

Abstract
Binuclear non-heme iron enzymes activate O2 for diverse chemistries that include oxygenation of organic substrates and hydrogen atom abstraction. This process often involves the formation of peroxo-bridged biferric intermediates, only some of which can perform electrophilic reactions. To elucidate the geometric and electronic structural requirements to activate peroxo reactivity, the active peroxo intermediate in 4-aminobenzoate N-oxygenase (AurF) has been characterized spectroscopically and computationally. A magnetic circular dichroism study of reduced AurF shows that its electronic and geometric structures are poised to react rapidly with O2. Nuclear resonance vibrational spectroscopic definition of the peroxo intermediate formed in this reaction shows that the active intermediate has a protonated peroxo bridge. Density functional theory computations on the structure established here show that the protonation activates peroxide for electrophilic/single-electron-transfer reactivity. This activation of peroxide by protonation is likely also relevant to the reactive peroxo intermediates in other binuclear non-heme iron enzymes.
Graphical Abstract
INTRODUCTION
Dioxygen is an essential component for metabolic processes in aerobic organisms, but requires activation to become reactive toward organic metabolites at ambient conditions.1 Binuclear non-heme iron (NH-Fe2) enzymes are a large class that activate O2 by two-electron reduction to form a peroxide intermediate.2,3 The peroxide generated is then either further reduced to a bridging oxo intermediate or more generally directly reacts with substrate. The reactions catalyzed are important in DNA and natural product biosynthesis, Fe uptake and release, and involve electrophilic aromatic substitution and hydrogen atom abstraction.4–7
These NH-Fe2 enzymes form peroxo-bridged antiferromagnetically coupled high-spin Fe(III)2 intermediates (Table 1), some of which, labeled P, display a prominent absorption band at ~700 nm (ε ≈ 1500 M−1 cm−1) and Mössbauer parameters of δ ≈ 0.66 and ΔEQ ≈ 1.5 mm/s,8–19 while there are species characterized by the lack of absorption at ~700 nm and decreased Mössbauer parameters of δ ≈ 0.52 and ΔEQ ≈ 0.55 mm/s.7,20–24 On the basis of resonance Raman (rR) spectroscopic analysis, P of class Ia ribonucleotide reductase (RNR) was determined to have a μ-1,2-peroxo bridge,13,25 while for others, several structures have been proposed (Figure 1). Based on a relatively low O–O stretch frequency (~790 cm−1), the peroxo intermediate of the arylamine oxygenase of the chloramphenicol biosynthesis (CmlI) was proposed to have a side-on/end-on peroxo-bridged structure.24 Deoxyhypusine hydroxylase (DOHH) has been characterized to have a μ-1,2-peroxo bridge by X-ray crystallography26 and rR spectroscopy,18 but shows a higher energy absorption feature and lower Mössbauer isomer shifts than other P type intermediates. These spectral changes have been attributed to the presence of an additional μ-hydroxo bridge.18,27 Recently, the crystal structure of toluene 4-monooxygenase (T4MO) has been obtained with O2 and substrate bound, and based on Fe–O distances, an Fe(II)Fe(III)-superoxide species rather than a peroxo biferric species has been suggested as the active intermediate.28
Table 1.
Peroxo-Level Intermediates Observed in Binuclear Non-heme Iron Enzymes
| enzyme | λ/nm (ε/M−1 cm−1) | δ(ΔEQ)/mm s−1 |
|---|---|---|
| AurF21,22 | 500 (500) | 0.55 (0.74); 0.61 (0.35) |
| CmlI24 | 500 (500) | 0.62 (−0.23); 0.54 (−0.68) |
| W48A RNR from E. coli20 | 500 (100) | 0.52 (0.55); 0.45 (1.53) |
| toluene-o-xylene monooxygenase (ToMO)7 | no absorption | 0.55 (0.67) |
| aldehyde deformylating oxygenase (ADO)23 | 450 (1200) | 0.48 (0.55); 0.49 (1.23) |
| wild-type RNR from mouse19 | 700 (1500) | 0.63 (1.74) |
| D84E RNR from E. coli8,13 | 700 (1500) | 0.63 (1.58) |
| stearoyl-acyl carrier protein Δ9 desaturase9,10 | 700 (1200) | 0.68 (1.90); 0.64 (1.06) |
| T201S ToMO15 | 675 (1500) | 0.67 (1.51) |
| soluble methane monooxygenase (sMMO)11,12,14,16,17 | 725 (1800) | 0.69 (1.55) |
| deoxyhypusine hydroxylase (DOHH)18 | 630 (2800) | 0.55 (1.16); 0.58 (0.88) |
Figure 1.
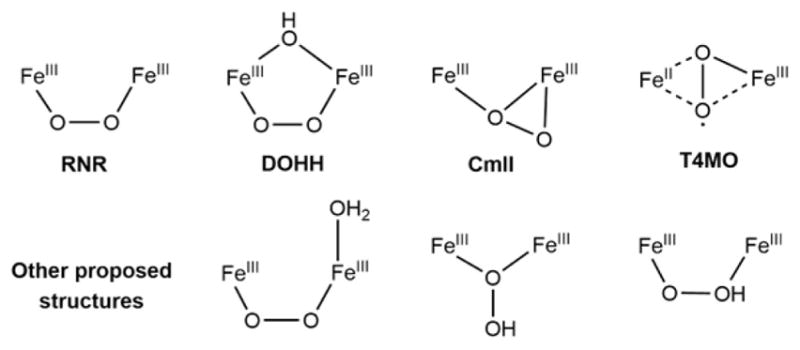
Possible structures of peroxo-level intermediates. The top four structures have been partially characterized in RNR, DOHH, CmlI, and T4MO, and the bottom three structures have been proposed in DFT calculations.
Importantly, the P sites appear to require activation in a number of the NH-Fe2 reactions. In RNR, P converts to an active peroxo intermediate, labeled P′, which is isolable in the W48A variant and enables the reductive formation of a high-valent oxo-bridged Fe(IV)Fe(III) intermediate X that abstracts an H atom from a nearby Tyr.20 A similar conversion was proposed for P in stearoyl-acyl carrier protein Δ9 desaturase (Δ9D)10 and CmlI,29 as the cis-μ-1,2-peroxo-bridged P does not initiate reaction with substrate. Thus, the conversion of P to P′ appears to be the peroxide activation step, and elucidating the geometric and electronic structural differences between these two intermediates is key to understanding the activation of O2 at the peroxide level. Computational searches for P′ in RNR and Δ9D proposed protonated peroxo structures with either a μ-1,1- or μ-1,2-hydroperoxo bridge or a non-protonated peroxo structure having an additional water ligand (Figure 1).30–32
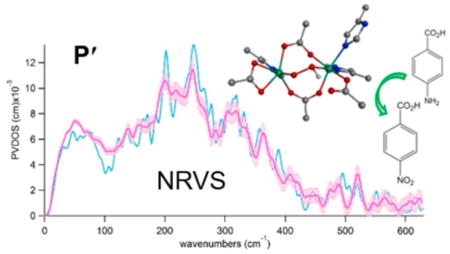
The 4-aminobenzoate N-oxygenase (AurF) forms a P′-type peroxo intermediate that can directly react with 4-aminobenzoic acid to produce 4-nitrobenzoic acid (Figure 2).21,22 This intermediate is diamagnetic with antiferromagnetically coupled ferric centers and thus not accessible by para-magnetism-based techniques. However, it is relatively stable in the absence of substrate, and a high-yield sample (>70%) with 57Fe can be prepared based on the rapid O2 reaction of the 57Fe(II)2 state (within 0.01 s at ~0.6 mM O2 and 20 °C) and the slow decay of P′ in the absence of substrate (no noticeable decay on the 100-s time scale).21,22 This 57Fe-peroxo intermediate in AurF is suitable for defining the nature of the P′ intermediate via nuclear resonance vibrational spectroscopy (NRVS). This method probes the vibrational side bands of the 57Fe Mössbauer transition and thus all vibrations involving significant Fe displacement are observed.33–37 This rich information on the vibrations of the 57Fe site can further limit the possible structures of P′ previously proposed.
