

Abstract
Numerous studies show a direct relationship between circulating autoantibodies, characteristic of systemic autoimmune disorders, and primary hypertension in humans. Whether these autoantibodies mechanistically contribute to the development of hypertension remains unclear. Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterized by aberrant immunoglobulin production, notably pathogenic autoantibodies, and is associated with prevalent hypertension, renal injury, and cardiovascular disease. Because plasma cells produce the majority of serum immunoglobulins and are the primary source of autoantibodies in SLE, we hypothesized that plasma cell depletion using the proteasome inhibitor bortezomib would lower autoantibody production and attenuate hypertension. Thirty week old female SLE (NZBWF1) and control (NZW) mice were injected i.v. with vehicle (0.9% saline) or bortezomib (0.75 mg/kg) twice weekly for four weeks. Bortezomib treatment significantly lowered the percentage of bone marrow plasma cells in SLE mice. Total plasma IgG and anti-dsDNA IgG levels were higher in SLE mice as compared to control mice, but were lowered by bortezomib treatment. Mean arterial pressure (MAP; mmHg) measured in conscious mice by carotid artery catheter was higher in SLE mice than in control mice, but MAP was significantly lower in bortezomib-treated SLE mice. Bortezomib also attenuated renal injury, as assessed by albuminuria and glomerulosclerosis, and reduced glomerular immunoglobulin deposition and B and T lymphocytes infiltration into the kidneys. Taken together, these data show that the production of autoantibodies by plasma cells mechanistically contributes to autoimmune associated hypertension, and suggests a potential role for patients with primary hypertension who have increased circulating immunoglobulins.
Keywords: hypertension, autoimmunity, systemic lupus erythematosus, autoantibodies, plasma cells
Introduction
Mounting evidence suggests that increased immunoglobulin production may contribute to the pathogenesis of hypertension. Studies from as early as the 1970s indicate that patients with untreated or treated essential hypertension have higher levels of circulating IgG and IgM as compared to normotensive individuals1–3. In addition, multiple studies by Kristensen and colleagues measured the levels of autoantibodies to a variety of autoantigens and found that hypertensive individuals were more likely to have circulating autoantibodies4–6. Taken together, these clinical studies suggest a link between autoantibodies and the development of hypertension. In support of this concept, patients with autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) have prevalent autoantibody production7, 8 and high rates of hypertension9–15. SLE is a prototypic systemic autoimmune disease that predominately affects women of childbearing age. It is characterized by a loss of tolerance to self-antigens that results in the production of autoantigen-specific B and T lymphocytes, which leads to pathogenic autoantibody production, especially against nuclear components. These autoantibodies form immune complexes that deposit in tissues such as the kidneys leading to chronic inflammation and end-organ damage. However, it is unclear whether the autoantibodies produced in SLE disease mechanistically contribute to the development of hypertension in these patients.
Animal models of autoimmune diseases such as SLE are an important tool to understand the link between autoantibodies and hypertension. The female NZBWF1 mouse mimics many of the characteristics of SLE disease, including autoantibody production, immune complex mediated renal injury, and hypertension16, 17. Recent studies by our laboratory showed that long term depletion of B cells using anti-CD20 resulted in decreased autoantibody production and prevented the development of hypertension in SLE mice18. In addition, chronic treatment with the immunosuppressive drug mycophenolate mofetil selectively depleted B cells and attenuated hypertension in SLE mice19. Taken together, these studies clearly demonstrate an association between B cells, autoantibodies and the development of hypertension; however, these treatments were only effective when started before disease onset. Similarly, therapies that target B cells in humans, such as anti-CD20 (Rituximab) have had limited success in large controlled clinical trials20–22. It has been suggested that the limited efficacy is at least partially due to the persistence of long-lived plasma cells that are not targeted by B cell therapies23.
Plasma cells, which differentiate from germinal center or memory B cells, reside in the bone marrow and spleen for months to years and are responsible for the majority of serum immunoglobulin production24, 25, including SLE autoantibodies26. Bortezomib (Velcade™) is a potent and selective inhibitor of the 26S proteasome that is currently used in the treatment of multiple myeloma, a plasma cell neoplasia27. Neubert et al. reported that treatment of NZBWF1 mice, an established female mouse model of SLE, with bortezomib effectively depleted plasma cells and ameliorated symptoms of lupus nephritis28. In addition, clinical studies have shown that bortezomib is effective at lessening disease severity in patients with refractory SLE and persistent autoantibody titers29. However, the effect of plasma cell depletion on the development of hypertension remains unknown. In the present study, we demonstrate a mechanistic role for plasma cells and autoantibody production in the pathogenesis of hypertension associated with autoimmunity.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Adult (30 week old) female NZBWF1 (SLE, n=24) and NZW/LacJ (control, n=26) mice (Jackson Laboratories, Bar Harbor, ME) were used in this study as published previously16. Mice were maintained on a 12 hour light/dark cycle in temperature controlled rooms with access to chow and water ad libitum. All studies were performed with the approval of the University of Mississippi Medical Center Institutional Animal Care and Use Committee and in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Bortezomib Administration
Bortezomib (Velcade; Millennium Pharmaceuticals, Cambridge, MA) was dissolved in sterile 0.9% NaCl. Mice were administered 0.75 mg/kg 2X week iv for 4 weeks in a volume of 50 μL. Mice not receiving BTZ were injected with 50 μL 0.9% NaCl (vehicle).
Blood pressure
Mean arterial pressure (MAP, mmHg) was recorded via indwelling carotid artery catheters in freely moving conscious mice as previously described by our laboratory18, 30–33.
Preparation of cells for flow cytometry
Bone marrow cells
Bone marrow cells were isolated from the femur and tibia as previously described34. Briefly, the femurs were dissected from the surrounding muscle and rinsed in sterile Hank’s balanced salt solution (HBSS). Both ends of the bone were trimmed to expose the marrow shaft. The marrow was flushed with 10 mL of sterile HBSS and large marrow particles were allowed to settle and were removed. The resulting cell suspension was used for flow cytometric analyses.
Peripheral blood leukocytes (PBL)
Blood was collected from the retroorbital plexus from bortezomib or vehicle-treated animals at 34 or 35 weeks (4 weeks bortezomib) of age. The blood was centrifuged at 350 × g to isolate plasma. Erythrocytes were lysed by adding 10X volume of 1X PharmLyse (BD Biosciences, San Jose, CA). After incubation for 5 min at room temperature, blood was centrifuged at 200 × g for 5 min. The pelleted cells were washed 1 X PBS, 2% FCS and centrifuged at 350 × g for 5 min. The purified PBL were suspended in 90% FCS, 10% DMSO and stored at −80°C until use.
