

Abstract
Cocaine abuse increases the risk of cardiovascular (CV) mortality and morbidity; however, the underlying molecular mechanisms remain elusive. By using a mouse model for cocaine abuse/use, we found that repeated cocaine injection led to increased blood pressure (BP) and aortic stiffness in mice associated with elevated levels of reactive oxygen species (ROS) in the aortas, a phenomenon similar to that observed in hypertensive humans. This ROS elevation was correlated with downregulation of malic enzyme 1 (Me1), an important redox molecule that counteracts ROS generation, and upregulation of microRNA (miR)-30c-5p that targets Me1 expression by directly binding to its 3’UTR. Remarkably, lentivirus-mediated overexpression of miR-30c-5p in aortic smooth muscle cells (SMCs) recapitulated the effect of cocaine on Me1 suppression, which in turn led to ROS elevation. Moreover, in vivo silencing of miR-30c-5p in SMCs resulted in Me1 upregulation, ROS reduction, and significantly suppressed cocaine-induced increases in BP and aortic stiffness—a similar effect to that produced by treatment with the antioxidant N-acetyl cysteine. Discovery of this novel cocaine-↑miR-30c-5p-↓Me1-↑ROS pathway provides a potential new therapeutic avenue for treatment of cocaine abuse-related CV disease.
Keywords: blood pressure, aortic stiffness, microRNA, reactive oxygen species, cocaine
INTRODUCTION
Cocaine addiction inflicts enormous health and economic costs to individuals, families and society. Although significant research has focused on the neurobiological consequences of chronic cocaine abuse1-3, cocaine intake is also associated with potentially fatal cardiovascular (CV) events such as arrhythmias, myocardial infarction, and stroke4-7. Long-term exposure to cocaine can lead to hypertension (HTN), aortic stiffness, and atherosclerosis8-11. Although the clinical consequences of cocaine-induced CV toxicities are well documented, the molecular mechanisms underlying these effects of cocaine remain poorly understood.
Accumulating evidence suggests that excessive oxidative stress plays an important role in the pathogenesis of HTN, since increased levels of reactive oxygen species (ROS) have been found in aortas of hypertensive animals and humans12-17. Intracellular ROS is generated by oxidation of NADPH into NADP+ (NADPH + 2O2 → NADP+ + 2O2- + H+)18-20. This process, however, is counteracted by antioxidants and redox molecules, such as the malic enzyme 1 (Me1), which encodes a NADP(+)-dependent enzyme that turns NADP+ into NADPH when catalyzing the oxidative decarboxylation of malate into pyruvate18-20. Me1 expression is regulated by microRNAs (miRNAs), the short, non-coding RNA molecules that bind to the 3’UTR of target mRNAs leading to their translational repression and/or mRNA degradation21, 22. MiRNA target prediction programs, such as TargetScan (targetscan.org)23, 24 and Miranda (microRNA.org)25, have identified highly conserved binding sites for the miR-30c-5p/384-5p and miR-153-3p families in the Me1 3’UTR. Among these, the miR-30c-5p/384-5p family are predicted to have a more stable binding to the Me1 3’UTR (position 552-559) with a lower context++ and mirSVR score23-25. Although the involvement of miRNAs in cardiac development and pathogenesis of CV disease (CVD) has been reported21, 22, the role of miR-30c-5p in cocaine-driven CVD is unknown.
We sought to understand whether the miR-30c-5p-Me1-ROS molecular axis is involved in mediating the effect of cocaine on inducing CVD. By using a cocaine abuse/use mouse model combined with aortic smooth muscle cell (SMC) cultures, we examined the interrelationship between components of the miR-30c-5p-Me1 pathway as well as how modulating these components affects cocaine-induced CV phenotypes through a set of gain- and loss-of-function experiments.
METHODS
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Male C57BL/6 mice (8-10 weeks old, The Jackson Laboratory) were housed 4 animals per cage, under a regular 12/12 hour light/dark cycle with ad libitum access to food and water. Mice received a daily intraperitoneal (i.p.) injection of cocaine, cocaine methiodide (CM, 20 mg/kg body weight each, NIDA Drug Supply) or saline for 10 consecutive days, a treatment regimen that has been validated in multiple animal models of addiction26. Cocaine and CM were dissolved in 0.9% sterile saline prior to injection.
In parallel experiments, mice were injected 3 times on alternate days via the tail vein with 2×105 IU/mouse lentiviral constructs encoding a miR-30c-5p antagomir (miR-Zip30c) or no gene (empty vector control) under driven by a SMC specific promoter, Sm22a (Biosettia). Subsequently, the mice were injected with cocaine as above. One day prior and daily during cocaine injection, a separate group of mice received i.p. injection of N-acetyl cysteine (NAC, Sigma Aldrich) at 250 mg/kg body weight dissolved in phosphate buffered saline (PBS). All experiments were approved by the Institutional Animal Care and Use Committee at the University of Miami.
Blood pressure (BP) and aortic stiffness
Systolic and diastolic BP was measured using a noninvasive tail-cuff CODA BP monitor following the manufacturer’s protocols (Kent Scientific). BP was measured 1 day before the first injection (baselines) and one hour (half-life of cocaine) after each daily injection in a 10 day-treatment course. Pulse wave velocity (PWV) measuring aortic stiffness was determined 1 day before the first injection and 2 days after the last injection (day 12). Briefly, mice were anesthetized with 2% isoflurane through a face mask and laid on a platform in the supine position with all legs taped to electrocardiography electrodes for heart rate monitoring. Aortic arch velocity signal was assessed by placing a focused 420-40 MHz Doppler probe (Vevo 770, VisualSonics) to the right of the upper sternum, angling the sound beam to align with the direction of the aortic arch, and capturing the aortic shape velocity signal moving away from the probe. At least three measurements were recorded for each mouse.
Immunohistochemistry
Immediately after aortic stiffness measurements, mice were euthanized by carbon dioxide and isolated aortas were flash frozen in optimum cutting temperature freezing medium on dry ice. Aortas were sliced using a CM 1850 microtome (Leica Biosystems) to produce 10μm thick sections. Cryosections were fixed in cold 4% paraformaldehyde in PBS for 10 minutes, washed with PBS, and incubated with 0.3% Triton in PBS for 10 minutes. Subsequently, sections were blocked with 5% BSA in PBS for one hour and incubated with rat anti-mouse Me1 antibody conjugated with Alexa Fluor 594 (PharMingen) overnight at 4oC. After washing with PBS, sections were incubated with Hoechst blue nuclear dye (Molecular Probes) for 1 hour at room temperature in the dark. At least 5 consecutive sections were examined for each aorta. Fluorescent signal was imaged on the Zeiss LSM710 Confocal AxioObserver Inverted Automated Microscope and quantified by Image J software (NIH).
