imido complex is capable of C—H bond activation and nitrene transfer reactions.
In summary, we have reported the first synthetic mononuclear nonheme iron(V)-imido complex that is capable of C—H functionalization of hydrocarbons and the sulfimidation of sulfur-containing substrates. 1 is more reactive than [(N4Py)FeIV(NTs)]2+ in C—H functionalization,6c which is expected for such an electrophilic reaction given the higher effective charge on the Fe. However, interestingly, 1 is less reactive than [(N4Py)FeIV(NTs)]2+ in sulfimidation,6c which is also an electrophilic reaction. Future studies will focus on understanding this difference in reactivity, defining the detailed mechanism of the C—H bond activation reaction (e.g., nitrogen rebound versus nitrogen nonrebound parallel to oxygen reactivity in nonheme metal-oxo systems),12 and understanding the reactivity differences between nonheme iron(V)-imido complexes and their iron(V)-oxo analogs in C—H bond activation, nitrene group versus oxygen atom transfer, and electron transfer reactions.
Supplementary Material
Acknowledgments
This work was supported by NRF of Korea through CRI (NRF-2012R1A3A2048842 to W.N.), GRL (NRF-2010-00353 to W.N.), MSIP (NRF-2017R1C1B2002037 to S.H.), and Sookmyung Women’s University Grants (1-1603-2054 to S.H.). Funding for this work was also provided by the NIH (GM-40392 to E.I.S.). T.O. acknowledges the support of “Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation” and Grant-in-Aid for Scientific Research (No. 15H00960) by JSPS. Use of the SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. DOE under Contract No. DE-AC02-76SF00515.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b04695.
Experimental details (PDF)
References
- 1.(a) Hohenberger J, Ray K, Meyer K. Nat Commun. 2012;3:720. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]; (b) Nam W, Lee YM, Fukuzumi S. Acc Chem Res. 2014;47:1146. doi: 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]; (c) Cook SA, Borovik AS. Acc Chem Res. 2015;48:2407. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Puri M, Que L., Jr Acc Chem Res. 2015;48:2443. doi: 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Engelmann X, Monte-Pérez I, Ray K. Angew Chem, Int Ed. 2016;55:7632. doi: 10.1002/anie.201600507. [DOI] [PubMed] [Google Scholar]
- 2.(a) Saouma CT, Peters JC. Coord Chem Rev. 2011;255:920. doi: 10.1016/j.ccr.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Che CM, Lo VKY, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]; (c) Abu-Omar MM. Dalton Trans. 2011;40:3435. doi: 10.1039/c0dt01341b. [DOI] [PubMed] [Google Scholar]
- 3.(a) Eikey RA, Abu-Omar MM. Coord Chem Rev. 2003;243:83. [Google Scholar]; (b) Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y, Guan X, Wong ELM, Liu P, Huang JS, Che CM. J Am Chem Soc. 2013;135:7194. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]; (d) Leeladee P, Jameson GNL, Siegler MA, Kumar D, de Visser SP, Goldberg DP. Inorg Chem. 2013;52:4668. doi: 10.1021/ic400280x. [DOI] [PubMed] [Google Scholar]; (e) Gouré E, Avenier F, Dubourdeaux P, Sénèque O, Albrieux F, Lebrun C, Clémancey M, Maldivi P, Latour JM. Angew Chem, Int Ed. 2014;53:1580. doi: 10.1002/anie.201307429. [DOI] [PubMed] [Google Scholar]; (f) Gouré E, Senthilnathan D, Coin G, Albrieux F, Avenier F, Dubourdeaux P, Lebrun C, Maldivi P, Latour JM. Angew Chem, Int Ed. 2017;56:4305. doi: 10.1002/anie.201612472. [DOI] [PubMed] [Google Scholar]