Figure 2.
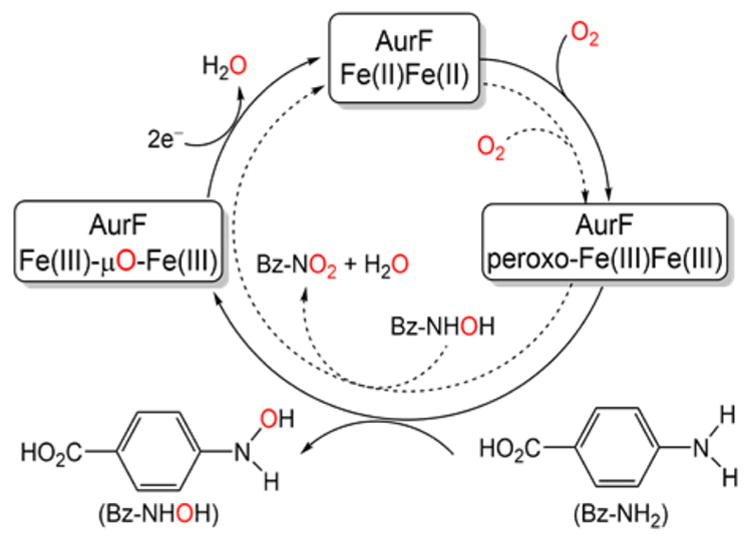
Reaction scheme for 4-aminobenzoate N-oxygenase (AurF). AurF is a monooxygenase that catalyzes the conversion of 4-aminobenzoic acid to 4-nitrobenzoic acid via 2+4 electron oxidation steps, each indicated by solid and dashed arrows, respectively.
We have also used near-infrared (NIR) circular dichroism (CD), magnetic circular dichroism (MCD), and variable-temperature, variable-field (VTVH) MCD spectroscopic techniques2,38 to elucidate the structure of reduced AurF that is poised for the rapid reaction with O2. The reduced AurF structure thus obtained was used to generate possible structures of P′ using density functional theory (DFT) computations. Among the possibilities, only one structure could reproduce the NRVS spectra and this structure has been used to evaluate the reactivity of P′ relative to P in N-oxygenation.
MATERIALS AND METHODS
Sample Preparation
Reduced AurF was obtained by degassing the HEPES–D2O (pD ≈ 7.4) solution of the AurF protein prepared as previously described21 and reducing it using sodium dithionite. This solution was mixed with glycerol-d3 to form a glass for low-temperature CD and MCD studies. The NRVS samples of P′ in AurF were prepared by growing E. coli cells in 57FeSO4-containing media and trapping P′ as described before.21,22 The yield and quantity of [57Fe] in P′ was assured by Mössbauer spectroscopy.
Spectroscopy
A Jasco J730 spectropolarimeter with an InSb detector cooled with liquid nitrogen was used for NIR (600–2000 nm), CD, and MCD studies. For the latter, an Oxford Instruments SM4000 7T superconducting magnet provided up to 7 T magnetic field. NRVS data were collected at third-generation synchrotron beamlines, BL09XU of SPring-8 in Japan and 3-ID of APS in Argonne National Laboratory, under He(l)-cooled cryostat conditions. Reproducibility of each scan was ensured and multiple scans were collected until the combined data achieved an acceptable signal-to-noise ratio. These accumulated raw data were processed using the PHOENIX program to extract the partial vibrational density-of-states (PVDOS) spectra.34–36 The details of the data processing processes are given in refs 34–36.
Computation
The initial active-site geometry of AurF was obtained by truncating the X-ray crystal structure of the resting biferric AurF (PDB entry 3CHU)39 using a scheme previously developed for NRVS analysis.40 Based on our previous NRVS study of peroxy biferric model complexes,41 the 90% Becke–Perdew86/10% HF hybrid functional and the 6-31G* basis set for geometry optimizations and frequency calculations, and the 6-311G* basis set for single-point energy calculations42,43 were used with the Gaussian 09 (g09) package.44–58 NRVS spectra were predicted from g09 frequency calculations using the GenNRVS program, while electronic59,60 and Mössbauer spectroscopic properties61 were calculated with the ORCA 2.9 package, using BP86/def2-TZVP for Fe’s and BP86/def2-SVP for other atoms in time-dependent (TD) DFT computations62 and BP86/CP(PPP) for Fe’s and BP86/DZP for other atoms in Mössbauer parameter calculations.63 The potential energy surfaces of the AurF P′ and hypothetical P intermediates were constructed in two dimensions along peroxy O–O bond and substrate N–peroxy O distance. From the saddle points of these surfaces, transition states were searched and validated using g09 intrinsic reaction coordinate calculations.
RESULTS AND ANALYSIS
Reduced AurF
Since the Fe(II)2 state of AurF reacts rapidly with O2,21 elucidating the structure of reduced AurF and its reaction with O2 limits the possible structures for P′. The NIR CD and MCD spectroscopic data in Figure 3a show four bands, each corresponding to a ligand-field transition from the lowest-energy Fe-t2-3d-based molecular orbital (MO) into an Fe-e-3d-based MO. Given that each Fe(II) center can display up to two d-d transitions in the NIR region,2 the observation of the four bands indicates that the two Fe(II) centers in reduced AurF have significantly different coordination environments.
Figure 3.
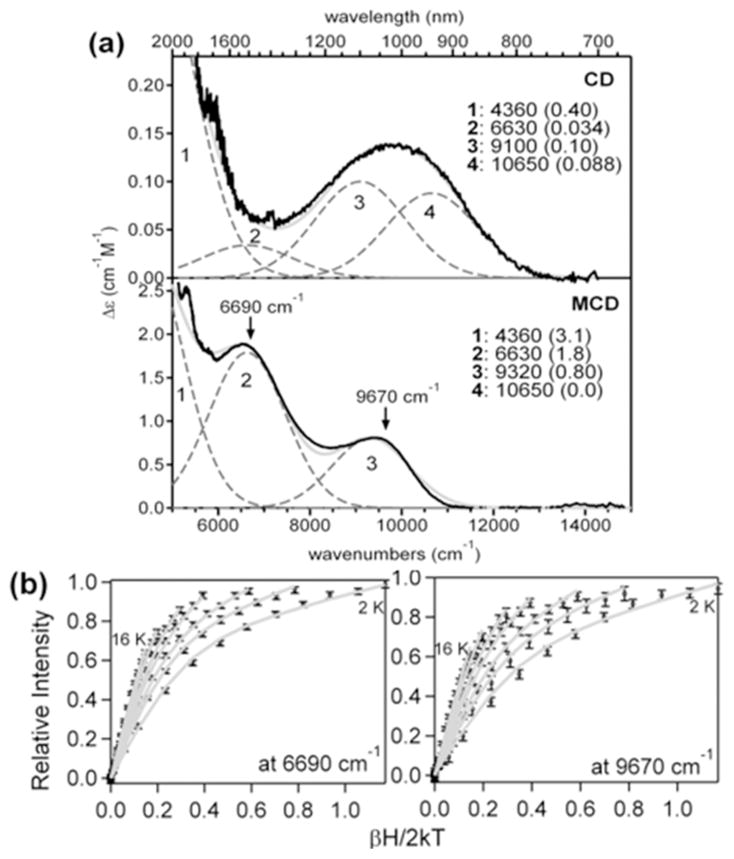
NIR CD, MCD, and VTVH MCD data for reduced AurF. (a) 278 K CD and 7 T, 4 K MCD spectra (solid black) of the reduced AurF and their Gaussian fits (dashed gray for Gaussian deconvolutions and solid gray for the sum). The fit parameters on the right include the energies and extinction coefficients (in parentheses) of the individual bands. (b) VTVH MCD data collected on bands 2 and 3 and their spin-Hamiltonian fits (gray). Both VTVH MCD data sets are fit with J = −3.1 cm−1, D1= −12.4 cm−1, E1/D1 = 0.29, D2 = 10.6 cm−1, and E2/D2 = 0.32. Spin Hamiltonian projections of each Fe on the dimer wave functions show that the transitions associated with bands 2 and 3 originate on the same Fe center, which has a negative D value.