Renal immune cells
One kidney was homogenized in 5 mL RPMI media containing 200 U/mL DNase and 10 mg/mL collagenase IV using the GentleMACS Octo Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) using a user-defined protocol for mouse kidney. The resulting homogenate was filtered through a 70 μM cell strainer and washed with 1X PBS containing 2% FCS and 2 mM EDTA. The single cell suspension was centrifuged at 300 × g for 10 min. The resulting cell pellet was then resuspended in 1X PBS, 2% FCS. Lymphocytes were isolated from the kidney cell suspension using Lymphoprep (Accurate Chemical, Westbury, NY) according to the manufacturer’s instructions.
Flow cytometric analyses
For all flow cytometric analyses, cells were first washed and resuspended in 1X PBS, 2% FCS, and 0.9% sodium azide at a concentration of 2 × 107 cells/mL. 1×106 cells (50 μL) were aliquoted into a flow cytometry tube and incubated with 0.25 μg of anti-mouse CD32/CD16 (FcR block, BD Biosciences) for 5 min. on ice. For bone marrow plasma cells, cells were stained with anti-CD138-APC (clone 281-2, BD Biosciences) followed by fixation and permeabilization using BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. After washing with Perm/Wash buffer, cells were stained with rat anti-kappa light chain (clone 187.1, BD Biosciences). For staining of PBL lymphocytes, cells were stained with either isotype control antibodies, a T cell subset antibody cocktail (BD Biosciences), which contains anti-mouse CD3e-PE-Cy7 (clone 145-2C11), CD4-PE (clone RM4-5), and CD8-APC (clone 53-6.7), or anti-CD45R-PE-Cy7 (clone RA3-6B2). For kidney lymphocytes, cells were stained with anti-CD45-FITC as well as anti-mouse CD3e-PE-Cy7, CD4-PE, and CD8-APC, or anti-CD45R-PE-Cy7. Kidney lymphocytes were gated on live cells as well as on CD45+ cells. Cells were incubated on ice for 30 minutes and protected from light. All samples were analyzed on a Gallios (Becton Dickinson, Franklin Lakes, NJ) flow cytometer at the UMMC Flow Cytometry core facility. A total of 100,000 events were acquired for each sample. Data were analyzed using Kaluza software.
Autoantibodies and total IgG
Anti-dsDNA IgG was detected in plasma at 34 weeks of age (SLE mice) or 35 weeks of age (control mice) using the anti-dsDNA IgG ELISA (Alpha Diagnostic International, San Antonio, TX) per the manufacturer’s instructions and as previously described by our laboratory18, 30, 32, 33. Total plasma IgG concentrations were determined at the same ages using the mouse IgG ELISA kit (Alpha Diagnostic International) according to the manufacturer’s instructions.
Renal Injury
Urinary albumin was monitored weekly by dipstick analysis (Albustix; Siemens). Animals were considered positive for albuminuria at ≥100 mg/dL18, 30, 31, 33. Urinary albumin excretion rate (mg/day) was assessed by ELISA (Alpha Diagnostic International) using overnight urine samples collected at the conclusion of the study as previously described18, 30, 31, 33. Glomerulosclerosis scoring was assessed by investigators blinded to the sample as previously described by our laboratory18, 19.
Immunofluorescence
Four micron frozen kidney sections were first washed in PBS, 3×5 minutes each. The blocking solution was applied for 30 minutes at room temperature. The primary antibody was then added for 2 hours at room temperature. The sections were washed for 5 minutes in PBS three times. FITC primary antibody (anti-IgG, Sigma Aldrich) was used. After antibody incubation, slides were washed in PBS 3×5 minutes and then viewed and imaged with confocal microscopy.
Statistical Analysis
Data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism 7. A two-way ANOVA was used to analyze treatment (bortezomib vs. vehicle) or group (SLE vs. control) interactions. One-way ANOVA was used to analyze individual differences between groups and Tukey’s post hoc test for multiple comparisons was used to compare groups. An unpaired T test was used to analyze differences between renal lymphocytes or circulating lymphocytes in vehicle and bortezomib-treated SLE mice. A p value of less than 0.05 was considered statistically significant.
Results
Because bortezomib depletes plasma cells in both mice and humans28, we analyzed bone marrow plasma cells in a subset of control and SLE mice treated with vehicle or bortezomib using flow cytometry. Figure 1A shows representative dot plots of SLE mice treated with vehicle or bortezomib, and Figure 1B indicates the percentages of plasma cells in control and SLE mice. Both vehicle and bortezomib-treated control mice had low levels of plasma cells (control-vehicle: 0.12±0.03%, control-bortezomib: 0.12±0.08%) (Figure 1B), consistent with previous studies that show a low level (<0.5% of total bone marrow cells) of plasma cells in young, non-immunized mice35. Vehicle-treated SLE mice had significantly higher levels of plasma cells as compared to both groups of control animals (1.8±0.1%, p<0.0001 vs. control-vehicle and control-bortezomib), and SLE mice treated with bortezomib for four weeks had significantly lower percentages of plasma cells (1.8±0.1 vs. 0.94±0.1%, p<0.0001). To analyze any potential effects of bortezomib on other circulating immune cell populations, we examined circulating B and T lymphocytes in vehicle- and bortezomib-treated SLE mice. No differences were detected in the percentages of circulating CD45R+ B cells, CD3+CD4+ T cells, or CD3+CD8+ T cells (Figure 1C–E).
Figure 1.
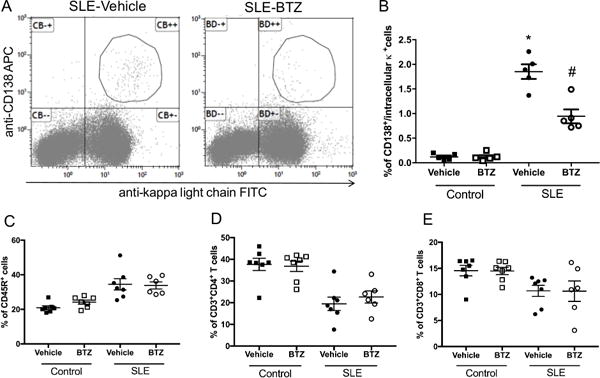
Effect of bortezomib treatment on bone marrow plasma cells in control and SLE mice. A, Representative dot plots of SLE mice treated with vehicle or bortezomib. Cells were isolated from femurs at the conclusion of the study and stained with anti-mouse CD138 and anti-mouse kappa light chain mAbs. Plasma cells are indicated in the figure. B, Percentage of plasma cells in vehicle and bortezomib-treated SLE and control mice. The percentages of plasma cells are higher in SLE mice as compared to control mice and were lowered by bortezomib treatment. *, p<0.0001 vs. all other groups; #, p<0.001 vs. control-vehicle and control-bortezomib. C, Percentage of circulating CD45R+ B cells in vehicle and bortezomib-treated SLE and control mice. D, Percentage of circulating CD3+CD4+ T cells in vehicle and bortezomib-treated SLE and control mice. E, Percentage of circulating CD3+CD8+ T cells in vehicle and bortezomib-treated SLE and control mice.