ROS detection
Intracellular O2-, H2O2, and peroxynitrite (ONOO-) were detected by incubating aorta cryosections with 10μM nonfluorescent dye dihydroethidium (DHE), chloromethyl-2’,7’-dichlorofluorescein diacetate (CM-H2DCFDA, all from Sigma Aldrich), and aminophenyl fluorescein (APF, Molecular Probes), respectively, for 5 to 30 minutes at 37°C in the dark. The fluorescent signal was imaged and quantified as described above. The level of nitrotyrosine and prostaglandin in cultured cells were detected using specific ELISA kits (Cell Biolabs) following the manufacturer’s protocols.
Cell cultures and 3’UTR reporter assay
Mouse aorta derived SMCs were cultured following the manufacturer’s protocol (Cell Biologics). SMCs were transduced by lentiviruses encoding miR30c, miR-Zip30c or miR-scrambled control (miR-Ctr) (System Biosciences) overnight to generate stable cell lines that over expressed or suppressed miR-30c-5p expression. Transduction efficiency was monitored by green fluorescent protein expression and transduced cells were selected by treatment with 10μg/ml puromycin.
HEK293 cells (Sigma Aldrich) were transfected using Lipofectamine 2000 Reagent (Promega) with miR-30c precursor or miR-Ctr constructs (System Biosciences) along with either a wild type 3’UTR-Me1 luciferase reporter construct or a construct in which the putative miR-30c-5p’s binding sites were mutated from TGAGaaagctcactgctgtTTAC to GAGTaaagctcactgctgtCATT (GeneCopoeia). Gaussia luciferase activity in cell medium was determined 48 hours after transfection using a Centro XS3 LB 960 Microplate Luminometer (Berthold Technologies) and normalized to the activities of secreted alkaline phosphatase.
Quantitative reverse transcription and polymerase chain reaction (QRT-PCR)
One μg of total RNA was reversed transcribed using the SuperScript IV VILO Master Mix (Invitrogen). Me1 gene expression was measured by qPCR on 5ng of cDNA using the iQ SYBR Green Supermix (Bio-Rad) with specific forward (5’-gacccgcatctcaacaagga-3’) and reverse (5’-gtcgaagtcagagttcagtcgt-3’) primers. Vales were normalized to the level of a house-keeping gene, Gapdh (Forward: 5’-tgcatcctgcaccaccaact-3’, Reverse: 5’-acgccacagctttccagagg-3’). MiR-30c-5p expression was assessed using Taqman MicroRNA Assays (Thermofisher Scientific) and normalized to the level of small nucleolar RNA, following the manufacturer’s protocols.
Statistics
Five mice were included in each treatment group at each time point. For cultured cells, 3-6 samples (wells) were included in each treatment group with duplicate measurements for each sample, and experiments were repeated at least twice independently using different batches of cells. The sample size was selected based on results from pilot studies. All data were presented as mean ± standard deviation. Difference between two groups was compared by One Way ANOVA followed by Tukey’s post-hoc test (GraphPad Prism7) with a minimal 80% power and significance set at p ≤ 0.05.
RESULTS
1. Cocaine and CM treatment increases BP, aortic stiffness, and ROS levels in the aorta
Mice were treated with daily injection of saline, cocaine, or CM for 10 consecutive days. CM, a cocaine analog that does not enter the brain, was used to corroborate the non-CNS effect of cocaine. There were no differences in baseline (day 0) BPs between treatment groups. However, a significant increase in systolic (Fig 1A) and diastolic (Fig 1B) BP was observed in cocaine and CM treated animals throughout the treatment course compared to saline treatment. Comparable baseline levels of PWV were also found across the groups prior to the initiation of treatment (Fig 1C). However, PWV was significantly elevated in cocaine and CM treated mice at day 12 compared with saline treatment.
Figure 1. Cocaine and CM treatment increases BP and aortic stiffness in mice.
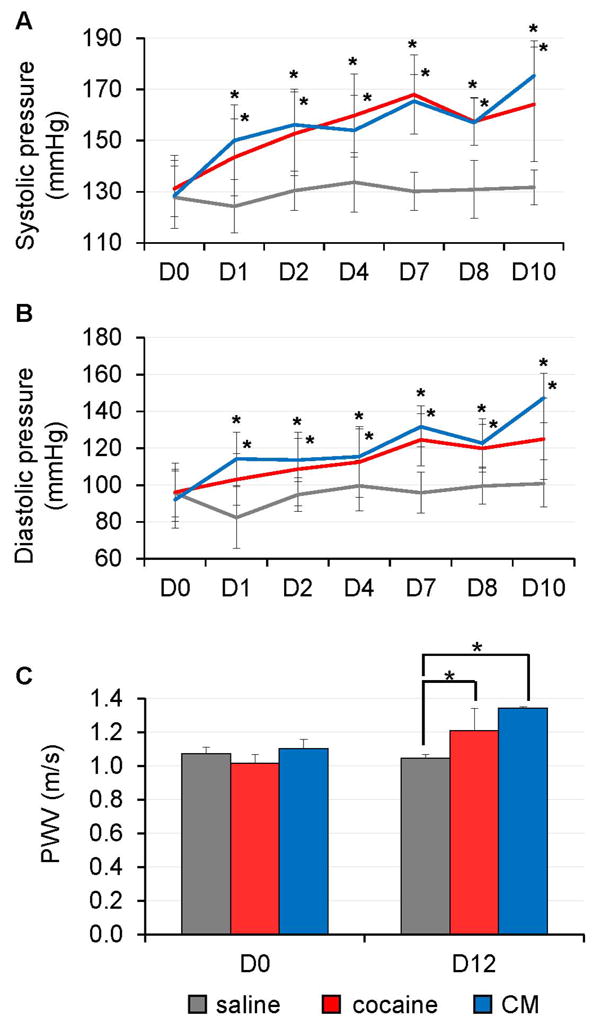
C57/BL6 mice (n=5) were injected with cocaine, CM, or saline each day for 10 consecutive days. (A-B) Cocaine or CM-treated mice showed elevated systolic and diastolic BP compared to saline-treated mice with the BP steadily increasing over the treatment course (*: p < 0.05 vs. saline at each time point). (C) Aortic stiffness (measured by PWV) was elevated in the cocaine or CM-treated mice 2 days after the last injection (*: p < 0.05 vs. saline at D12).
Staining for intracellular ROS in aorta cryosections revealed that cocaine and CM treatment significantly increased the levels of O2-, H2O2, and ONOO- compared to saline treatment (Fig 2A-B). There were no significant differences between cocaine and CM treated mice in terms of BP, PWV, and ROS levels.
Figure 2. Cocaine and CM treatment leads to increased ROS levels in the mouse aorta.