To determine which two bands arise from each Fe(II) center, the magnetization behavior of the MCD signal at the energies indicated in Figure 3a was determined by using VTVH MCD spectroscopy. The nested saturation curves obtained (Figure 3b) were fit using a spin Hamiltonian analysis (Supporting Information), which determined the exchange coupling J between the two Fe(II) centers and the axial and rhombic zero-field splitting parameters D and E for each Fe(II) center.2,38,64 The negative J value (−3 cm−1) shows that the Fe(II)2 centers are weakly antiferromagnetically coupled via μ-1,3-carboxylato bridges.2 Opposite-signed D values (−12.4 and +10.6 cm−1) with significant rhombicity indicate that each Fe(II) center exhibits a different magnetization behavior. Bands 2 and 3 exhibit the same VTVH MCD behavior and thus are associated with one Fe(II) center, which should have a five-coordinate (5C) trigonal bipyramidal (TBP) structure with a weak axial ligand, given its d-d transition energies, 6630 and ~9200 cm−1.38 Then, bands 1 and 4 are associated with the second Fe(II) center (the low signal-to-noise ratio of the VTVH MCD data on band 1 precludes reliable fits), and their energies at 4360 and 10650 cm−1 reflect a 5C square pyramidal (SP) structure.38 With both Fe(II) centers being 5C, a side-on binding of O2 to reduced AurF is not likely to give the fast kinetics observed in the formation of the peroxo intermediate (kobs ≈ 147 s−1).21
A DFT model that matches the spectroscopically defined reduced AurF structure could be obtained by truncating the crystal structure of the biferric resting state AurF,39 removing the μ-oxo bridge, setting Fe centers to be ferrous, and performing geometry optimization with the backbone frozen but allowing the side chains to move freely to reproduce MCD-defined structural features (Figure 4). The two Fe(II) centers bridged by E227 and E136 in μ-1,3 binding modes were calculated using the broken-symmetry formulation65 to have an exchange coupling J ≈ −2 cm−1 with Fe1 (MS = +2) and Fe2 (MS = −2). The Fe2(II) center with one histidine (H230) optimizes to a 5C TBP structure with a weak axial ligand trans to H230 from the bidentate E196. The Fe1(II) center is ligated by two histidines, H139 axial and H223 in the equatorial position of the 5C SP structure. The TD-DFT calculation of this optimized reduced AurF structure predicts d-d transitions at 10 562 and 13 365 cm−1 for Fe2 and 8900 and 14 104 cm−1 for Fe1, a qualitative energy order consistent with the MCD data.
Figure 4.
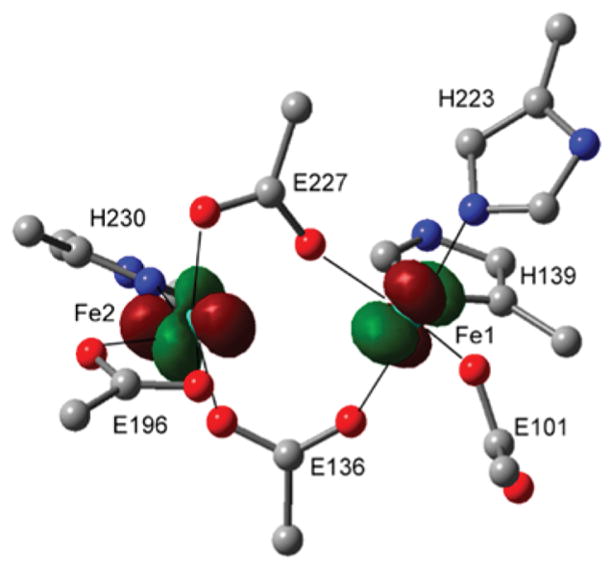
Optimized geometry and redox-active molecular orbitals of reduced AurF. For clarity, only the first-sphere ligands are depicted without hydrogens and –CH2– side chains. The Fe1- and Fe2-based highest-occupied molecular orbitals that will donate electrons to O2 are overlaid. H139 on Fe1 is axial and oriented into the page.
For fast formation of a peroxo-bridged biferric intermediate, the highest-occupied molecular orbital (HOMO) of each Fe(II) center should interact with one of the two orthogonal π* lowest-energy unoccupied molecular orbitals (LUMOs) of O2. Thus, to facilitate the two, one-electron transfers from each Fe(II) center, the HOMOs of the two Fe(II) centers should be perpendicular to each other.66–69 This orientation is achieved in reduced AurF (Figure 4). With the μ-1,3-carboxylato bridges that provide only limited superexchange pathways for the ferrous centers,2,64 the spin-up and spin-down d-based HOMOs are localized on Fe1 and Fe2, respectively. The HOMO of Fe2 can interact with the π*-MO of O2 that will be in the Fe–(O22−)–Fe plane upon formation of a peroxo bridge, while that of Fe1 can interact with the out-of-plane π*-MO. Thus, this good frontier MO overlap rationalizes the rapid O2 reaction of reduced AurF to form P′.
Intermediate P′ of AurF
To determine the structure of P′ in AurF, NRVS spectra were obtained for P′ generated with 16O2 and 18O2 (Figure 5). These active-site vibrational spectra can be divided into three regions: region (1) at energies below 180 cm−1 is featureless; region (2) displays four distinct features at 200, 246, 315, and 363 cm−1 with comparable NRVS intensities; and region (3) shows two oxygen-isotope-sensitive peaks at energies above 375 cm−1. On the basis of our previous NRVS study of μ-1,2-peroxo-bridged Fe(III)2 models,41 these regions include features associated with the vibrations of the bridged Fe(III)2 core given in Figure 6. The peaks that shift upon 18O labeling in region (3) from 409 and 520 cm−1 in the 16O data to 390 and 495 cm−1 in the 18O data are assigned to the Fe–O stretching vibrations. These peaks have a large energy separation of ~110 cm−1, compared to ~40 cm−1 found in the rR study of the cis-μ-1,2-peroxo-bridged P in RNR.13,25
Figure 5.