To analyze the downstream effects of depletion of immunoglobulin-secreting plasma cells, levels of anti-dsDNA IgG and total plasma IgG were measured at the conclusion of the study. Vehicle-treated SLE mice have significantly higher levels of anti-dsDNA IgG autoantibodies as compared to control mice (0.25±0.06 vs. 1.23±0.20 OD450, p<0.001) (Figure 2A); however, autoantibody production was lower in bortezomib-treated SLE mice compared to vehicle-treated animals (1.23±0.2 vs. 0.40±0.09 OD450, p<0.01). In addition, vehicle-treated SLE mice had significantly higher concentrations of total plasma IgG as compared to control mice (2.1±0.2 vs. 5.0±1.2 mg/mL, p<0.05), (Figure 2B) which has been previously reported36. Bortezomib treatment significantly lowered plasma IgG in SLE mice as compared to vehicle-treated animals (1.5±0.48 vs. 5.0±1.2 mg/mL, p<0.01).
Figure 2.
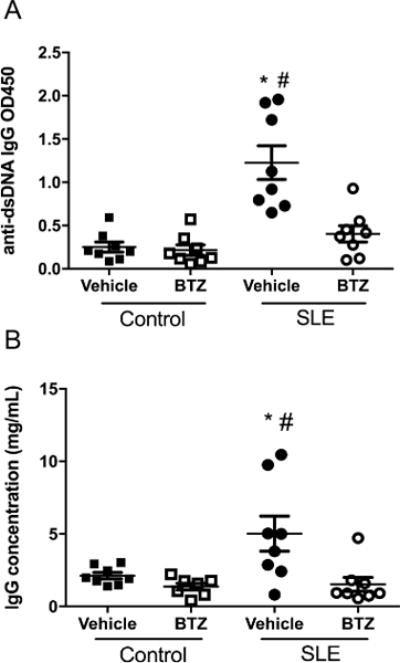
Effect of bortezomib treatment on plasma IgG and anti-dsDNA IgG in control and SLE mice. A, Anti-dsDNA IgG measured at 34 weeks of age in control and SLE mice. Plasma levels of anti-dsDNA IgG were higher in SLE mice and were lowered by bortezomib treatment. *, P<0.001 vs. control-vehicle and control-bortezomib; #, P<0.01 vs. SLE-bortezomib. B, Plasma IgG concentrations measured at 34 weeks of age in control and SLE mice. IgG concentrations were higher in SLE mice as compared to control mice, but were significantly lower after bortezomib treatment. *, P<0.05 vs. control-vehicle; #, P<0.01 vs. control-bortezomib and SLE-bortezomib.
To assess the role of plasma cells in the development of hypertension, mean arterial pressure (MAP) was measured in conscious freely moving mice at the conclusion of the study. Vehicle-treated SLE mice had elevated MAP as compared to vehicle-treated control mice (142±5 vs. 118±3 mmHg, p<0.0001) (Figure 3). Blood pressure was significantly lower in SLE mice treated with bortezomib (120±3 vs. 142±5 mmHg p<0.001), but bortezomib treatment did not affect blood pressure in control mice (118±3 vs. 114±1 mmHg).
Figure 3.
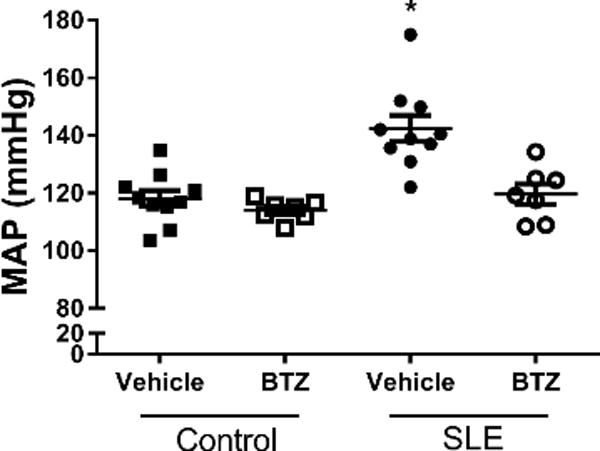
Effect of bortezomib treatment on MAP in control and SLE mice. MAP was significantly higher in SLE mice as compared to control mice. Bortezomib significantly lowered MAP in SLE mice, but had no effect in control mice. *, P<0.001 vs. all other groups.
NZBWF1 mice excrete large amounts of albumin in their urine and develop renal injury. Thirty-three percent of the vehicle treated SLE mice developed albuminuria, while only one mouse treated with bortezomib developed albuminuria (Figure 4A). Urinary albumin excretion was measured by ELISA at the conclusion of the study (Figure 4B). Vehicle-treated SLE mice had higher levels of urinary albumin excretion as compared to control mice (16.8±10 vs. 0.015±0.002 mg/day) which was lower in bortezomib treated mice (SLE-bortezomib: 0.24±0.16 mg/day), although the differences did not reach statistical significance. Bortezomib treatment did not affect urinary albumin in control animals (Control-vehicle: 0.015±0.002 mg/day vs. control-bortezomib: 0.057±0.03 mg/day). SLE mice had increased gomerulosclerosis as compared to control animals (Figure 4C–D), as measured by glomerulosclerosis index (0.13±0.07 vs. 0.90±0.2, p=0.11); however, SLE mice treated with bortezomib trended towards decreased glomerulosclerosis (SLE-bortezomib: 0.61±0.4). In addition, SLE mice had increased fibrosis as compared to control mice (0.28±0.12 vs. 1.0± 0.35%), but it was decreased in SLE mice treated with bortezomib (SLE-bortezomib: 0.27±0.064%) (Figure S1A–B). Deposition of IgG autoantibodies and immune complexes in the kidneys, as well as direct binding of IgG autoantibodies to in glomerular antigens, contributes to renal injury by initiating downstream inflammatory and fibrotic processes37. Because of the marked decrease in total IgG and anti-dsDNA autoantibodies in the plasma after treatment with bortezomib, we assessed the presence of IgG in the glomeruli of control and SLE mice using immunofluorescence staining with anti-mouse IgG FITC (Figure 5A). Control animals, regardless of treatment, have low levels of IgG staining in their glomeruli, while vehicle treated SLE animals had high levels of IgG staining. Treatment with bortezomib decreased the levels of IgG staining in SLE mice to levels similar to that of control animals. Quantification of the fluorescence within the glomeruli is shown in Figure 5B.
Figure 4.