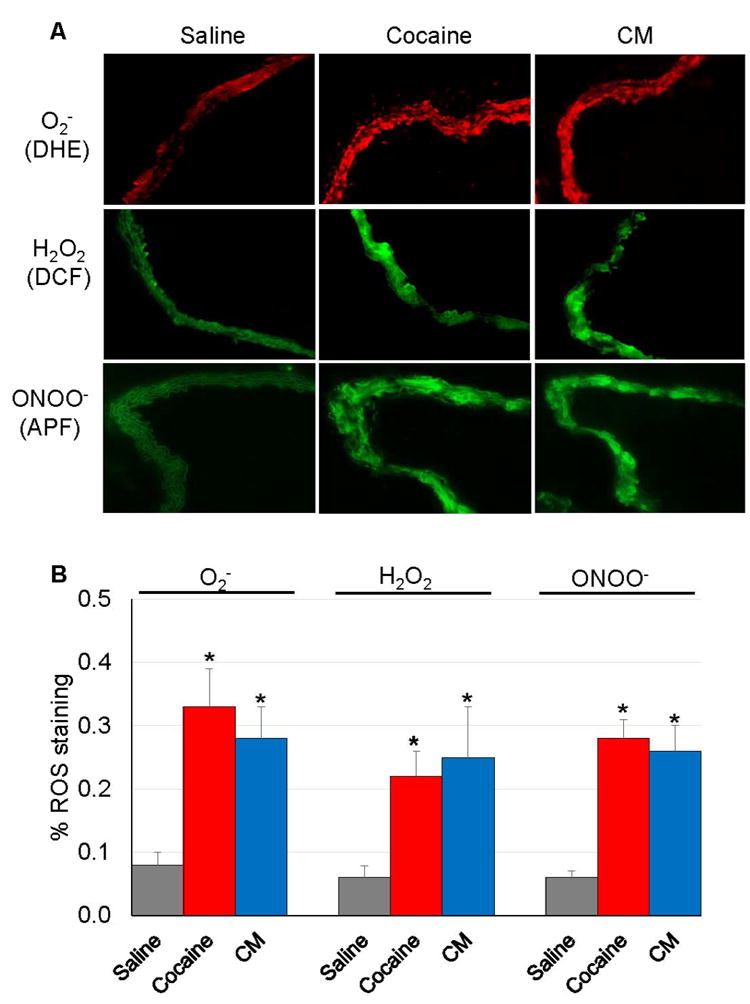
Aortas were obtained from mice (n=5) that received repeated injections of cocaine, CM, or saline. (A) Intracellular O2-, H2O2, and ONOO- were examined by incubating aorta cryosections with DHE, CM-H2DCFDA, and APF dye, respectively. (B) Fluorescent signal was quantified using Image J software. Cocaine or CM treatment significantly increased ROS levels compared to saline treatment (*: p < 0.05 vs. saline in each ROS).
2. Cocaine and CM exposure affects the expression of miR-30c-5p and Me1 in the aorta
QRT-PCR was performed to assess the expression of Me1 and its predicted regulatory miRNA, miR-30c-5p, in the mouse aorta. Significantly reduced Me1 mRNA expression but elevated miR-30c-5p expression was detected upon cocaine or CM treatment compared to saline treatment (Fig 3A). Immunohistochemistry staining of aorta cryosections also confirmed a 67% and 58% decrease in Me1 protein expression following cocaine and CM exposure, respectively, compared to saline (Fig 3B-C).
Figure 3. Cocaine and CM treatment increases miR-30c-5p expression but decreases Me1 expression in the mouse aorta.
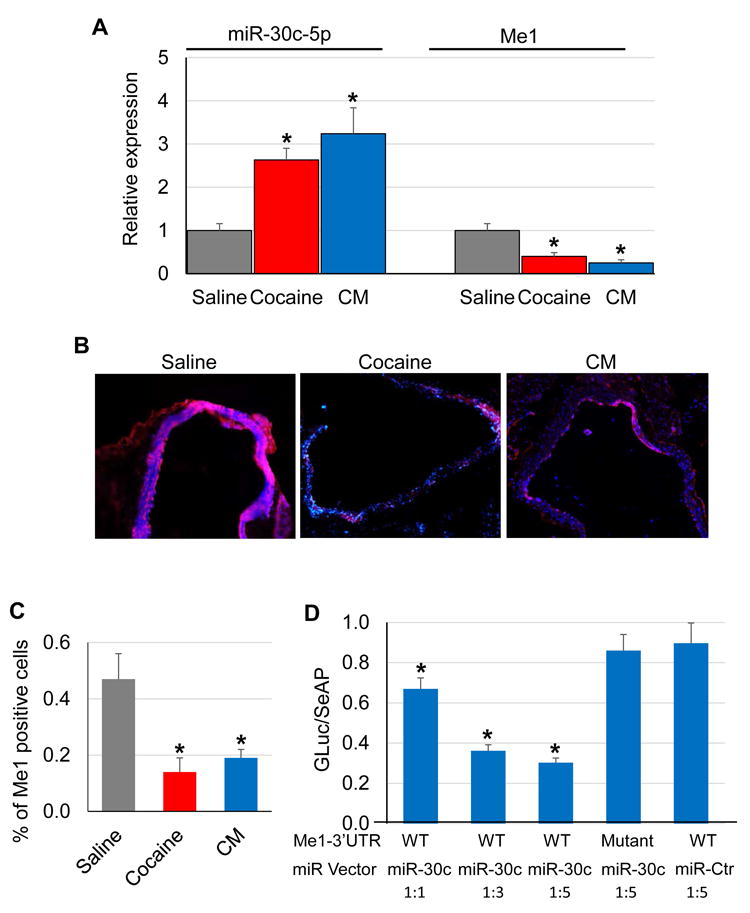
Total RNAs were extracted from aortas of mice (n=5) after injection with cocaine, CM, or saline for 10 consecutive days. (A) QRT-PCR analyses showed significantly increased miR-30c-5p levels and decreased Me1 expression (normalized to small nucleolar RNA and Gapdh, respectively) by cocaine treatment (*: p < 0.05 vs. saline). (B-C) Immunohistochemistry analysis showed cocaine- and CM- induced decreases in Me1 protein expression compared to saline. The relative number of Me1-positive cells was quantified using Image J software (*: p < 0.05 vs. saline). (D) 3’UTR reporter assays showed that transfection with miR-30c-5p but not miR-Ctr decreased luciferase activities (normalized to secreted alkaline phosphatase activities) of reporter constructs containing the wild type (WT), but not mutant (with disrupted binding sites for miR-30c-5p), 3’UTR of Me1 gene in a dose-dependent manner (*: p < 0.05 vs. WT Me1-3’UTR + miR-Ctr at 1:5).
3’UTR reporter assays were performed in HEK293 cells to determine if miR-30c-5p can directly silence Me1 expression. As shown in Fig 3D, the luciferase activity of the wild type (wt) Me1-3’UTR was significantly decreased by miR-30c-5p but not miR-Ctr in a dose-dependent manner. In contrast, the luciferase activity of the mutant (mt, with disrupted miR-30c-5p binding site) Me1-3’UTR was not affected by miR-30c-5p, suggesting that Me1 is a direct target of miR-30c-5p.
