

Abstract
Stiffening of the vasculature with aging is a strong predictor of adverse cardiovascular events, independent of all other risk factors including blood pressure, yet no therapies target this process. Mineralocorticoid receptors in smooth muscle cells have been implicated in the regulation of vascular fibrosis but have not been explored in vascular aging. Comparing smooth muscle cell mineralocorticoid receptors-deleted male mice to mineralocorticoid receptors-intact littermates at 3, 12 and 18 months of age, we demonstrated that aging-associated vascular stiffening and fibrosis are mitigated by mineralocorticoid receptor deletion in smooth muscle cells. Progression of cardiac stiffness and fibrosis and the decline in exercise capacity with aging were also mitigated by mineralocorticoid receptor deletion in smooth muscle cell. Vascular gene expression profiling analysis revealed that mineralocorticoid receptor deletion in smooth muscle cell is associated with recruitment of a distinct anti-fibrotic vascular gene expression program with aging. Moreover, long-term pharmacological inhibition of mineralocorticoid receptor in aged mice prevented the progression of vascular fibrosis and stiffness and induced a similar anti-fibrotic vascular gene program. Finally, in a small trial in elderly male humans, short-term mineralocorticoid receptor antagonism produced an anti-fibrotic signature of circulating biomarkers similar to that observed in the vasculature of smooth muscle cell mineralocorticoid receptors-deleted mice. These findings suggest that smooth muscle cell mineralocorticoid receptor contributes to vascular stiffening with aging and is a potential therapeutic target to prevent the progression of aging-associated vascular fibrosis and stiffness.
Keywords: aging, vascular stiffening, vascular fibrosis, mineralocorticoid receptors, smooth muscle cell
Introduction
Aging is a potent and universal risk factor for cardiovascular diseases (CVD) including heart attack, stroke and heart failure.1 Mortality from CVD increase exponentially with age, accounting for over 40% of deaths in people aged 65–74 and 60% in those over 85 years.2 As previous anti-aging therapies have shown only modest benefits in CVD,3,4 further understanding of the aging process is needed to identify effective treatments to prevent or reverse cardiovascular aging.
Increased vascular stiffness is a prominent consequence of the aging process. Although the presence of multiple CVD risk factors, particularly hypertension, may accelerate vascular stiffening, a previous study found that increased vascular stiffness precedes hypertension.5 Pulse wave velocity (PWV) is a well validated noninvasive measure of vascular stiffness and substantial evidence supports that PWV significantly increases with age in humans and contributes to systolic hypertension.6–8 Increased PWV with aging has been linked to structural changes in the vasculature, including increased collagen accumulation, reduced elastin content and vascular calcification.7 Vascular stiffening increases the workload required by the heart to maintain adequate blood flow and in this way may promote cardiac hypertrophy and fibrosis and ultimately contribute to the high incidence of heart failure with aging.9,10 Moreover, vascular stiffness is a strong predictor of future myocardial infarction (MI), stroke or CVD mortality independent of any other traditional CVD risk factors including blood pressure (BP).6,8,11
The renin-angiotensin-aldosterone system (RAAS) is a hormonal cascade that plays a critical role in BP regulation. Previous studies revealed that RAAS component expression and angiotensin signaling in the arterial wall increase with aging and contribute to vascular structural alterations.2,8,12,13 The terminal step of the RAAS is the activation of mineralocorticoid receptors (MR) by the hormone aldosterone. MR is a hormone-activated transcription factor that modulates BP by regulating renal genes that promote sodium retention.14 MR is also expressed in extra-renal cells including vascular smooth muscle cells (SMC), endothelial cells (EC), cardiomyocytes, and macrophages and substantial evidence from animal studies reveals that extra-renal MR contributes to CVD.15 Similarly in human clinical trials, MR antagonists significantly reduce mortality in patients with heart failure or hypertension out of proportion to changes in BP, supporting extra-renal roles.16,17
Prior studies support that MR activation contributes to the regulation of vascular structure and that this may be mediated by direct effects on the vasculature.18–20 In rodent models, MR antagonist drugs attenuate vascular stiffness and fibrosis in the setting of CVD risk factors despite minimal effects on BP.21,22 Also in humans with chronic MR activation due to primary aldosteronism, vascular stiffness is significantly increased compared to BP-matched hypertensive patients with normal aldosterone levels.23 Moreover, in patients with hypertension, treatment with the MR antagonist eplerenone for one year significantly decreased vascular stiffness when compared to treatment to the same blood pressure goal with a beta blocker.24 These studies suggest that MR can contribute to vascular stiffness and fibrosis independent of renal effects to lower BP.