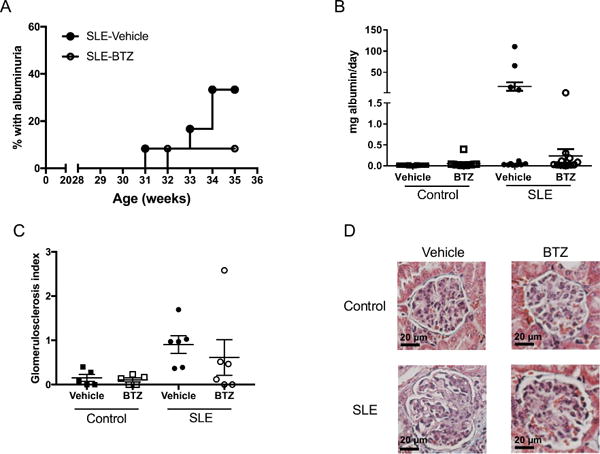
Effect of bortezomib on albuminuria and glomerulosclerosis in control and SLE mice. A, Weekly percentage of SLE mice with positive urinary albumin as measured by dipstick assay. B, Urine albumin in control and SLE mice as measured by albumin ELISA at the conclusion of the study. Urine albumin was similar in control mice treated with vehicle or bortezomib. Albumin was higher in SLE mice as compared to control mice, but bortezomib treatment lowered albuminuria. C, Glomerulosclerosis index assessed in control and SLE mice administered vehicle or bortezomib. D, Representative pictures of glomerulosclerosis (40X) from paraffin-embedded kidneys stained with Masson’s trichrome.
Figure 5.
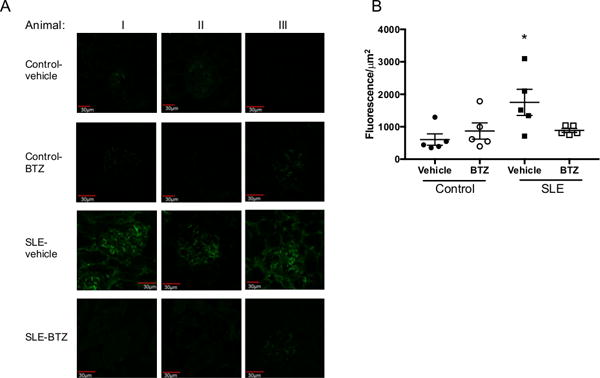
Effect of bortezomib on glomerular IgG deposition in SLE and control mice. A, Control and SLE mice kidney sections were stained with anti-mouse IgG FITC. Representative glomeruli are shown from three different mice in each treatment group. B, Quantification of fluorescence intensity per μm2 in each glomerulus (5 mice/group). *, p<0.05 vs. Control-vehicle. SLE mice have increased glomerular IgG staining as compared to control mice, but it is decreased after treatment with bortezomib.
Renal lymphocyte infiltration is mechanistically linked with several models of experimental hypertension38–41, including the female NZBWF1 mous19. To assess the effect of plasma cell depletion on the infiltration of immune cells into the kidneys, we isolated renal leukocytes, stained with T cell (CD3/CD4/CD8) and B cell (CD45R) specific antibodies and performed flow cytometric analyses (Figure 6A–C). SLE-vehicle treated mice had significantly higher levels of CD3+CD4+ T cells (9.7±3.0 vs. 2.1±0.3%, p<0.05), CD3+CD8+ T cells (3.3±0.5 vs. 0.90±0.2%, p<0.001), and CD45R+ B cells (5.7±0.7 vs. 2.2±0.6%, p<0.01) as compared to SLE mice treated with bortezomib. Because renal lymphocyte infiltration can lead to downstream inflammatory cytokine production, we also analyzed renal inflammatory cytokines using a flow cytometric bead assay and vehicle-treated SLE mice had higher levels of TNF-α, MCP-1, and IL-6 (Figure S2A–C). The data suggest that these cytokines and chemokines are lower in bortezomib treated SLE mice.
Figure 6.
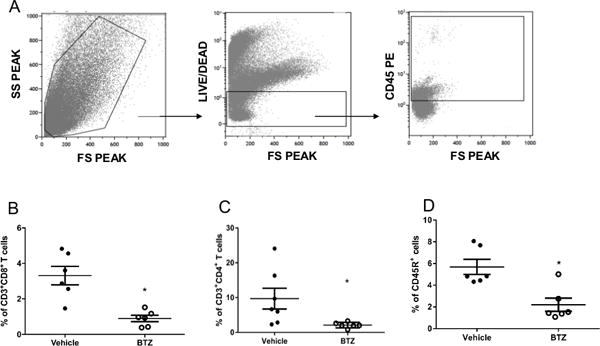
Effect of bortezomib on renal lymphocyte infiltration in SLE mice. Cells were stained with anti-mouse-CD3 PeCy7, anti-mouse-CD4 PE, and anti-mouse-CD8 APC or with anti-mouse- CD45R PE-Cy7. SLE-vehicle treated mice have lower levels of renal B and T lymphocytes. A, Representative scatter profile of leukocytes isolated from the kidney. B, Percentage of CD3+CD4+ T cells in SLE mice treated with vehicle and bortezomib. *, p<0.05 vs. SLE-vehicle. C, Percentage of CD3+CD8+ T cells in SLE mice treated with vehicle and bortezomib. *, p<0.001 vs. SLE-vehicle. D, Percentage of CD45R+ B cells in SLE mice treated with vehicle and bortezomib. *, p<0.01 vs. SLE-vehicle.
Discussion
The prevalence of hypertension is markedly increased in patients with SLE, an autoimmune disorder driven by the production of autoantibodies, and numerous studies show that circulating autoantibodies are increased in patients with primary hypertension. The present study directly tested whether depletion of plasma cells, which are responsible for the vast majority of autoantibody production, would attenuate the development of hypertension in an established female mouse model of SLE. The major new findings of this study are that plasma cell depletion 1) effectively lowers circulating autoantibodies and ameliorates autoimmune-associated hypertension independent of changes in circulating T and B lymphocytes, 2) prevents IgG deposition and subsequently the renal infiltration of inflammatory cells that are known to mechanistically contribute to the pathogenesis of hypertension, and 3) prevents the development of renal injury, as evidenced by decreased albuminuria and glomerulosclerosis. These data advance our understanding of the mechanisms increasing the prevalence of hypertension in patients with autoimmune disorders, and has broad implications for understanding the pathogenesis of primary hypertension in patients with increased circulating immunoglobulins.