3. The miR-30c-5p—Me1 axis mediates cocaine-induced ROS elevation in mouse aortic SMCs
To determine if modulating the miR-30c-5p–Me1 pathway alters ROS levels, SMCs were transduced with lentiviral vectors that express miR-30c-5p, miR-Zip30c, or miR-Ctr. Overexpression of miR-30c-5p led to a significant decrease in Me1 levels compared to miR-Ctr treated cells (Fig 4A-B). Conversely, SMCs with suppressed miR-30c-5p levels by miR-Zip30c had elevated Me1 expression relative to miR-Ctr (Fig 4A-B). Cocaine treatment further potentiated the miR-30c-5p-induced Me1 reduction in a dose-dependent manner (Fig 4B). In contrast, cocaine exposure produced no significant changes in Me1 expression in miR-Zip30c transduced SMCs (Fig 4B), suggesting that cocaine induces its effect on Me1 expression through miR-30c-5p.
Figure 4. Perturbing the miR-30c-5p/Me1 axis affects cocaine-induced ROS elevation in mouse aortic SMCs.
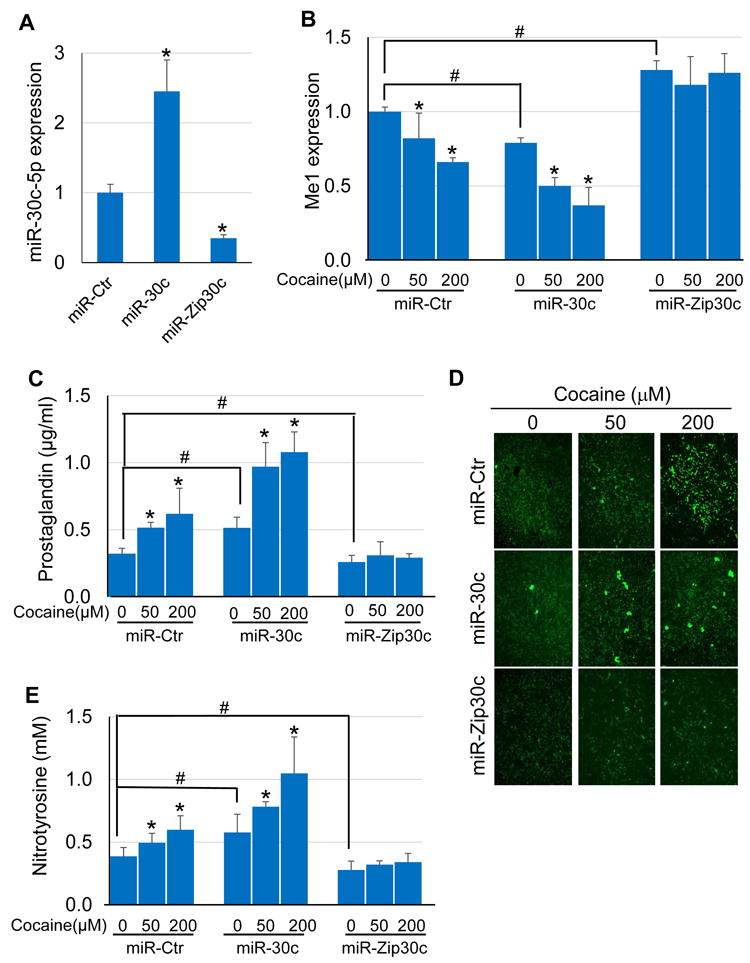
SMCs were stably transduced with lentiviral vectors expressing miR-30c-5p, miR-Zip30c, or miR-Ctr. (A) miR-30c-5p expression was increased in the miR-30c-5p transduced cells and decreased in the miR-Zip30c transduced cells (*: p < 0.05 vs. miR-Ctr). (B) Cocaine dose-dependently decreased Me1 expression, an effect that was exacerbated by overexpression of miR-30c-5p. MiR-Zip30c transduction led to increased Me1 expression, which was unaltered after cocaine treatment. (C) Prostaglandin levels were elevated in miR-30c-5p transduced cells, and were further elevated by cocaine in a dose-dependent manner. MiR-Zip30c not only decreased the basal level of prostaglandin but also abrogated the effect of cocaine on increasing prostaglandin. (D-E) Overexpression of miR-30c-5p exacerbated the effect of cocaine on increasing H2O2 (detected by DCF staining) and nitrotyrosine levels. Conversely, silencing miR-30c-5p eliminated cocaine-induced increases in H2O2 and nitrotyrosine levels (*: p < 0.05 vs. respective miR-transduced SMC line alone; #: p < 0.05 vs. miR-Ctr-transduced SMCs alone).
ROS detection showed that overexpression of miR-30c-5p in SMCs increased prostaglandin (an indicator of peroxides) levels compared to miR-Ctr treated cells (Fig 4C). Cocaine had an additive effect on prostaglandin upregulation in miR-30c-5p transduced cells (Fig 4C). In contrast, miR-Zip30c treatment reduced prostaglandin to levels below that seen in miR-Ctr treated cells, and cocaine exposure failed to increase prostaglandin levels in miR-Zip30c treated cells (Fig 4C). In addition, increased staining for H2O2 was observed in cells overexpressing miR-30c-5p, while reduced H2O2 staining was seen in miR-Zip30c transduced SMCs compared to miR-Ctr treated cells (Fig 4D). Cocaine treatment further increased H2O2 in miR-Ctr and miR-30c-5p transduced SMCs, but had no significant effects on H2O2 in miR-Zip30c transduced SMCs (Fig 4D). A similar pattern of results was seen for nitrotyrosine– the peroxynitrite modification of tyrosine residues to 3-nitrotyrosine (Fig 4E). Overexpression of miR-30c-5p enhanced nitrotyrosine levels which were further increased by cocaine in a dose-dependent manner compared to miR-Ctr treated cells (Fig 4E). MiR-Zip30c treatment, however, reduced nitrotyrosine levels to that below those seen in miR-Ctr treated cells even in the presence of cocaine (Fig 4E).
To show that the miR-30c-5p—Me1 axis mediated cocaine-increased ROS. SMCs were transduced with lentiviral vectors encoding a miR-30c-5p resistant version of Me1 lacking the 3’UTR (Me1-del 3’UTR) or an empty vector Ctr. Treatment with miR-30c-5p or cocaine inhibited endogenous Me1 expression (Fig 5A). However, SMCs transduced with Me1-del 3’UTR showed elevated Me1 expression compared to cells treated with vector Ctr. Neither miR-30c-5p nor cocaine significantly affected Me1 expression in Me1-del 3’UTR transduced cells (Fig 5A). Moreover, Me1-del 3’UTR transduction abrogated the effects of miR-30c-5p or cocaine on increasing prostaglandin (Fig 5B) or nitrotyrosine (Fig 5C) levels. In contrast, the vector Ctr transduced cells showed both prostaglandin and nitrotyrosine elevation when treated with miR-30c-5p overexpression or cocaine (Fig 5C).