MR is expressed in human and mouse vascular SMC where it directly regulates vascular gene transcription and SMC function.25,26 Recent studies using mice with MR specifically deleted from SMC (SMC-MR-KO) revealed that SMC-MR contributes to vascular remodeling and fibrosis in response to wire injury20 and to vascular stiffening induced by the aldosterone/salt model of hypertension26. However the role of SMC-MR in cardiovascular stiffening with aging has not been explored. MR expression has been shown to increase with aging in rat aortic SMC27 and in mouse resistance vessels.28 Also, aged SMC-MR-KO mice are protected from the modest rise in BP, enhanced vascular constriction and oxidative stress observed in aged MR-intact littermates,18 supporting a direct role for SMC-MR in the process of aging. Hence, the aim of this study was to characterize the role of SMC-MR in cardiovascular tissue stiffness, fibrosis and vascular gene regulation in aging mice and to explore whether targeting MR would inhibit SMC-MR driven pathways to attenuate vascular aging in mice and in humans. The life expectancy of C57BL6 mice in captivity is 24–36 months. Prior studies exploring the vascular aging phenotype demonstrate that endothelial stiffness increases from 3 to 15 months of age29 and aortic SMC hypertrophy and expression of fibrosis genes increases from 3 to 12 months of age with a further increase by 24 months.13,30 Since these prior studies demonstrate vascular aging changes as early as at 12 months of age in mice, here we characterized the vascular and cardiac phenotype of SMC-MR-KO and MR-intact littermates at 12 and 18 months of age compared to 3-month old controls. We further determined changes in the vascular gene expression profile at 12 compared to 3 months of age to explore early aging effects on gene expression that might be causative and potentially modifiable by SMC-MR deletion or by treatment with a MR antagonist drug in mice and in humans.
Methods
Detailed methods are available in the Data Supplement.
Complete microarray data are available at National Center for Biotechnology Information Gene Expression Omnibus (accession number: GSE102397). All other data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
All mice were handled in accordance with US National Institutes of Health standards and all procedures were approved by the Tufts Medical Center Institutional Animal Care and Use Committee. Creation of tamoxifen-inducible SMC-MR-KO mice and confirmation of inducibility and specificity of SMC-MR recombination has been described previously.18 Wild-type (WT) male mice (12-month old) were implanted in the subscapular region with pellets containing either placebo or spironolactone (20 mg kg−1 day−1) for 120 days.
Human serum
Detailed protocol for collecting human serum is available in the Data Supplement. All studies were approved by the Institutional Review Board at all institutions involved.
Statistical Analysis
Values are presented as mean ± SEM. Means were compared by 1- or 2-factor ANOVA as appropriate, with Tukey post-hoc test using SigmaPlot statistical software. p<0.05 was considered significant.
Results
Deletion of MR in SMC prevents vascular stiffness and fibrosis with aging
Vascular stiffness was measured by quantifying aortic PWV in male MR-intact and SMC-MR-KO littermates at 3, 12 and 18 months of age. PWV increased with aging from 3 to 12 months with a non-significant trend towards a further increase from 12 to 18 months of age (p=0.08) in MR-intact mice but not in SMC-MR-KO mice. Thus, SMC-MR-KO mice had significantly lower PWV at 12 and 18 months of age compared to age-matched MR-intact mice (Fig. 1A). Vascular fibrosis, a driver of vascular stiffness, was quantified in three different sized vessels; aorta, carotid artery and renal arteriole. In MR-intact mice, vascular fibrosis increased with aging from 3 to 12 months in all three vascular beds (Fig. 1B to D). In the aorta, the vessel from which stiffness is measured by PWV, fibrosis was prevented in SMC-MR-KO (Fig. 1B). In carotid arteries, fibrosis was decreased in SMC-MR-KO mice at 12- and 18- months relative to age-matched MR-intact mice (Fig. 1C). Finally, in renal arterioles, fibrosis was attenuated in SMC-MR-KO at 18 months of age compared to age-matched MR-intact mice (Fig. 1D). These results indicate that SMC-MR is necessary for aging-associated vascular stiffening and contributes to fibrosis in both conduit arteries and arterioles of aging mice.
Fig. 1. Deletion of SMC-MR attenuates vascular stiffness and fibrosis with aging.