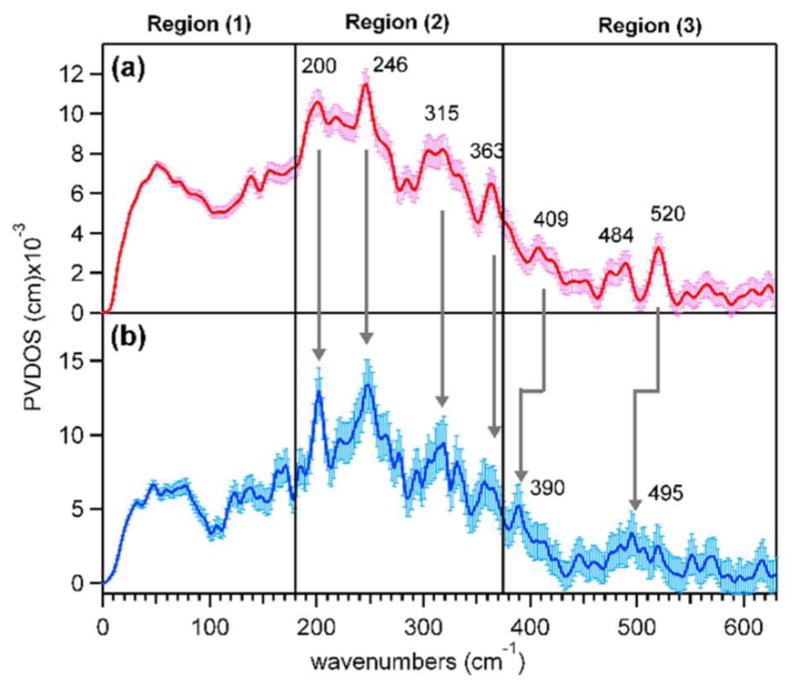
NRVS spectra of P′ in AurF obtained from the reaction of reduced AurF with (a) 16O2 and (b) 18O2.
Figure 6.
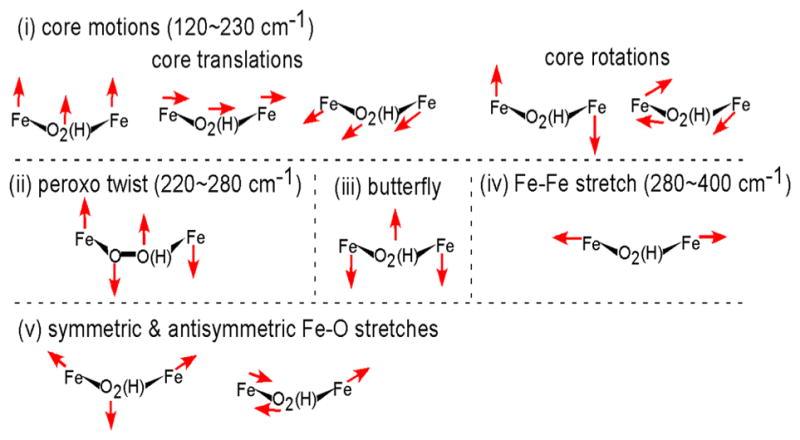
Vibrations of the {Fe2-(hydro)peroxo} core contributing to NRVS spectra.
To search for possible structure(s) that can reproduce the NRVS data of P′, various peroxo structures were constructed by adding O2 to the DFT-optimized reduced AurF structure (Figure 4) and allowing for the possible uptake of a proton and/or a water molecule as proposed earlier.30–32 28 different structures were obtained (5 O2-only O models, 14 protonated h models, 6 water-associated w models, and 3 water-and-proton-associated wh models; Figure S1) that include variations in the bridging peroxo ligand (μ-1,2-end-on; μ-η1, η2-end-on, side-on; μ-η1, η2-side-on peroxide; and μ-1,1- and μ-1,2-hydroperoxide), the number of carboxylato bridges (one or two), the conformations of the terminal carboxylate ligands (syn or anti; mono- or bidentate), and the hydrogen-bonding interactions.
The NRVS spectral/structural correlations observed in the 28 models narrowed the possible structures for P′ (Figures S2–S6). For example, Figure 7 includes the DFT-simulated NRVS spectra and structures of 6 representative models, each of which has the lowest energy among the models with the same structural type. All peroxo models, including O1, O5, and w1, show a small energy splitting of ~45 cm−1 between 18O-sensitive peaks in region (3), and thus these can be ruled out. There are three Fe–O stretches for the μ-η1,η2-end-on, side-on peroxo-bridged model O5, but one of the Fe-η2O stretches is considerably low in energy and thus appears in region (2). The μ-1,1-hydroperoxo-bridged model h9 is also ruled out because it is predicted to have one Fe–O stretch in region (2) and the other in region (3) with low NRVS intensity due to minimal Fe displacements involved in the normal modes. Both of the h3 and w5 models show an increased energy separation between the 18O-sensitive peaks compared to peroxy models. However, w5 shows a three-peak pattern in region (2), and thus h3 with the four-peak pattern in region (2) is the best among the six models presented in Figure 7.
Figure 7.

NRVS data and DFT-assisted assignment of P′ in AurF. (a) (1) The NRVS spectra of P′ obtained with 16O2 (red) and 18O2 (gray). The four major features in region (2) and the noise level are indicated by solid vertical and dashed horizontal lines, respectively. The features with arrows on the 18O data appear upon isotope labeling. (2–7) The DFT-predicted NRVS spectra of representative model structures given in (b). To assess chances of seeing predicted NRVS features, the experimental noise level, ~10% of the maximum intensity, is also noted for each model. The μ-1,2-hydroperoxo-bridged h3 is the only structure that reproduces the NRVS data, and its normal modes (i–iv) are given in Figure 6. (b) DFT-calculated structures of μ-1,2-hydroperoxo-bridged h3, μ-1,1-hydroperoxo-bridged h9, μ-1,2-peroxo-bridged O1, μ-η1,η2-end-on, side-on peroxo-bridged O5, μ-1,2-peroxo-bridged w1 with a terminal water ligand, and μ-hydroxo-μ-1,2-hydroperoxo-bridged w5 models. h3 was used to simulate the NRVS data in Figure 3a.
Similarly, in consideration of all 28 models, μ-1,2-end-on peroxo-bridged structures and μ-η1,η2-end-on, side-on peroxo-bridged structures with the η1-O atom out of the Fe1–η2O–Fe2 plane are calculated to have the symmetric and antisymmetric Fe–O stretch vibrations split in energy by less than 60 cm−1. This small energy splitting of 18O-sensitive peaks excluded nine models (O1, O3–O5, h6, h7, w1, w3, and w4). The combinations of low-energy bends and torsions create the translational and rotational motions of the {Fe2-(hydro)-peroxo} core (Figure 6), which contribute to NRVS intensity in a broad energy range of ~120–260 cm−1. Having no prominent peak in region (1) in the experimental NRVS spectrum of P′ further limits the possible structures; the bis-bridged structures with one carboxylate bridge exhibit an intense peak in region (1) due to the less constrained core relative to the tris-bridged structures, and thus models O2 and w2 are further excluded (Figures S2–S6).
A majority of the remaining structures could not reproduce the four peaks with comparable intensity in region (2). In all, 15 out of 17 structures show fewer than four resolved peaks with a major broad band at ~200–240 cm−1. Only the hydroperoxo-bridged structures h3 and h10 (i.e., μ-1,2- and μ-1,1-hydroperoxo-bridged structures, respectively) exhibit multiple peaks with comparable intensities in region (2). However, the energy splitting of 18O-sensitive peaks in the predicted NRVS spectrum of the μ-1,1-hydroperoxo-bridged structure h10 is too large (166 cm−1, Table S1), with the symmetric stretch contributing in region (2) (consistent with the peak at 322 cm−1 in the rR spectrum of a μ-1,1-hydroperoxo-2Cu(II) complex70). Thus, h10 is excluded. (See Table S2 for detailed NRVS comparisons of all models.)