Figure 5. Expression of a miRNA-resistant form of Me1 abrogates cocaine-induced ROS elevation in mouse aortic SMCs.
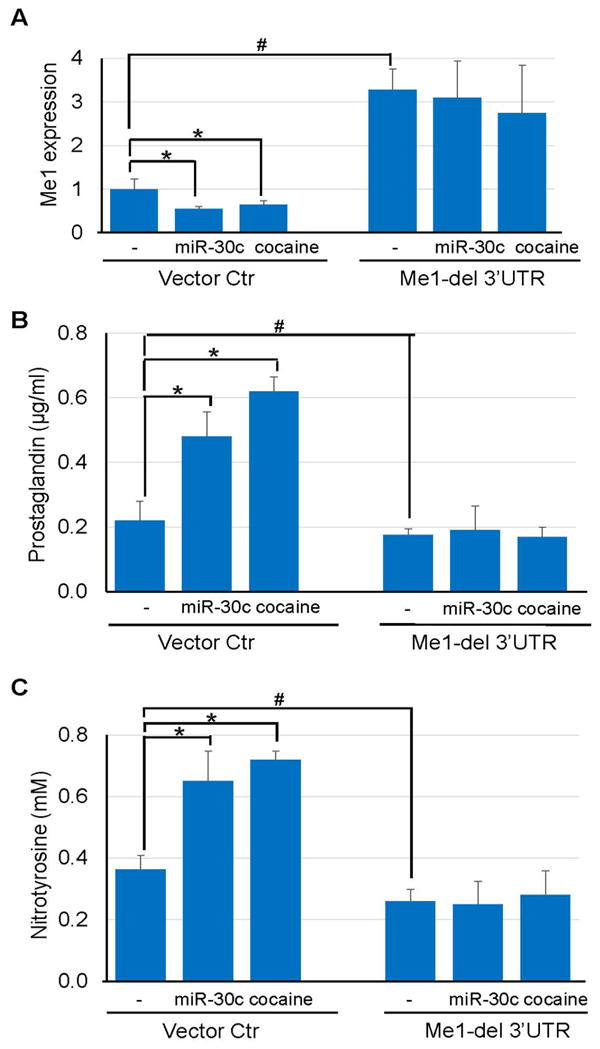
(A) Transduction of SMCs with lentiviral vectors expressing Me1 lacking the 3’UTR (Me1-del 3’UTR) overcame the inhibitory effect of exogenous miR-30c-5p or cocaine treatment on Me1 expression. (B-C) Unlike vector Ctr-transduced cells, Me1-del 3’UTR-transduced SMCs showed decreased levels of prostaglandin and nitrotyrosine, and that were unchanged by miR-30c-5p overexpression or cocaine treatment. (*: p < 0.05 vs. vector Ctr alone; #: p < 0.05 vs. Me1-del 3’UTR alone).
4. Perturbing the miR-30c-5p—Me1—ROS pathway suppresses cocaine-induced increases in BP and aortic stiffness
To validate the cocaine—↑miR-30c-5p—↓Me1—↑ROS axis in vivo, mice were injected with lentiviral vectors encoding miR-Zip30c or no gene (vector Ctr) driven by a SMC-specific promoter, Sm22a, prior to cocaine exposure. Additional mice were pretreated with NAC that acts a scavenger of free oxygen radicals27. Consistent with the in vitro results, cocaine treatment for 10 days, which decreased Me1 expression (Fig S1A) and elevated O2- levels in the aortas (Fig S1B-C), led to a significant increase in both systolic (Fig 6A) and diastolic (Fig 6B) BP compared to saline treated mice. However, these cocaine-induced changes in Me1 expression and O2- levels were significantly suppressed by pretreatment with miR-Zip30c (Fig S1A-C), which reduced miR-30c-5p expression in the aortas (Fig S1D). Reduced BP levels were also seen from day 3 to 10 in miR-Zip30c pretreated animals following cocaine exposure (Fig 6A-B). In contrast, pretreatment with vector Ctr had no effects on cocaine-induced Me1 expression, O2- levels (Fig S1A-C), and BP changes (Fig 6A-B). Pretreatment with NAC also significantly inhibited the increases in O2- and BP after cocaine exposure, without affecting the cocaine-induced reduction of Me1 (Fig S1A-C). Similarly, pretreatment with vector Ctr did not affect cocaine-induced increases in aortic stiffness measured by PWV, while pretreatment with miR-Zip30c or NAC dampened the cocaine-induced PWV increases by ~20% (Fig 6C).
Figure 6. SMC-specific knockdown of miR-30c-5p suppresses cocaine-induced increases in BP and aortic stiffness.
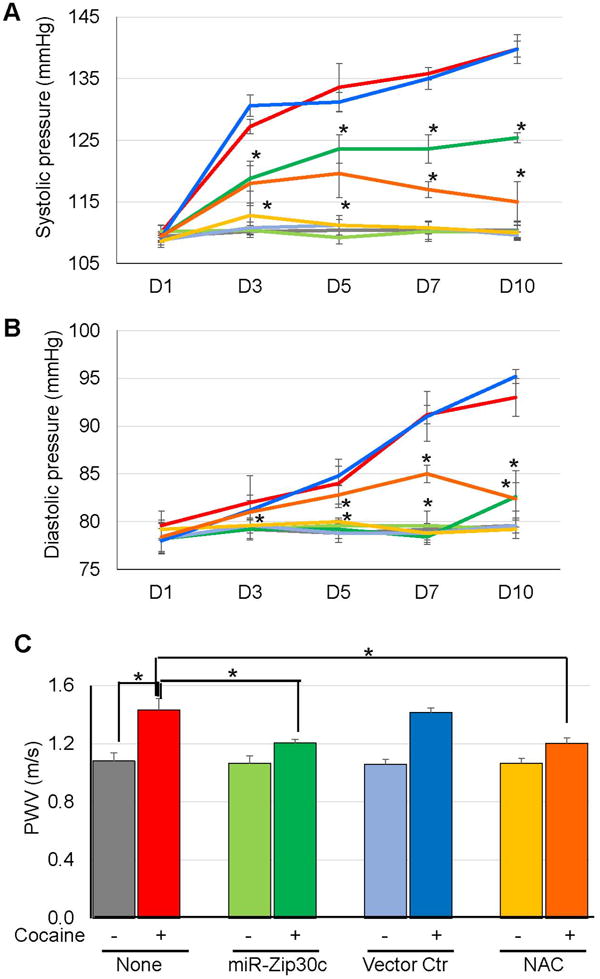
Mice (n=5) were injected with cocaine or saline each day for 10 consecutive days. In parallel experiments, mice were injected with lentiviral vectors expressing miR-Zip30c or no gene (vector Ctr) under driven by a Sm22a promoter, or antioxidant NAC, prior to cocaine exposure. (A-B) Cocaine exposure elevated systolic and diastolic BP levels compared to saline or vector Ctr- treated mice, while miR-Zip30c or NAC pretreatment partially reduced the cocaine-induced BP elevation from day 3 to 10 (*: p < 0.05 vs. cocaine treatment alone at each time point). (C) Similar to BP results, pretreatment of miR-Zip30c or NAC significantly decreased cocaine-induced increases in PWV levels (*: p < 0.05 vs. cocaine treatment alone at D12). Line/bar colors, red: cocaine; gray: saline; light green: miR-Zip30c; green: miR-Zip30c + cocaine; light blue: vector Ctr; blue: vector Ctr + cocaine; yellow: NAC; orange: NAC + cocaine.