Plasma cell depletion
Previous studies have shown that bortezomib is effective in the treatment of several rodent models of autoimmunity. For example, bortezomib depleted plasma cells and alleviated symptoms in a rat model of myasthenia gravis characterized by autoantibodies to the acetylcholine receptor (AChR) of skeletal muscle42. In addition, Lee et al. used bortezomib for treatment of a mouse model of collagen-induced arthritis. The therapy reduced arthritis severity and lowered the levels of inflammatory cytokines TNF-⍺, IL-1β, and IL-6 within inflamed joints43. Neubert and colleagues were the first to report that bortezomib could also deplete autoreactive plasma cells produced during SLE and attenuate lupus nephritis in NZBWF1 mice28. The authors reported a significant decrease in IgG- and anti-dsDNA IgG secreting cells in the spleen and bone marrow after eight weeks of bortezomib treatment, as measured by ELISPOT analyses. This depletion of plasma cells corresponded with a near complete disappearance of anti-dsDNA autoantibodies and a 50% decrease in plasma IgG concentrations. In the present study, a four-week treatment with bortezomib starting at 30 weeks of age resulted in a 50% decrease in the percentage of bone marrow plasma cells in SLE animals that corresponded with a 67% reduction in circulating anti-dsDNA IgG and a 70% reduction in plasma IgG levels (Figure 2). The lack of an effect of bortezomib in control animals may be due to the generally low percentages of plasma cells, making it difficult to identify changes via flow cytometry.
There are several mechanisms by which bortezomib can deplete plasma cells. First, due to their excessive immunoglobulin synthesis, plasma cells produce large amounts of unfolded proteins and defective ribosomal products, which accumulate in the ER. While a certain amount of the unfolded protein response is needed for plasma cell survival, proteasome inhibition rapidly induces ER stress and activates the terminal unfolded protein response, leading to the expression of the proapoptotic protein Chop and caspase activation44, 45. Previous studies have shown that bortezomib rapidly induces robust Chop mRNA expression in plasma cells, but not in splenic T and B cells28. This aligns with results obtained in our study in which no changes in circulating T and B lymphocytes were observed after treatment with bortezomib (Figure 1C–E).
Hypertension
Previous studies by our laboratory implicate an important role for B cells in the development of hypertension; however, B cells have multiple pathogenic roles that could contribute to hypertension. These include antigen presentation to autoreactive T cells46, 47 and direct contributions to local inflammation48. Therefore, the question of whether autoantibodies can mechanistically drive the development of hypertension remains unclear. The current study significantly advances our understanding because plasma cells do not serve these additional functions, allowing for a direct test of the role for autoantibodies in the development of hypertension in SLE.
At least 180 different autoantibodies have been identified in the serum of patients with SLE, many of which have been linked to disease manifestations49. The pathogenic actions of autoantibodies produced during SLE disease include immune complex formation and deposition, direct cell surface binding, interaction with cell surface autoantigens and apoptotic cells, and binding to cross-reactive extracellular molecules50, 51. Each of these pathogenic actions has the potential to contribute to the development of hypertension in SLE. For example, agonistic autoantibodies specific for the angiotensin II type I receptor (AT1R-AA), which have been identified in hypertensive individuals52–57, are present in some patients with SLE. A small clinical study revealed that SLE patients who tested positive for the AT1R-AA had higher blood pressure than those that were negative for the autoantibody58. In addition, autoantibodies directed against endothelial cells are present in up to 80% of patients with SLE, depending on the cohort59–61. While the effects of these autoantibodies are not fully characterized, they have been shown to enhance leukocyte adhesion and inflammation in vitro62 and have been associated with vascular injury and endothelial dysfunction in clinical studies63.
The present work is the first to demonstrate a mechanistic role for plasma cells in autoimmune-associated hypertension. A previously published study showed that bortezomib attenuated angiotensin II mediated hypertension after two weeks in male Sprague Dawley rats64. The authors suggested that bortezomib’s beneficial effects resulted from proteasome inhibition to suppress reactive oxygen species generation and inhibit angiotensin II-induced vascular smooth muscle cell proliferation. However, this study did not examine the effect of bortezomib on plasma cells or antibodies, which have recently been suggested to have a role in angiotensin II hypertension in mice65. In addition, the vascular hypertrophy and reactive oxygen species mirror the changes in blood pressure making it difficult to discern whether these changes are pressure dependent or independent. Importantly, bortezomib does not appear to universally reduce blood pressure in experimental models given that treatment of Dahl SS rats on a high salt diet did not significantly reduce systolic blood pressure66.
Renal mechanisms
In the present study, SLE mice treated with bortezomib have decreased renal injury, as measured by albuminuria and assessment of glomerular injury. These results are in agreement with previous studies in which bortezomib protected NZBWF1 mice from renal injury28, 67. We also examined glomerular IgG deposition using immunofluorescence microscopy. The presence of IgG is greatly reduced in the glomeruli of SLE mice treated with bortezomib (Figure 5). Anti-dsDNA autoantibodies of all IgG isotypes, which were measured in the present study, have been shown to be involved in the pathogenesis of lupus nephritis and other renal manifestations of the disease37. Current evidence indicates that anti-dsDNA IgG can bind to exposed chromatin fragments at the glomerular basement membrane (GBM)68; interact directly with GBM components such as α-actinin69, laminin70, and entactin71; and form anti-dsDNA immune complexes in the circulation which deposit in glomeruli68. The presence of CD45R+ B lymphocytes as well as CD3+CD4+ and CD3+CD8+ T lymphocytes in the kidneys was markedly reduced in bortezomib-treated SLE mice (Figure 6). This is consistent with previous studies by our laboratory, in which treatment with the immunosuppressive drug mycophenolate mofetil reduced the numbers of renal CD4+ T cells and CD45R+ B cells19. Both B and T lymphocytes contribute to the progression of renal injury during SLE. B lymphocytes and plasma cells promote intrarenal autoantibody production72, and both B and T lymphocytes promote renal inflammation73. Thus, the decrease in B and T lymphocytes in the kidneys likely reduces renal inflammation, which is an important factor in the development of hypertension. Furthermore, analyses of renal inflammatory cytokines using a flow cytometry based cytokine bead array indicated that SLE mice treated with bortezomib had decreased levels of the inflammatory cytokines TNF-α, MCP-1, and IL-6 (Figure S2). However, it is unlikely that the hypertension in the NZBWF1 mouse model is secondary to renal injury caused by infiltrating immune cells. This is supported by our data showing that all of the vehicle-treated SLE mice develop hypertension whereas only one-third of the vehicle-treated SLE mice in the present study developed albuminuria. In addition, we provide data (Figure S3A–B) showing that no correlation exists between hypertension and urinary albumin excretion or glomerulosclerosis. These data are consistent with studies reported in human SLE patients suggesting a dissociation of renal injury from blood pressure14.