Only h3, which has two μ-1,3-carboxylato bridges (E227 and E136) and a μ-1,2-hydroperoxo bridge with the proton on the peroxo oxygen coordinated to the 2His-coordinated Fe1 and H-bonded to E101 on Fe1 (Figure 8), reproduces four bands with comparable intensity in the NRVS spectrum in region (2) (Figure 5). The four bands predicted at 209, 245, 315, and 356 cm−1 (Figure 7a, (2), h3) agree well with the experimental features at 200, 246, 315, and 363 cm−1 (Figure 7a, (1)) and are assigned as the core motions, peroxo twist, butterfly, and Fe–Fe stretch, respectively (Figure 6). The moderately intense feature predicted at 180 cm−1 matches the broad feature at ~165 cm−1 in Figure 7a, (1). For h3, the two 18O-sensitive features are predicted at 389 and 463 cm−1, which correspond to the rather localized Fe1–OH and Fe2–O stretches, respectively, of the hydroperoxo bridge, with the former lower in energy due to the protonation. These DFT-predicted energies underestimate experiment by 20 and 57 cm−1. In our previous study of peroxo-bridged biferric models, the DFT methodology used here underestimated the energies of the symmetric and antisymmetric Fe–peroxo stretches by 14 and 42 cm−1 on average.41 This leads to the underestimation of the energy separation between the two Fe–O stretches by 30 cm−1. Given this correction factor, the 74 cm−1 separation calculated for h3 reasonably matches with the experimental observation of ~110 cm−1, showing that the increased energy separation between the Fe–O stretches of P′ in AurF compared to P in RNR is due to the protonation of the peroxide.
Figure 8.
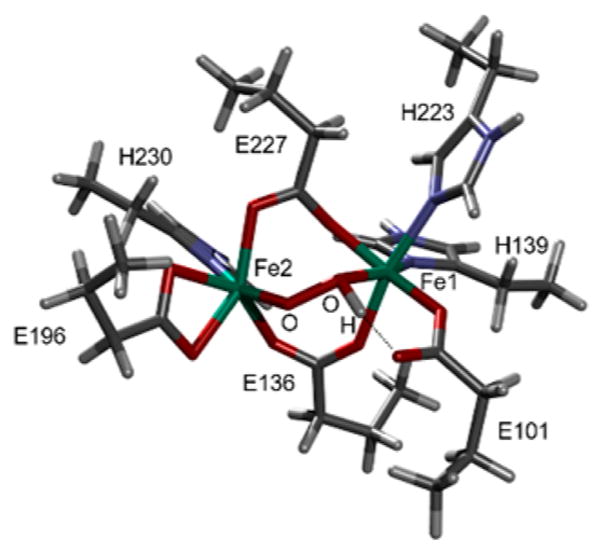
Structure of AurF P′ determined by NRVS analysis. This structure h3 was used to simulate the spectrum of Figure 7a, (2). H, C, N, O, and Fe are depicted in white, gray, blue, red, and green, respectively.
h3 also reproduces the absorption and Mössbauer spectral differences between the P′ and P sites. Compared to a computational model constructed for P in RNR, h3 reproduces the lack of the ~700 nm absorption band and the decreased Mössbauer isomer shifts (Figure S8 and Table S3), suggesting that the spectral differences between the P′ and P sites result from the diminished interactions between the peroxide and the Fe(III) centers due to protonation of the peroxo bridge. Protonation stabilizes the filled peroxo π*-based MOs, shifting the peroxo-to-Fe(III) charge-transfer band to higher energy with lower intensity, and diminishes electron donation to the Fe(III) centers, thereby diminishing the isomer shifts. The DFT-predicted O–O stretches of h3 and the P model for RNR are at 918 and 992 cm−1, respectively, which parallel the experimental data of peroxo intermediates in CmlI (791 cm−1)24 for P′ and in RNR (868 cm−1)25 for P. The DFT-calculated and rR-observed frequencies of P in RNR (992 and 868 cm−1) give a correction scaling factor of 0.88, which can be applied to predict the experimental O–O stretch of P′ in AurF to be about ~800 cm−1. Furthermore, among the 28 models tested, h3 (in Figure 8) is the most stable structure. An analogous structure with the proton on the peroxo oxygen coordinated to the 1His-coordinated Fe2 (h1) is 5 kcal/mol less stable than h3, suggesting that the extra His coordination increases the basicity of the peroxide. In general, structures with a hydroperoxo bridge that is H-bonded to a terminal carboxylate ligand are more energetically favored than peroxo-bridged structures (ΔΔG in Table S1). Thus, E101, which used to be in an anti conformation in the reduced state, rotates around to form a H bond to the proton of the hydroperoxide. This structural variation may imply that E101 could play a role in deliverying a proton upon peroxide formation.
Reaction Coordinate
To examine the viability of h3 in the N-oxygenation of 4-aminobenzoic acid, the potential energy surface (PES) was constructed along the N(substrate)–O(hydroperoxo) and O(hydroperoxo)–O(hydroperoxo) distances. From the substrate-docked structure (Figure 9a, Rh),39 the reaction initiates as the O–O bond cleaves (Figure 9b, lower right). This reaction involves a ΔG‡ ≈ 12.0 kcal/mol, consistent with the experimental observation (ΔG‡ < 14 kcal/mol from kobs ≈ 150 ± 20 s−1 in the reaction of 0.16 mM P′ with 1 equiv of substrate). This O–O bond cleavage (Rh → TS1h → Inth) occurs at a considerable N(substrate)–O(hydroperoxo) distance of ~3.4 Å, and involves electron density transfer to fill the hydroperoxo σ*-based LUMO; starting from the antiferromagnetically coupled Fe1 (MS = +5/2) and Fe2 (MS = −5/2) in Rh, spin-up electron density is donated into the hydroperoxo σ*-based MO by the substrate, while spin-down electron density is transferred from the Fe2 d-based HOMO (Table S4), which is the dz2 orbtial oriented toward the peroxide oxygen. This substrate oxidation results in an Fe1(III)–OH/Fe2(IV)═O intermediate (Figure 10, Inth), and the reaction is exergonic by −11.4 kcal/mol relative to Rh. An alternative mechanism of O–O bond cleavage that involves H-atom abstraction from the substrate is calculated to be unfavorable, as ΔG‡ ≈ 17.7 kcal/mol and the reaction is endergonic by +7.9 kcal/mol relative to Rh. Note that while aminobenzoic acid is relatively easily oxidized and thus undergoes single electron transfer (SET) in the calculated reaction coordinate, this behavior reflects a general electrophilic character for P′ (vide infra).
Figure 9.