DISCUSSION
Current evidence suggests that cocaine impacts the CVS directly and indirectly through complex mechanisms. For example, cocaine acts directly on cardiomyocytes to block the voltage-gated sodium and potassium channels in the sinoatrial node and myocardium, leading to impaired contractility, arrhythmia, and decreased left ventricular function28-30. Cocaine is also known to potentiate the effect of norepinephrine (NE) on cardiovascular function by blocking NE transporters (NET) from transporting NE back to the pre-synaptic neurons1-3. Moreover, Scheduler et al31 reported that cocaine directly elevates BP and heart rate (HR) at doses that do not potentiate NE, and administration of various doses of cocaine or CM could not enhance NE’s pressor effect in anesthetized animals, implicating that additional mechanisms other than blockade of NE reuptake may mediate the CV effects of cocaine. In this study, we examined the molecular bases by which cocaine affects CVS using a mouse model that recapitulates the cocaine-induced increases in ROS and BP as seen in humans. Although miRNAs play important roles in cardiomyocyte dysfunction-mediated myocardial infarction32, 33, EC and SMC senescence and apoptosis associated with biological aging34-36, and cocaine-seeking behaviors in the CNS37, no reports have defined the involvement of miRNAs in cocaine-driven CVD. Our results revealed a novel role for miR-30c-5p in regulating cocaine-induced increases in BP and aortic stiffness by elevating ROS production through directly suppressing Me1 in aortic SMCs. We also found that inhibiting NET or adding exogenous NE had no effect on cocaine-induced upregulation of miR-30c-5p expression in SMCs (Fig S2), supporting the hypothesis that NE-independent mechanisms may be operating to mediate the effect of cocaine on the CVS.
As a putative miRNA predicted to target Me1 expression23, 24, miR-30c-5p, has not been previously linked to CVD. In cocaine or CM treated mice that exhibit increased BP and aortic stiffness, we first reported a significant upregulation of miR-30c-5p in the aortas. Moreover, Me1 was confirmed to be a direct target of miR-30c-5p which recognized specific miRNA binding sites in the Me1 3’UTR. Unlike miR-30c-5p, several lines of evidence suggest an association of Me1 with CVD. A meta-analysis of genome-wide gene expression studies in the spontaneously hypertensive and Lyon hypertensive rats found differential Me1 expression between onset and progressive stages of HTN38. A locus on chromosome 6q12-16 encompassing the Me1 gene was linked to autosomal dominant dilated cardiomyopathy in a French family with 9 affected individuals39. Our study provides additional evidence linking Me1 to CVD, since perturbing Me1 expression in SMCs through miR-30c-5p regulation affects cocaine-induced BP and aortic stiffness.
Growing evidence supports the importance of ROS in CVD pathogenesis. A significant link between cocaine exposure and increased oxidative stress was found in mitochondrial dysfunction of cardiomyocytes that leads to cardiomyopathy, arrhythmias, and heart failure40-42. Elevated ROS have been observed in spontaneous hypertensive rats13, animals with renovascular HTN14, salt-sensitive HTN15, and obesity-induced HTN16. Human HTN also displays signs of increased ROS12, 17, 43. Moreover, ROS affects cell proliferation, apoptosis, senescence, and lipid oxidation, processes key to vascular aging44. Based on our data, the increased BP and arterial stiffness seen by repeated administration of cocaine or CM could also be directly linked to ROS elevation, since treatment with antioxidant NAC markedly inhibited these cocaine induced CV phenotypes. While miR-Zip30c transduction almost completely abolished the effect of cocaine on increasing ROS in SMC cultures, the in vivo tail vein injection of miR-Zip30c led to a significant suppression but not complete reversal of cocaine-increased BP and aortic stiffness in mice. This is not unexpected since it is far more difficult to achieve high levels of miR-30c-5p silencing in vivo than it is in cultured cells. Moreover, it is possible that additional, as of yet unidentified, biological pathways are functioning in vivo to mediate the effect of cocaine on BP and PWV.
SMCs, the major cellular component of the media aorta wall that controls blood flow by contracting or relaxing in response to external stimuli, were the focus of this study. The precise mechanism(s) of how ROS elevation in SMCs leads to HTN or aortic stiffness are, however, incompletely understood. ROS-dependent aortic stiffness may be associated with ERK1/2 phosphorylation45 or osteopontin46-mediated osteogenic differentiation of vascular SMCs that leads to aortic medial calcification. ROS overproduction may also increase angiotensin II activity leading to increased SMC proliferation, migration, and Ca2+ release, events associated with HTN development47, 48. Other than aorta wall, impairment of the endothelial layer of the aorta may participate in the cocaine-induced HTN and aortic stiffness. For example, cocaine mediated ROS elevation leads to loss of the tight junction protein Zo-1 and increases endothelial cell (EC) monolayer permeability in human pulmonary artery EC49. EC release of nitric oxide (NO) is also affected by ROS50, and the formation of ONOO- by reactions between O2- and NO not only reduces the bioavailability of NO but also induces SMC-mediated vasoconstriction51. With this in mind, cocaine may cause dysfunction of both SMCs and ECs that act synergistically to mediate the effect of cocaine on inducing HTN and aortic stiffness. Moreover, structural changes of the aortas associate with their pathogenic phenotypes. Consistent with previous findings52, we observed thickening intima-media of aortas from cocaine-treated mice (Fig S3A-B), indicating their contribution to peripheral resistance control in the pathogenesis of HTN. The thickened and stiffer (increased PWV) aortas in cocaine-treated mice may suggest a dampened contractibility of SMCs due to cocaine exposure. Although both peripheral resistance and cardiac output contribute to the development of HTN, we did not detect increased HR in cocaine-treated mice (data not shown). This could be associated with the cocaine dosage used in our animal model, since previous studies showed that lower doses of cocaine produced an immediate increase in HR while HR increases were delayed to be observed with higher cocaine doses31.