Perspectives
Autoantibody-secreting plasma cells present a therapeutic problem in SLE and other autoimmune diseases because they are resistant to treatment with traditional therapies such as cyclophosphamide74 or mycophenolate mofetil (MMF)75. Moreover, plasma cells are resistant to the effects of B cell depletion agents such as anti-CD20 (rituximab) and anti-BAFF (belimumab)76. Previous studies by our laboratory have shown that depletion of B cells using anti-CD20 can prevent the development of hypertension when treatment starts before disease onset18, but is less effective once SLE disease is established. The present study supports the concept that autoreactive plasma cells are responsible for the majority of autoantibody production in established SLE disease, and that these autoantibodies play a mechanistic role in the hypertension during SLE. Hypertension affects a significant portion of patients with SLE, and autoantibodies are associated with primary hypertension. Therefore, continued study is warranted to further understand the role of specific antibodies in the pathogenesis of hypertension not only for patients with autoimmune disorders like SLE, but also for the population of individuals with primary hypertension and increased circulating immunoglobulins.
Supplementary Material
Novelty and Significance.
What is new?
This is the first study to investigate the effect of plasma cell depletion on autoimmune-associated hypertension.
Using a mouse model of systemic lupus erythematosus (SLE), a prototypic systemic autoimmune disorder, we showed that a short-term treatment with the proteasome inhibitor bortezomib was effective at depleting plasma cells in the bone marrow and significantly lowering circulating autoantibodies.
Treatment with borteozmib ameliorated hypertension in SLE mice and prevented renal immune complex deposition and immune cell infiltration into the kidney.
What is relevant?
Hypertension is prevalent in patients with autoimmune disease, which confers significant risk for cardiovascular disease.
SLE patients have elevated levels of circulating immunoglobulins and autoantibodies. In this study, we show that the autoantibodies produced by plasma cells play a mechanistic role in the development of hypertension during SLE.
Continued study is needed to further understand the role of specific antibodies in the pathogenesis of hypertension not only for patients with autoimmune disorders like SLE, but also for the population of individuals with primary hypertension and increased circulating immunoglobulins.
Summary.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterized by the production of autoantibodies by autoreactive plasma cells. Treatment with the proteasome inhibitor bortezomib in mice with SLE depleted plasma cells and lowered autoantibody production. This effectively lowered blood pressure and attenuated renal injury while reducing the infiltration of B and T lymphocytes into the kidneys.
Acknowledgments
Sources of funding:
E.B.T. was supported by an American Heart Association postdoctoral fellowship (17POST33410862), an individual NIH National Research Service Award (F32HL137393), and an NIH National Heart, Lung, and Blood Institute (NHLBI) T32HL105324-05. This work was supported by Veteran’s Administration Merit award (BX002604-01A2) to M.J.R. and NIH NHLBI awards PO1HL051971, P20GM104357 to UMMC-Department of Physiology and Biophysics. D.W.P. was supported by National Institute of Health (NIH) grant R01AR063124.
Footnotes
Conflict(s) of Interest/Disclosure(s): None
References
- 1.Ebringer A, Doyle AE. Raised serum igg levels in hypertension. Br Med J. 1970;2:146–148. doi: 10.1136/bmj.2.5702.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suryaprabha P, Padma T, Rao UB. Increased serum igg levels in essential hypertension. Immunol Lett. 1984;8:143–145. doi: 10.1016/0165-2478(84)90067-1. [DOI] [PubMed] [Google Scholar]
- 3.Hilme E, Herlitz H, Soderstrom T, Hansson L. Increased secretion of immunoglobulins in malignant hypertension. J Hypertens. 1989;7:91–95. [PubMed] [Google Scholar]
- 4.Kristensen BO. Autoantibodies in untreated and treated essential hypertension: Relationship to histocompatability leucocyte antigen-b15 and vascular complications. Clin Sci (Lond) 1979;57(Suppl 5):287s–290s. doi: 10.1042/cs057287s. [DOI] [PubMed] [Google Scholar]
- 5.Kristensen BO, Andersen PL. Autoantibodies in untreated and treated essential hypertension. I. Acta Med Scand. 1978;203:55–59. doi: 10.1111/j.0954-6820.1978.tb14831.x. [DOI] [PubMed] [Google Scholar]
- 6.Kristensen BO, Andersen PL, Wiik A. Autoantibodies and vascular events in essential hypertension: A five-year longitudinal study. J Hypertens. 1984;2:19–24. doi: 10.1097/00004872-198402000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in sle patients. Semin Arthritis Rheum. 2004;34:501–537. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Mewar D, Wilson AG. Autoantibodies in rheumatoid arthritis: A review. Biomed Pharmacother. 2006;60:648–655. doi: 10.1016/j.biopha.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Budman DR, Steinberg AD. Hypertension and renal disease in systemic lupus erythematosus. Archives of internal medicine. 1976;136:1003–1007. [PubMed] [Google Scholar]
- 10.Mandell BF. Cardiovascular involvement in systemic lupus erythematosus. Semin Arthritis Rheum. 1987;17:126–141. doi: 10.1016/0049-0172(87)90035-7. [DOI] [PubMed] [Google Scholar]
- 11.Petrin J, Rozman B, Dolenc P, Logar D, Bozic B, Vizjak A, Ferluga D, Jezersek P. The dissociation of arterial hypertension and lupus glomerulonephritis in systemic lupus erythematosus. Blood Press. 1993;2:108–112. doi: 10.3109/08037059309077537. [DOI] [PubMed] [Google Scholar]
- 12.Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, Manzi S. Vascular stiffness in women with systemic lupus erythematosus. Hypertension. 2001;37:1075–1082. doi: 10.1161/01.hyp.37.4.1075. [DOI] [PubMed] [Google Scholar]
- 13.Panoulas VF, Metsios GS, Pace AV, John H, Treharne GJ, Banks MJ, Kitas GD. Hypertension in rheumatoid arthritis. Rheumatology (Oxford) 2008;47:1286–1298. doi: 10.1093/rheumatology/ken159. [DOI] [PubMed] [Google Scholar]
- 14.Shaharir SS, Mustafar R, Mohd R, Mohd Said MS, Gafor HA. Persistent hypertension in lupus nephritis and the associated risk factors. Clinical rheumatology. 2015;34:93–97. doi: 10.1007/s10067-014-2802-0. [DOI] [PubMed] [Google Scholar]
- 15.Sabio JM, Vargas-Hitos JA, Navarrete-Navarrete N, Mediavilla JD, Jimenez-Jaimez J, Diaz-Chamorro A, Jimenez-Alonso J, Grupo Lupus Virgen de las N Prevalence of and factors associated with hypertension in young and old women with systemic lupus erythematosus. J Rheumatol. 2011;38:1026–1032. doi: 10.3899/jrheum.101132. [DOI] [PubMed] [Google Scholar]