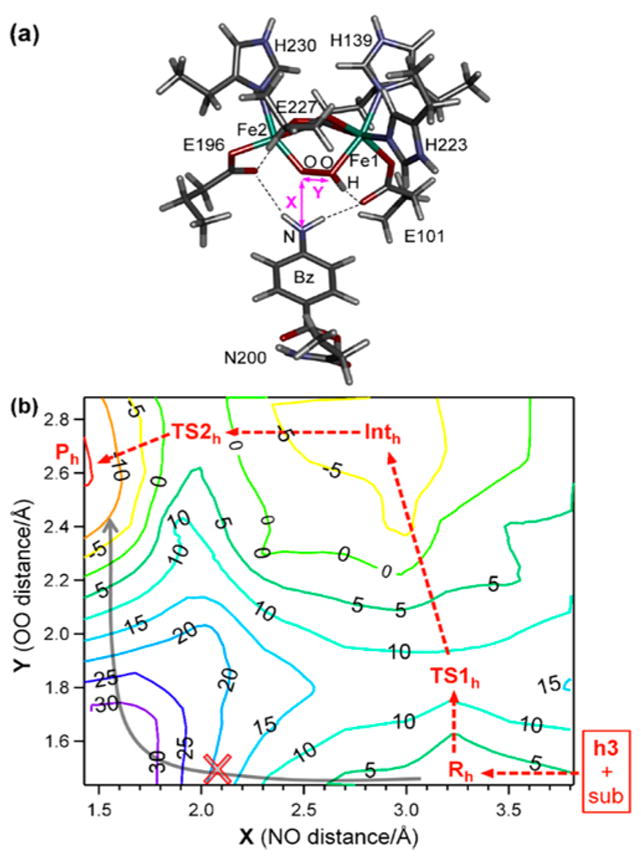
Potential energy surface (PES) calculations for N-oxygenation in AurF. (a) NRVS-derived structure of intermediate P′ in AurF complexed with the 4-aminobenzoic acid substrate (Rh). (b) PES in ΔG along OO bond cleavage and NO bond formation. The minimum energy pathway indicates that reaction coordinate starts at lower right and proceeds along the red dashed arrow (SET pathway) rather than the gray solid arrow (direct electrophilic attack on substrate).
Figure 10.
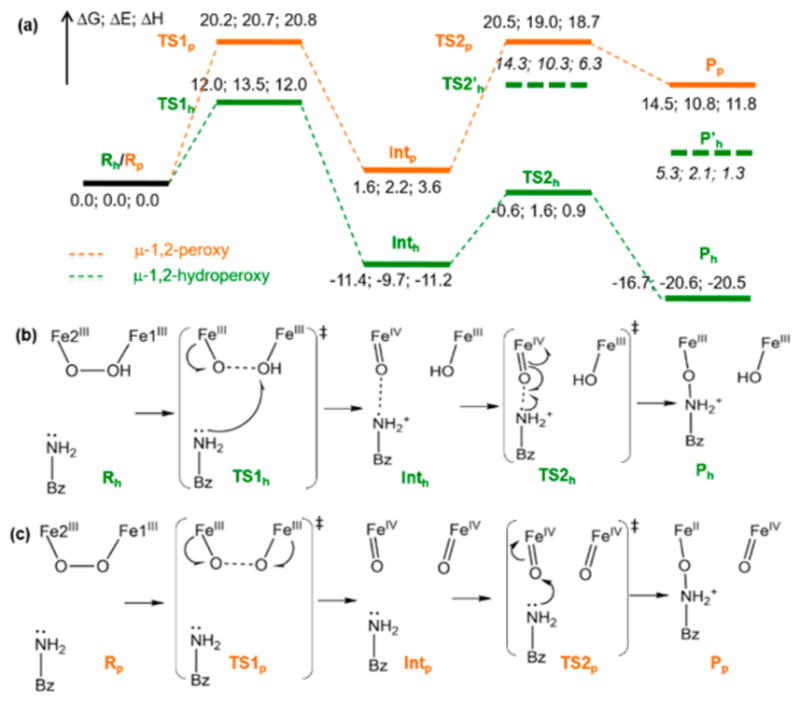
Reaction coordinate for N-oxygenation in AurF. (a) Energy profile (ΔG; ΔE; ΔH) of the AurF μ-1,2-hydroperoxo-bridged P′ intermediate (green) compared to that of a hypothetical μ-1,2-peroxo-bridged P intermediate (orange) for the N-oxygenation of 4-aminobenzoic acid. The dashed energy levels (TS2′h and P′h) are for an alternative unfavorable reaction pathway where the amino group of the substrate forms a bond with the hydroxide on Fe1. (b) Reaction scheme for the P′ intermediate. The NO and OO distances are 3.25 and 1.43 Å in Rh, 3.38 and 1.81 Å in TS1h, 2.66 and 2.82 Å in Inth, 2.00 and 2.86 Å in TS2h, and 1.43 and 2.64 Å in Ph. (c) Reaction scheme for the P intermediate. The NO and OO distances are 3.08 and 1.36 Å in Rp, 3.00 and 1.75 Å in TS1p, 3.01 and 2.60 Å in Intp, 1.80 and 2.79 Å in TS2p, and 1.42 and 2.79 Å in Pp.
The Fe2(IV)═O moiety of Inth resulting from the SET pathway can then form the Nsub–O bond (Figure 10a, b) with a 10.8 kcal/mol free-energy barrier from Inth and back-electron-transfer from the oxo moiety to the Fe2 center. This process generates the Fe(III)2 product Ph with a highly exergonic ΔG of −16.7 kcal/mol relative to Rh. The alternate reaction between the Fe1(III)–OH moiety of Inth and substrate (Figure 10a, dashed green) is highly unfavorable, involving ~26 kcal/mol energy barrier from the Inth state. The product Ph can be further stabilized by forming a μ-OH-bridged structure (ΔG = −21.3 kcal/mol). Note that the resting oxidized state of AurF has a μ-oxo bridge with E227 terminal, 39 and is calculated to be ΔG ≈ −1.3 kcal/mol relative to Rh after migrations of protons (Figure S12).71
To understand the difference in reactivity between the protonated peroxo P′ and the non-protonated peroxo P, the hypothetical P structure in AurF (O1) was evaluated (Figure 10a, orange). The PESs for the N-oxygenation of P (O1) and P′ (h3) show a similar trend (Figure S13), but the O–O bond cleavage of P involves a much higher energy barrier of ΔG‡ ≈ 20.2 kcal/mol (vs 12.0 kcal/mol for P′) and does not oxidize the substrate. Instead, both the spin-up and spin-down electron densities from the Fe(III) centers flow into the peroxo-σ*-based MO. The formation of the resulting (Fe(IV)═oxo)2 intermediate (Figure 10c, Intp) is endergonic by 1.6 kcal/mol, and its subsequent electrophilic attack on the substrate triggers a two-electron transfer from the substrate to this Fe(IV) center. This step is endergonic by 14.5 kcal/mol, involves a second activation barrier of 20.5 kcal/mol and produces an Fe(IV)═O/Fe(II) product (Figure 10c, Pp). Thus, protonation of the peroxo intermediate in AurF lowers the kinetic barrier for the reaction by ~8 kcal/mol, allowing the direct oxidation of substrate. Note that without the electron-donating substrate, the O–O bond cleavage of P′ is unlikely, because the formation of an Fe(IV)═O/Fe(IV)–OH species is not thermodynamically driven and thus involves a 6.3 kcal/mol additional energy barrier (Figure 11). This is consistent with the relatively long lifetime of the intermediate prepared in AurF without substrate.21
Figure 11.