In summary, using an animal model combined with ex vivo aortic tissue and primary SMC culture we demonstrate, for the first time, a critical role of the ↑miR-30c-5p-↓Me1-↑ROS pathway in mediating the cocaine-induced increases in BP and aortic stiffness. Future studies are needed to corroborate these findings in vascular cells and aortic tissues obtained from cocaine abusers and patients with CVD.
PERSPECTIVES
The results of this study suggest that modulating the miR-30c-5p–Me1 pathway may have therapeutic benefits for the attenuation of cocaine-induced HTN and aortic stiffness. More broadly, perturbing the miR-30c-5p–Me1 axis may control excessive ROS levels known to contribute to a wide range of CV complications.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
1. What is New?
The first report of a novel miR-30c-5p-Me1-ROS pathway that mediates cocaine-induced increases in BP and aortic stiffness.
2. What is Relevant?
Understanding the miR-30c-5p–Me1-ROS pathway could facilitate the identification of new molecular targets for intervention to combat cocaine abuse-related HTN and aortic stiffness.
3. Summary
The miR-30c-5p–Me1 pathway plays a critical role in mediating the effect of cocaine on inducing hypertension and aortic stiffness.
Acknowledgments
SOURCES OF FUNDING: This work was supported by a seed grant from the Miami Heart Research Institute.
Footnotes
CONFLICT(S) OF INTEREST/DISCLOSURE(S): None.
References
- 1.Gawin FH, Ellinwood EH., Jr Cocaine dependence. Annu Rev Med. 1989;40:149–161. doi: 10.1146/annurev.me.40.020189.001053. [DOI] [PubMed] [Google Scholar]
- 2.Heal DJ, Gosden J, Smith SL. Dopamine reuptake transporter (DAT) “inverse agonism”--a novel hypothesis to explain the enigmatic pharmacology of cocaine. Neuropharmacology. 2014;87:19–40. doi: 10.1016/j.neuropharm.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Majewska MD. Cocaine addiction as a neurological disorder: implications for treatment. NIDA Res Monogr. 1996;163:1–26. [PubMed] [Google Scholar]
- 4.Gurudevan SV, Nelson MD, Rader F, Tang X, Lewis J, Johannes J, Belcik JT, Elashoff RM, Lindner JR, Victor RG. Cocaine-induced vasoconstriction in the human coronary microcirculation: new evidence from myocardial contrast echocardiography. Circulation. 2013;128:598–604. doi: 10.1161/CIRCULATIONAHA.113.002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325–331. doi: 10.1016/0002-9343(90)90484-u. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558–2569. doi: 10.1161/CIRCULATIONAHA.110.940569. [DOI] [PubMed] [Google Scholar]
- 7.Qureshi AI, Suri MF, Guterman LR, Hopkins LN. Cocaine use and the likelihood of nonfatal myocardial infarction and stroke: data from the Third National Health and Nutrition Examination Survey. Circulation. 2001;103:502–506. doi: 10.1161/01.cir.103.4.502. [DOI] [PubMed] [Google Scholar]
- 8.Aquaro GD, Gabutti A, Meini M, Prontera C, Pasanisi E, Passino C, Emdin M, Lombardi M. Silent myocardial damage in cocaine addicts. Heart. 2011;97:2056–2062. doi: 10.1136/hrt.2011.226977. [DOI] [PubMed] [Google Scholar]
- 9.Kozor R, Grieve SM, Buchholz S, Kaye S, Darke S, Bhindi R, Figtree GA. Regular cocaine use is associated with increased systolic blood pressure, aortic stiffness and left ventricular mass in young otherwise healthy individuals. PLoS One. 2014;9:e89710. doi: 10.1371/journal.pone.0089710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma J, Rathnayaka N, Green C, Moeller FG, Schmitz JM, Shoham D, Dougherty AH. Bradycardia as a Marker of Chronic Cocaine Use: A Novel Cardiovascular Finding. Behav Med. 2016;42:1–8. doi: 10.1080/08964289.2014.897931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egashira K, Morgan KG, Morgan JP. Effects of cocaine on excitation-contraction coupling of aortic smooth muscle from the ferret. J Clin Invest. 1991;87:1322–1328. doi: 10.1172/JCI115135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacy F, Kailasam MT, O’Connor DT, Schmid-Schonbein GW, Parmer RJ. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension. 2000;36:878–884. doi: 10.1161/01.hyp.36.5.878. [DOI] [PubMed] [Google Scholar]
- 13.Wu L, Juurlink BH. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension. 2002;39:809–814. doi: 10.1161/hy0302.105207. [DOI] [PubMed] [Google Scholar]
- 14.Lerman LO, Nath KA, Rodriguez-Porcel M, Krier JD, Schwartz RS, Napoli C, Romero JC. Increased oxidative stress in experimental renovascular hypertension. Hypertension. 2001;37:541–546. doi: 10.1161/01.hyp.37.2.541. [DOI] [PubMed] [Google Scholar]
- 15.Trolliet MR, Rudd MA, Loscalzo J. Oxidative stress and renal dysfunction in salt-sensitive hypertension. Kidney Blood Press Res. 2001;24:116–123. doi: 10.1159/000054217. [DOI] [PubMed] [Google Scholar]
- 16.Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL. Oxidative stress in a rat model of obesity-induced hypertension. Hypertension. 2001;37:554–560. doi: 10.1161/01.hyp.37.2.554. [DOI] [PubMed] [Google Scholar]
- 17.Romero JC, Reckelhoff JF. State-of-the-Art lecture. Role of angiotensin and oxidative stress in essential hypertension. Hypertension. 1999;34:943–949. doi: 10.1161/01.hyp.34.4.943. [DOI] [PubMed] [Google Scholar]
- 18.Ratledge C. The role of malic enzyme as the provider of NADPH in oleaginous microorganisms: a reappraisal and unsolved problems. Biotechnol Lett. 2014;36:1557–1568. doi: 10.1007/s10529-014-1532-3. [DOI] [PubMed] [Google Scholar]
- 19.Wise EM, Jr, Ball EG. Malic Enzyme and Lipogenesis. Proc Natl Acad Sci U S A. 1964;52:1255–1263. doi: 10.1073/pnas.52.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rush GF, Gorski JR, Ripple MG, Sowinski J, Bugelski P, Hewitt WR. Organic hydroperoxide-induced lipid peroxidation and cell death in isolated hepatocytes. Toxicol Appl Pharmacol. 1985;78:473–483. doi: 10.1016/0041-008x(85)90255-8. [DOI] [PubMed] [Google Scholar]