- 16.Ryan MJ, McLemore GR., Jr Hypertension and impaired vascular function in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. 2007;292:R736–742. doi: 10.1152/ajpregu.00168.2006. [DOI] [PubMed] [Google Scholar]
- 17.Rudofsky UH, Dilwith RL, Roths JB, Lawrence DA, Kelley VE, Magro AM. Differences in the occurrence of hypertension among (nzb x nzw)f1, mrl-lpr, and bxsb mice with lupus nephritis. Am J Pathol. 1984;116:107–114. [PMC free article] [PubMed] [Google Scholar]
- 18.Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension. 2014;64:792–800. doi: 10.1161/HYPERTENSIONAHA.114.04006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor EB, Ryan MJ. Immunosuppression with mycophenolate mofetil attenuates hypertension in an experimental model of autoimmune disease. J Am Heart Assoc. 2017;6 doi: 10.1161/JAHA.116.005394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamal A, Khamashta M. The efficacy of novel b cell biologics as the future of sle treatment: A review. Autoimmun Rev. 2014;13:1094–1101. doi: 10.1016/j.autrev.2014.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, Maciuca R, Zhang D, Garg JP, Brunetta P, Appel G, Group LI Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012;64:1215–1226. doi: 10.1002/art.34359. [DOI] [PubMed] [Google Scholar]
- 22.Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anolik JH. B cell biology: Implications for treatment of systemic lupus erythematosus. Lupus. 2013;22:342–349. doi: 10.1177/0961203312471576. [DOI] [PubMed] [Google Scholar]
- 24.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 25.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 26.Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, Manz RA. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in nzb/w mice. J Exp Med. 2004;199:1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Field-Smith A, Morgan GJ, Davies FE. Bortezomib (velcadetrade mark) in the treatment of multiple myeloma. Ther Clin Risk Manag. 2006;2:271–279. doi: 10.2147/tcrm.2006.2.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, Voll RE. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 29.Alexander T, Sarfert R, Klotsche J, Kuhl AA, Rubbert-Roth A, Lorenz HM, Rech J, Hoyer BF, Cheng Q, Waka A, Taddeo A, Wiesener M, Schett G, Burmester GR, Radbruch A, Hiepe F, Voll RE. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015;74:1474–1478. doi: 10.1136/annrheumdis-2014-206016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension. 2012;59:673–679. doi: 10.1161/HYPERTENSIONAHA.111.190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathis KW, Venegas-Pont M, Masterson CW, Wasson KL, Ryan MJ. Blood pressure in a hypertensive mouse model of sle is not salt-sensitive. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1281–1285. doi: 10.1152/ajpregu.00386.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venegas-Pont M, Mathis KW, Iliescu R, Ray WH, Glover PH, Ryan MJ. Blood pressure and renal hemodynamic responses to acute angiotensin ii infusion are enhanced in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1286–1292. doi: 10.1152/ajpregu.00079.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathis KW, Venegas-Pont M, Flynn ER, Williams JM, Maric-Bilkan C, Dwyer TM, Ryan MJ. Hypertension in an experimental model of systemic lupus erythematosus occurs independently of the renal nerves. Am J Physiol Regul Integr Comp Physiol. 2013;305:R711–719. doi: 10.1152/ajpregu.00602.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amend SR, Valkenburg KC, Pienta KJ. Murine hind limb long bone dissection and bone marrow isolation. J Vis Exp. 2016 Apr 14;:110. doi: 10.3791/53936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minges Wols HA, Witte PL. Plasma cell purification from murine bone marrow using a two-step isolation approach. J Immunol Methods. 2008;329:219–224. doi: 10.1016/j.jim.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monneaux F, Dumortier H, Steiner G, Briand JP, Muller S. Murine models of systemic lupus erythematosus: B and t cell responses to spliceosomal ribonucleoproteins in mrl/fas(lpr) and (nzb x nzw)f(1) lupus mice. Int Immunol. 2001;13:1155–1163. doi: 10.1093/intimm/13.9.1155. [DOI] [PubMed] [Google Scholar]
- 37.Yung S, Chan TM. Mechanisms of kidney injury in lupus nephritis - the role of anti-dsdna antibodies. Front Immunol. 2015;6:475. doi: 10.3389/fimmu.2015.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson RJ, Rodriguez-Iturbe B, Nakagawa T, Kang DH, Feig DI, Herrera-Acosta J. Subtle renal injury is likely a common mechanism for salt-sensitive essential hypertension. Hypertension. 2005;45:326–330. doi: 10.1161/01.HYP.0000154784.14018.5f. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: All for one and one for all. Am J Physiol Renal Physiol. 2004;286:F606–616. doi: 10.1152/ajprenal.00269.2003. [DOI] [PubMed] [Google Scholar]
- 40.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin ii infusions. Am J Physiol Renal Physiol. 2007;292:F330–339. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomez AM, Vrolix K, Martinez-Martinez P, Molenaar PC, Phernambucq M, van der Esch E, Duimel H, Verheyen F, Voll RE, Manz RA, De Baets MH, Losen M. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J Immunol. 2011;186:2503–2513. doi: 10.4049/jimmunol.1002539. [DOI] [PubMed] [Google Scholar]
- 43.Lee SW, Kim JH, Park YB, Lee SK. Bortezomib attenuates murine collagen-induced arthritis. Ann Rheum Dis. 2009;68:1761–1767. doi: 10.1136/ard.2008.097709. [DOI] [PubMed] [Google Scholar]
- 44.Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann M, Jack HM, Voll RE. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007;67:1783–1792. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 45.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune t cell responses. J Immunol. 1994;152:1453–1461. [PubMed] [Google Scholar]
- 47.Lin RH, Mamula MJ, Hardin JA, Janeway CA., Jr Induction of autoreactive b cells allows priming of autoreactive t cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with b cells but lacking serum antibody reveals an antibody-independent role for b cells in murine lupus. J Exp Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, Komisar O, Slonimsky E, Klang E, Lotan E, Welt M, Marai I, Shina A, Amital H, Shoenfeld Y. A volcanic explosion of autoantibodies in systemic lupus erythematosus: A diversity of 180 different antibodies found in sle patients. Autoimmun Rev. 2015;14:75–79. doi: 10.1016/j.autrev.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Rekvig OP, Putterman C, Casu C, Gao HX, Ghirardello A, Mortensen ES, Tincani A, Doria A. Autoantibodies in lupus: Culprits or passive bystanders? Autoimmun Rev. 2012;11:596–603. doi: 10.1016/j.autrev.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 51.Gualtierotti R, Biggioggero M, Penatti AE, Meroni PL. Updating on the pathogenesis of systemic lupus erythematosus. Autoimmun Rev. 2010;10:3–7. doi: 10.1016/j.autrev.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 52.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin at1 receptor. J Clin Invest. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. At(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382–2387. doi: 10.1161/01.cir.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 54.Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against at1-receptor and alpha1-adrenergic receptor in patients with hypertension. Hypertens Res. 2002;25:641–646. doi: 10.1291/hypres.25.641. [DOI] [PubMed] [Google Scholar]