- 4.(a) Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4956. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bart SC, Lobkovsky E, Bill E, Chirik PJ. J Am Chem Soc. 2006;128:5302. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]; (c) Tangen E, Conradie J, Ghosh A. J Chem Theory Comput. 2007;3:448. doi: 10.1021/ct600318n. [DOI] [PubMed] [Google Scholar]; (d) Nieto I, Ding F, Bontchev RP, Wang H, Smith JM. J Am Chem Soc. 2008;130:2716. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]; (e) Searles K, Fortier S, Khusniyarov MM, Carroll PJ, Sutter J, Meyer K, Mindiola DJ, Caulton KG. Angew Chem, Int Ed. 2014;53:14139. doi: 10.1002/anie.201407156. [DOI] [PubMed] [Google Scholar]; (f) Hennessy ET, Betley TA. Science. 2013;340:591. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]; (g) Iovan DA, Betley TA. J Am Chem Soc. 2016;138:1983. doi: 10.1021/jacs.5b12582. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Bucinsky L, Breza M, Lee WT, Hickey AK, Dickie DA, Nieto I, DeGayner JA, Harris TD, Meyer K, Krzystek J, Ozarowski A, Nehrkorn J, Schnegg A, Holldack K, Herber RH, Telser J, Smith JM. Inorg Chem. 2017;56:4751. doi: 10.1021/acs.inorgchem.7b00512. [DOI] [PubMed] [Google Scholar]
- 5.Kaizer J, Klinker EJ, Oh NY, Rohde JU, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L., Jr J Am Chem Soc. 2004;126:472. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 6.(a) Klinker EJ, Jackson TA, Jensen MP, Stubna A, Juhász G, Bominaar EL, Münck E, Que L., Jr Angew Chem, Int Ed. 2006;45:7394. doi: 10.1002/anie.200602799. [DOI] [PubMed] [Google Scholar]; (b) Vardhaman AK, Barman P, Kumar S, Sastri CV, Kumar D, de Visser SP. Angew Chem, Int Ed. 2013;52:12288. doi: 10.1002/anie.201305370. [DOI] [PubMed] [Google Scholar]; (c) Kumar S, Faponle AS, Barman P, Vardhaman AK, Sastri CV, Kumar D, de Visser SP. J Am Chem Soc. 2014;136:17102. doi: 10.1021/ja508403w. [DOI] [PubMed] [Google Scholar]; (d) Vardhaman AK, Lee YM, Jung J, Ohkubo K, Nam W, Fukuzumi S. Angew Chem, Int Ed. 2016;55:3709. doi: 10.1002/anie.201600287. [DOI] [PubMed] [Google Scholar]
- 7.(a) de Oliveira FT, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Jr, Bominaar EL, Münck E, Collins TJ. Science. 2007;315:835. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]; (b) Mills MR, Weitz AC, Hendrich MP, Ryabov AD, Collins TJ. J Am Chem Soc. 2016;138:13866. doi: 10.1021/jacs.6b09572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Panda C, Debgupta J, Díaz DD, Singh KK, Gupta SS, Dhar BB. J Am Chem Soc. 2014;136:12273. doi: 10.1021/ja503753k. [DOI] [PubMed] [Google Scholar]; (d) Kwon E, Cho KB, Hong S, Nam W. Chem Commun. 2014;50:5572. doi: 10.1039/c4cc01058b. [DOI] [PubMed] [Google Scholar]
- 8.de Visser SP, Rohde JU, Lee YM, Cho J, Nam W. Coord Chem Rev. 2013;257:381. [Google Scholar]
- 9.Mehn MP, Brown SD, Jenkins DM, Peters JC, Que L., Jr Inorg Chem. 2006;45:7417. doi: 10.1021/ic060670r. [DOI] [PubMed] [Google Scholar]
- 10.It is worth noting that iron-nitrido complexes exhibited Fe—N vibrational energy at ~860–1100 cm−1; see: Wagner WD, Nakamoto K. J Am Chem Soc. 1989;111:1590.Scepaniak JJ, Fulton MD, Bontchev RP, Duesler EN, Kirk ML, Smith JM. J Am Chem Soc. 2008;130:10515. doi: 10.1021/ja8027372.
- 11.Gorelsky SI, Basumallick L, Vura-Weis J, Sarangi R, Hodgson KO, Hedman B, Fujisawa K, Solomon EI. Inorg Chem. 2005;44:4947. doi: 10.1021/ic050371m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho KB, Hirao H, Shaik S, Nam W. Chem Soc Rev. 2016;45:1197. doi: 10.1039/c5cs00566c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.