- 21.Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11–18. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian J, An X, Niu L. Role of microRNAs in cardiac development and disease. Exp Ther Med. 2017;13:3–8. doi: 10.3892/etm.2016.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015:4. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sartor GC, Powell SK, Brothers SP, Wahlestedt C. Epigenetic Readers of Lysine Acetylation Regulate Cocaine-Induced Plasticity. J Neurosci. 2015;35:15062–15072. doi: 10.1523/JNEUROSCI.0826-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mokhtari V, Afsharian P, Shahhoseini M, Kalantar SM, Moini A. A Review on Various Uses of N-Acetyl Cysteine. Cell J. 2017;19:11–17. doi: 10.22074/cellj.2016.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Przywara DA, Dambach GE. Direct actions of cocaine on cardiac cellular electrical activity. Circ Res. 1989;65:185–192. doi: 10.1161/01.res.65.1.185. [DOI] [PubMed] [Google Scholar]
- 29.Wu SN, Chang HD, Sung RJ. Cocaine-induced inhibition of ATP-sensitive K+ channels in rat ventricular myocytes and in heart-derived H9c2 cells. Basic Clin Pharmacol Toxicol. 2006;98:510–517. doi: 10.1111/j.1742-7843.2006.pto_354.x. [DOI] [PubMed] [Google Scholar]
- 30.O’Leary ME, Chahine M. Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels. J Physiol. 2002;541:701–716. doi: 10.1113/jphysiol.2001.016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schindler CW, Tella SR, Katz JL, Goldberg SR. Effects of cocaine and its quaternary derivative cocaine methiodide on cardiovascular function in squirrel monkeys. Eur J Pharmacol. 1992;213:99–105. doi: 10.1016/0014-2999(92)90238-y. [DOI] [PubMed] [Google Scholar]
- 32.Care A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 33.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu S, Deng S, Ma Q, Zhang T, Jia C, Zhuo D, Yang F, Wei J, Wang L, Dykxhoorn DM, Hare JM, Goldschmidt-Clermont PJ, Dong C. MicroRNA-10A* and MicroRNA-21 modulate endothelial progenitor cell senescence via suppressing high-mobility group A2. Circ Res. 2013;112:152–164. doi: 10.1161/CIRCRESAHA.112.280016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng S, Wang H, Jia C, Zhu S, Chu X, Ma Q, Wei J, Chen E, Zhu W, Macon CJ, Jayaweera DT, Dykxhoorn DM, Dong C. MicroRNA-146a Induces Lineage-Negative Bone Marrow Cell Apoptosis and Senescence by Targeting Polo-Like Kinase 2 Expression. Arterioscler Thromb Vasc Biol. 2017;37:280–290. doi: 10.1161/ATVBAHA.116.308378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin X, Zhan JK, Wang YJ, Tan P, Chen YY, Deng HQ, Liu YS. Function, Role, and Clinical Application of MicroRNAs in Vascular Aging. Biomed Res Int. 2016;2016:6021394. doi: 10.1155/2016/6021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenny PJ. Epigenetics, microRNA, and addiction. Dialogues Clin Neurosci. 2014;16:335–344. doi: 10.31887/DCNS.2014.16.3/pkenny. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques FZ, Campain AE, Yang YH, Morris BJ. Meta-analysis of genome-wide gene expression differences in onset and maintenance phases of genetic hypertension. Hypertension. 2010;56:319–324. doi: 10.1161/HYPERTENSIONAHA.110.155366. [DOI] [PubMed] [Google Scholar]
- 39.Sylvius N, Tesson F, Gayet C, Charron P, Benaiche A, Peuchmaurd M, Duboscq-Bidot L, Feingold J, Beckmann JS, Bouchier C, Komajda M. A new locus for autosomal dominant dilated cardiomyopathy identified on chromosome 6q12-q16. Am J Hum Genet. 2001;68:241–246. doi: 10.1086/316929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. 2015;309:H1453–1467. doi: 10.1152/ajpheart.00554.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liaudet L, Calderari B, Pacher P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail Rev. 2014;19:815–824. doi: 10.1007/s10741-014-9418-y. [DOI] [PubMed] [Google Scholar]
- 42.Finsterer J, Ohnsorge P. Influence of mitochondrion-toxic agents on the cardiovascular system. Regul Toxicol Pharmacol. 2013;67:434–445. doi: 10.1016/j.yrtph.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Raij L. Nitric oxide in hypertension: relationship with renal injury and left ventricular hypertrophy. Hypertension. 1998;31:189–193. doi: 10.1161/01.hyp.31.1.189. [DOI] [PubMed] [Google Scholar]
- 44.Urao N, Ushio-Fukai M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic Biol Med. 2013;54:26–39. doi: 10.1016/j.freeradbiomed.2012.10.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brodeur MR, Bouvet C, Barrette M, Moreau P. Palmitic acid increases medial calcification by inducing oxidative stress. J Vasc Res. 2013;50:430–441. doi: 10.1159/000354235. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh MS, Zhong WB, Yu SC, Lin JY, Chi WM, Lee HM. Dipyridamole suppresses high glucose-induced osteopontin secretion and mRNA expression in rat aortic smooth muscle cells. Circ J. 2010;74:1242–1250. doi: 10.1253/circj.cj-09-0561. [DOI] [PubMed] [Google Scholar]
- 47.Ceravolo GS, Montezano AC, Jordao MT, Akamine EH, Costa TJ, Takano AP, Fernandes DC, Barreto-Chaves ML, Laurindo FR, Tostes RC, Fortes ZB, Chopard RP, Touyz RM, Carvalho MH. An interaction of renin-angiotensin and kallikrein-kinin systems contributes to vascular hypertrophy in angiotensin II-induced hypertension: in vivo and in vitro studies. PLoS One. 2014;9:e111117. doi: 10.1371/journal.pone.0111117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Yan SM, Zheng HL, Hu DH, Zhang YT, Guan QH, Ding QL. A mechanism underlying hypertensive occurrence in the metabolic syndrome: cooperative effect of oxidative stress and calcium accumulation in vascular smooth muscle cells. Horm Metab Res. 2014;46:126–132. doi: 10.1055/s-0033-1355398. [DOI] [PubMed] [Google Scholar]
- 49.Dalvi P, Wang K, Mermis J, Zeng R, Sanderson M, Johnson S, Dai Y, Sharma G, Ladner AO, Dhillon NK. HIV-1/cocaine induced oxidative stress disrupts tight junction protein-1 in human pulmonary microvascular endothelial cells: role of Ras/ERK1/2 pathway. PLoS One. 2014;9:e85246. doi: 10.1371/journal.pone.0085246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montezano AC, Neves KB, Lopes RA, Rios F. Isolation and Culture of Endothelial Cells from Large Vessels. Methods Mol Biol. 2017;1527:345–348. doi: 10.1007/978-1-4939-6625-7_26. [DOI] [PubMed] [Google Scholar]
- 51.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aalkjaer C, Heagerty AM, Petersen KK, Swales JD, Mulvany MJ. Evidence for increased media thickness, increased neuronal amine uptake, and depressed excitation--contraction coupling in isolated resistance vessels from essential hypertensives. Circ Res. 1987;61:181–186. doi: 10.1161/01.res.61.2.181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.