- 55.Dechend R, Muller DN, Wallukat G, Homuth V, Krause M, Dudenhausen J, Luft FC. At1 receptor agonistic antibodies, hypertension, and preeclampsia. Semin Nephrol. 2004;24:571–579. doi: 10.1016/s0270-9295(04)00128-7. [DOI] [PubMed] [Google Scholar]
- 56.Walther T, Wallukat G, Jank A, Bartel S, Schultheiss HP, Faber R, Stepan H. Angiotensin ii type 1 receptor agonistic antibodies reflect fundamental alterations in the uteroplacental vasculature. Hypertension. 2005;46:1275–1279. doi: 10.1161/01.HYP.0000190040.66563.04. [DOI] [PubMed] [Google Scholar]
- 57.Herse F, Dechend R, Harsem NK, Wallukat G, Janke J, Qadri F, Hering L, Muller DN, Luft FC, Staff AC. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension. 2007;49:604–611. doi: 10.1161/01.HYP.0000257797.49289.71. [DOI] [PubMed] [Google Scholar]
- 58.Xiong J, Liang Y, Yang H, Zhu F, Wang Y. The role of angiotensin ii type 1 receptor-activating antibodies in patients with lupus nephritis. Int J Clin Pract. 2013;67:1066–1067. doi: 10.1111/ijcp.12242. [DOI] [PubMed] [Google Scholar]
- 59.Cines DB, Lyss AP, Reeber M, Bina M, DeHoratius RJ. Presence of complement-fixing anti-endothelial cell antibodies in systemic lupus erythematosus. J Clin Invest. 1984;73:611–625. doi: 10.1172/JCI111251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belizna C, Duijvestijn A, Hamidou M, Tervaert JW. Antiendothelial cell antibodies in vasculitis and connective tissue disease. Ann Rheum Dis. 2006;65:1545–1550. doi: 10.1136/ard.2005.035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Westphal JR, Boerbooms AM, Schalwijk CJ, Kwast H, De Weijert M, Jacobs C, Vierwinden G, Ruiter DJ, Van de Putte LB, De Waal RM. Anti-endothelial cell antibodies in sera of patients with autoimmune diseases: Comparison between elisa and facs analysis. Clin Exp Immunol. 1994;96:444–449. doi: 10.1111/j.1365-2249.1994.tb06049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Florey OJ, Johns M, Esho OO, Mason JC, Haskard DO. Antiendothelial cell antibodies mediate enhanced leukocyte adhesion to cytokine-activated endothelial cells through a novel mechanism requiring cooperation between fc{gamma}riia and cxcr1/2. Blood. 2007;109:3881–3889. doi: 10.1182/blood-2006-08-044669. [DOI] [PubMed] [Google Scholar]
- 63.Perry GJ, Elston T, Khouri NA, Chan TM, Cameron JS, Frampton G. Antiendothelial cell antibodies in lupus: Correlations with renal injury and circulating markers of endothelial damage. Q J Med. 1993;86:727–734. [PubMed] [Google Scholar]
- 64.Li S, Wang X, Li Y, Kost CK, Jr, Martin DS. Bortezomib, a proteasome inhibitor, attenuates angiotensin ii-induced hypertension and aortic remodeling in rats. PLoS One. 2013;8:e78564. doi: 10.1371/journal.pone.0078564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, Tipping P, Vinh A, Samuel CS, Peter K, Guzik TJ, Kyaw TS, Toh BH, Bobik A, Drummond GR. Obligatory role for b cells in the development of angiotensin ii-dependent hypertension. Hypertension. 2015;66:1023–1033. doi: 10.1161/HYPERTENSIONAHA.115.05779. [DOI] [PubMed] [Google Scholar]
- 66.Ludwig A, Fechner M, Wilck N, Meiners S, Grimbo N, Baumann G, Stangl V, Stangl K. Potent anti-inflammatory effects of low-dose proteasome inhibition in the vascular system. J Mol Med (Berl) 2009;87:793–802. doi: 10.1007/s00109-009-0469-9. [DOI] [PubMed] [Google Scholar]
- 67.Hainz N, Thomas S, Neubert K, Meister S, Benz K, Rauh M, Daniel C, Wiesener M, Voll RE, Amann K. The proteasome inhibitor bortezomib prevents lupus nephritis in the nzb/w f1 mouse model by preservation of glomerular and tubulointerstitial architecture. Nephron Exp Nephrol. 2012;120:e47–58. doi: 10.1159/000334955. [DOI] [PubMed] [Google Scholar]
- 68.Fenton K, Fismen S, Hedberg A, Seredkina N, Fenton C, Mortensen ES, Rekvig OP. Anti-dsdna antibodies promote initiation, and acquired loss of renal dnase1 promotes progression of lupus nephritis in autoimmune (nzbxnzw)f1 mice. PLoS One. 2009;4:e8474. doi: 10.1371/journal.pone.0008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mostoslavsky G, Fischel R, Yachimovich N, Yarkoni Y, Rosenmann E, Monestier M, Baniyash M, Eilat D. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: A case for tissue injury by molecular mimicry. Eur J Immunol. 2001;31:1221–1227. doi: 10.1002/1521-4141(200104)31:4<1221::aid-immu1221>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 70.Amital H, Heilweil M, Ulmansky R, Szafer F, Bar-Tana R, Morel L, Foster MH, Mostoslavsky G, Eilat D, Pizov G, Naparstek Y. Treatment with a laminin-derived peptide suppresses lupus nephritis. J Immunol. 2005;175:5516–5523. doi: 10.4049/jimmunol.175.8.5516. [DOI] [PubMed] [Google Scholar]
- 71.Krishnan MR, Wang C, Marion TN. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. 2012;82:184–192. doi: 10.1038/ki.2011.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G, Berek C, Hiepe F, Krenn V, Radbruch A, Manz RA. Inflamed kidneys of nzb/w mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001;31:2726–2732. doi: 10.1002/1521-4141(200109)31:9<2726::aid-immu2726>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 73.Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, Bottinger EP, Ivashkiv L, Kretzler M, Davidson A. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J Immunol. 2011;186:4994–5003. doi: 10.4049/jimmunol.1003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller JJ, 3rd, Cole LJ. Resistance of long-lived lymphocytes and plasma cells in rat lymph nodes to treatment with prednisone, cyclophosphamide, 6-mercaptopurine, and actinomycin d. J Exp Med. 1967;126:109–125. doi: 10.1084/jem.126.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karnell JL, Karnell FG, 3rd, Stephens GL, Rajan B, Morehouse C, Li Y, Swerdlow B, Wilson M, Goldbach-Mansky R, Groves C, Coyle AJ, Herbst R, Ettinger R. Mycophenolic acid differentially impacts b cell function depending on the stage of differentiation. J Immunol. 2011;187:3603–3612. doi: 10.4049/jimmunol.1003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hiepe F, Radbruch A. Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat Rev Nephrol. 2016;12:232–240. doi: 10.1038/nrneph.2016.20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.