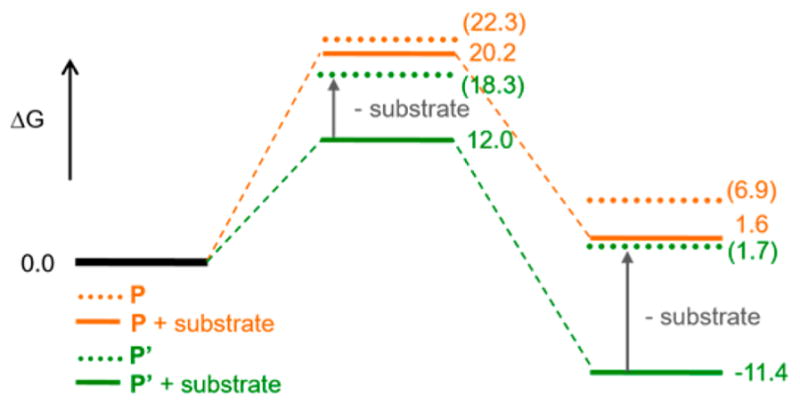
Substrate effect on the O–O bond cleavage. Energy profiles for the O–O bond cleavage of P (orange) and P′ (green) in the presence (solid) and absence (dotted) of 4-aminobenzoic acid substrate.
Comparison to a Mononuclear Fe-Hydroperoxide
Since electrophilic attack on substrates has also been observed for high-spin mononuclear Fe(III)-hydroperoxides, we evaluated the relative reactivity of mononuclear to binuclear iron hydroperoxo species. The [(TMC)Fe(III)-OOH]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacylotetradecane) complex72 that has an end-on hydroperoxide was chosen and tested for the reaction with 4-aminobenzoic acid. In principle, the lone-pair electrons on the amino group could directly attack the hydroperoxo-σ*-based MO to form the N–O bond. This two-electron pathway, however, is highly unfavorable in the μ-1,2-hydropero-bridged binuclear Fe(III) P′ structure (ΔG‡ ≈ 30 kcal/mol), because the hydroperoxo-σ*-based MO is oriented along the O–O vector, and in the cis-μ-1,2-hydroperoxo structure this is sterically too hindered to have overlap with the HOMO of the substrate (Figure 12a).
Figure 12.

Direct electrophilic attack on p-aminobenzoic acid by (a) a μ-1,2-hydroperoxo-bridged Fe dimer, (b) a μ-1,1-hydroperoxo-bridged Fe dimer, and (c) an end-on-hydroperoxo Fe monomer. At the left is the frontier MO interaction, and at the right is a schematic drawing for each reaction, for each panel.
Alternatively, in the mononuclear system, the hydroperoxo-σ*-based MO is now accessible and thus the two-electron electrophilic pathway can occur, but still has an energy barrier of ΔG‡ ≈ 21 kcal/mol (Figures 12c and S14). The high energy barrier, despite the lack of steric hindrance, is due to the transient formation of an unstable Fe(III)-oxo, where the substrate has transferred 2e− into the hydroperoxo-σ*-based MO in the hydroxylation.
The most favorable two-electron process is achieved in the binuclear Fe site when the μ-1,2-hydroperoxide bridge is rearranged into a μ-1,1-hydroperoxide (ΔG‡ ≈ 10 kcal/mol for this rearragement). The protonated oxygen of the μ-1,1-hydroperoxide can undergo an electrophilic attack with a total barrier comparable to SET (ΔG‡ ≈ 13 kcal/mol; Figures 12b and S15), as it forms an oxo-bridged biferric product. This comparable barrier to SET is understandable, because, compared to the peroxo-σ*-based MO of the μ-1,2-hydroperoxide, that of the μ-1,1-hydroperoxide is also significantly stabilized by the protonation but less sterically hindered for the electrophilic attack on substrates. Although the X-ray crystal structure of the resting biferric AurF displays an active site open enough to accommodate the substrate without “active site crowding”,39 conformational changes of the binuclear Fe site upon substrate binding cannot be excluded. The reaction coordinate involving the μ-1,1-hydroperoxide has been suggested for the electrophilic aromatic substitution reaction.3
CONCLUSIONS
The NRVS analysis of P′ in AurF has revealed this intermediate to be a protonated μ-1,2-peroxo-bridged Fe(III)2 complex. This structure reproduces the unusual absorption and Mössbauer spectroscopic features of P′ and is calculated to be viable in the electrophilic/SET N-oxygenation of the aminobenzoic acid substrate. The energy barrier for this process in P′ is ~8 kcal/mol lower than for a non-protonated P, because protonation increases the electron affinity and stabilizes the high-valent product of O–O bond cleavage. This protonation-enhanced electrophilicity of P′ and its conversion to an Fe(IV)Fe(III) intermediate in AurF may be relevant to the activation of P to form P′ for the conversion to X in RNR20 and the electrophilic chemistry of ToMO.7
Finally, while mononuclear Fe(III) hydroperoxides are also electrophilic, this reaction is more favorable in the binuclear Fe hydroperoxo system due to its stabilization of the oxo product.
Acknowledgments
Use of the synchrotron radiation at the BL09XU of SPring-8 was approved by JASRI (Proposal No. 2010B1569 and 2011A1326), and use of the Advanced Photon Source was supported by the Department of Energy, Office of Science, contract DE-AC-02-06CH11357. Financial support for this research was provided by grants from the National Science Foundation (MCB1404866 to E.I.S. and CHE-1058931 to J.M.B. and C.K.), the National Institutes of Health (GM40392 to E.I.S.), and JSPS KAKENHI (No. 24221005 to M. Seto).
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02997.
Detailed descriptions on VTVH MCD data fit; DFT model structures and their predicted NRVS, Abs, and Mössbauer spectroscopic data and assignments; DFT-calculated PES; and electronic structural information on transient and intermediate species calculated (PDF)
References
- 1.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 2.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Park K. JBIC, J Biol Inorg Chem. 2016;21:575. doi: 10.1007/s00775-016-1372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stubbe J. Curr Opin Chem Biol. 2003;7:183. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 5.Krebs C, Bollinger JM, Jr, Booker SJ. Curr Opin Chem Biol. 2011;15:291. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziehl M, He J, Dahse HM, Hertweck C. Angew Chem, Int Ed. 2005;44:1202. doi: 10.1002/anie.200461990. [DOI] [PubMed] [Google Scholar]
- 7.Murray LJ, Naik SG, Ortillo DO, Garcia-Serres R, Lee JK, Huynh BH, Lippard SJ. J Am Chem Soc. 2007;129:14500. doi: 10.1021/ja076121h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bollinger JM, Jr, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. J Am Chem Soc. 1998;120:1094. [Google Scholar]
- 9.Broadwater JA, Achim C, Münck E, Fox BG. Biochemistry. 1999;38:12197. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 10.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 11.Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 12.Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1995;117:10174. [Google Scholar]
- 13.Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr Biochemistry. 1998;37:14659. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 14.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr Science. 1997;275:515. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 15.Song WJ, Behan RK, Naik SG, Huynh BH, Lippard SJ. J Am Chem Soc. 2009;131:6074. doi: 10.1021/ja9011782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tinberg CE, Lippard SJ. Biochemistry. 2009;48:12145. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valentine AM, Stahl SS, Lippard SJ. J Am Chem Soc. 1999;121:3876. [Google Scholar]
- 18.Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que L. Proc Natl Acad Sci U S A. 2009;106:14814. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yun D, Garcia-Serres R, Chicalese BM, An YH, Huynh BH, Bollinger JM., Jr Biochemistry. 2007;46:1925. doi: 10.1021/bi061717n. [DOI] [PubMed] [Google Scholar]
- 20.Saleh L, Krebs C, Ley BA, Naik S, Huynh BH, Bollinger JM., Jr Biochemistry. 2004;43:5953. doi: 10.1021/bi036099e. [DOI] [PubMed] [Google Scholar]
- 21.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2009;131:13608. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li N, Korboukh VK, Krebs C, Bollinger JM., Jr Proc Natl Acad Sci U S A. 2010;107:15722. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pandelia ME, Li N, Nørgaard H, Warui DM, Rajakovich LJ, Chang W-c, Booker SJ, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2013;135:15801. doi: 10.1021/ja405047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Lipscomb JD. J Am Chem Soc. 2015;137:1608. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2004;126:8842. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 26.Han Z, Sakai N, Böttger LH, Klinke S, Hauber J, Trautwein AX, Hilgenfeld R. Structure. 2015;23:882. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Jasniewski AJ, Engstrom LM, Vu VV, Park MH, Que L. JBIC, J Biol Inorg Chem. 2016;21:605. doi: 10.1007/s00775-016-1373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acheson JF, Bailey LJ, Brunold TC, Fox BG. Nature. 2017;544:191. doi: 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- 29.Knoot CJ, Kovaleva EG, Lipscomb JD. JBIC, J Biol Inorg Chem. 2016;21:589. doi: 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen KP, Bell CB, III, Clay MD, Solomon EI. J Am Chem Soc. 2009;131:12155. doi: 10.1021/ja809983g. [DOI] [PubMed] [Google Scholar]
- 31.Srnec M, Rokob TA, Schwartz JK, Kwak Y, Rulíšek L, Solomon EI. Inorg Chem. 2012;51:2806. doi: 10.1021/ic2018067. [DOI] [PubMed] [Google Scholar]
- 32.Bochevarov AD, Li J, Song WJ, Friesner RA, Lippard SJ. J Am Chem Soc. 2011;133:7384. doi: 10.1021/ja110287y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seto M, Yoda Y, Kikuta S, Zhang XW, Ando M. Phys Rev Lett. 1995;74:3828. doi: 10.1103/PhysRevLett.74.3828. [DOI] [PubMed] [Google Scholar]
- 34.Sturhahn W, Toellner TS, Alp EE, Zhang X, Ando M, Yoda Y, Kikuta S, Seto M, Kimball CW, Dabrowski B. Phys Rev Lett. 1995;74:3832. doi: 10.1103/PhysRevLett.74.3832. [DOI] [PubMed] [Google Scholar]
- 35.Sturhahn W. J Phys: Condens Matter. 2004;16:S497. [Google Scholar]
- 36.Alp EE, Mooney TM, Toellner T, Sturhahn W. Hyperfine Interact. 1994;90:323. [Google Scholar]
- 37.Scheidt WR, Durbin SM, Sage JT. J Inorg Biochem. 2005;99:60. doi: 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 38.Solomon EI, Pavel EG, Loeb KE, Campochiaro C. Coord Chem Rev. 1995;144:369. [Google Scholar]
- 39.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci U S A. 2008;105:6858. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park K, Solomon EI. Can J Chem. 2014;92:975. doi: 10.1139/cjc-2014-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park K, Tsugawa T, Furutachi H, Kwak Y, Liu LV, Wong SD, Yoda Y, Kobayashi Y, Saito M, Kurokuzu M, Seto M, Suzuki M, Solomon EI. Angew Chem, Int Ed. 2013;52:1294. doi: 10.1002/anie.201208240. [DOI] [PubMed] [Google Scholar]
- 42.Kohn W, Sham LJ. Phys Rev. 1965;140:A1133. [Google Scholar]
- 43.Slater JC. Quantum Theory of Molecular and Solids. Vol. 4. McGraw-Hill; New York: 1974. The Self-Consistent Field for Molecular and Solids. [Google Scholar]
- 44.Becke AD. Phys Rev A: At, Mol, Opt Phys. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 45.Binning RC, Curtiss LA. J Comput Chem. 1990;11:1206. [Google Scholar]
- 46.Blaudeau JP, McGrath MP, Curtiss LA, Radom L. J Chem Phys. 1997;107:5016. [Google Scholar]
- 47.Ditchfield R, Hehre WJ, Pople JA. J Chem Phys. 1971;54:724. [Google Scholar]
- 48.Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA. J Chem Phys. 1982;77:3654. [Google Scholar]
- 49.Frisch MJ, Pople JA, Binkley JS. J Chem Phys. 1984;80:3265. [Google Scholar]
- 50.Gluckstern RL, Lin SR. Phys Rev. 1964;136:B859. [Google Scholar]
- 51.Gordon MS. Chem Phys Lett. 1980;76:163. [Google Scholar]
- 52.Hariharan PC, Pople JA. Theor Chim Acta. 1973;28:213. [Google Scholar]
- 53.Hariharan PC, Pople JA. Mol Phys. 1974;27:209. [Google Scholar]
- 54.Hehre WJ, Ditchfield R, Pople JA. J Chem Phys. 1972;56:2257. [Google Scholar]
- 55.Perdew JP. Phys Rev B: Condens Matter Mater Phys. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 56.Rassolov VA, Pople JA, Ratner MA, Windus TL. J Chem Phys. 1998;109:1223. [Google Scholar]
- 57.Rassolov VA, Ratner MA, Pople JA, Redfern PC, Curtiss LA. J Comput Chem. 2001;22:976. [Google Scholar]
- 58.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision B.01. Gaussian, Inc; Wallingford, CT: 2010. [Google Scholar]
- 59.Petrenko T, Kossmann S, Neese F. J Chem Phys. 2011;134:054116. doi: 10.1063/1.3533441. [DOI] [PubMed] [Google Scholar]
- 60.Neese F, Olbrich G. Chem Phys Lett. 2002;362:170. [Google Scholar]
- 61.Neese F. Inorg Chim Acta. 2002;337:181. [Google Scholar]
- 62.Schäfer A, Horn H, Ahlrichs R. J Chem Phys. 1992;97:2571. [Google Scholar]
- 63.The ORCA basis set “CoreProp” was used. This basis is based on the TurboMole DZ basis developed by Ahlrichs and co-workers and obtained from the basis set library under ftp.chemie.unikarlsru-he.de/pub/basen.
- 64.Yang YS, Broadwater JA, Pulver SC, Fox BG, Solomon EI. J Am Chem Soc. 1999;121:2770. [Google Scholar]
- 65.Noodleman L. J Chem Phys. 1981;74:5737. [Google Scholar]
- 66.Mitić N, Schwartz JK, Brazeau BJ, Lipscomb JD, Solomon EI. Biochemistry. 2008;47:8386. doi: 10.1021/bi800818w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwartz JK, Wei P-p, Mitchell KH, Fox BG, Solomon EI. J Am Chem Soc. 2008;130:7098. doi: 10.1021/ja800654d. [DOI] [PubMed] [Google Scholar]
- 68.Wei P-p, Skulan AJ, Mitić N, Yang YS, Saleh L, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2004;126:3777. doi: 10.1021/ja0374731. [DOI] [PubMed] [Google Scholar]
- 69.Yang YS, Broadwater JA, Pulver SC, Fox BG, Solomon EI. J Am Chem Soc. 1999;121:2770. [Google Scholar]
- 70.Root DE, Mahroof-Tahir M, Karlin KD, Solomon EI. Inorg Chem. 1998;37:4838. doi: 10.1021/ic980606c. [DOI] [PubMed] [Google Scholar]
- 71.Jayapal P, Rajaraman G. Phys Chem Chem Phys. 2012;14:9050. doi: 10.1039/c2cp40874k. [DOI] [PubMed] [Google Scholar]
- 72.Liu LV, Hong S, Cho J, Nam W, Solomon EI. J Am Chem Soc. 2013;135:3286. doi: 10.1021/ja400183g. [DOI] [PMC free article] [PubMed] [Google Scholar]