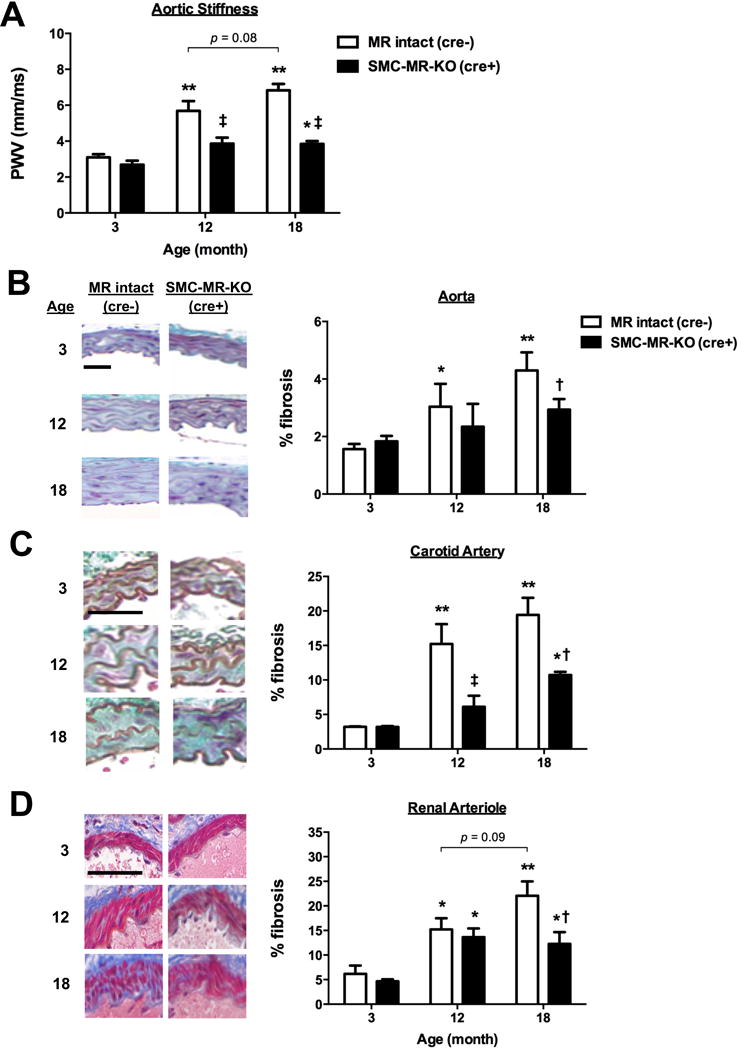
(A) Abdominal aortic pulse wave velocity (PWV) was measured, as an indicator for vascular stiffness, in 3-, 12- and 18-month old MR-intact and SMC-MR KO mice using echocardiography. n=7–8 mice per group. Fibrosis was quantified in sections stained with Masson’s Trichrome in (B) aortas, (C) carotid arteries and (D) renal arterioles from 3-, 12- and 18-month old MR-intact and SMC-MR KO mice. n=3–5 mice per age/genotype. Scale bar=10 μm. *p<0.05 and **p<0.01 compared to genotype-matched 3-month old mice; †p<0.05 and ‡p<0.01 compared to age-matched MR-intact mice.
SMC-MR deletion protects from aging-associated cardiac stiffness and fibrosis
We next examined the effect of SMC-MR deletion on end organ function in aging mice. Cardiac structure and function was assessed by cardiac ultrasound, in vivo hemodynamic measurements and histology. With aging to 12 months in MR-intact mice, cardiac hypertrophy developed (increased left ventricular (LV) mass/tibia length), followed by chamber dilation at 18 months (increased LV end-systolic and end-diastolic diameter), in association with a modest but significant reduction in LV ejection fraction and fractional shortening at 18 months of age (Table S1). These significant aging-related changes in cardiac hypertrophy, dilation, and dysfunction did not occur in SMC-MR-KO mice. Invasive hemodynamic measures of intrinsic cardiac function (stroke volume, peak LV pressure change in systole, diastolic relaxation time) did not significantly change with aging in either genotype (Table S1). However, aging was associated with a significant increase in LV diastolic and systolic stiffness in MR-intact mice that was prevented in SMC-MR-KO mice, as assessed by the slope of the LV end-diastolic or -systolic pressure-volume relationship (EDPVR (Fig. 2A) and ESPVR (Fig. 2B), respectively).
Fig. 2. Deletion of SMC-MR mitigates the development of cardiac stiffness and fibrosis and the decline in exercise capacity with aging.
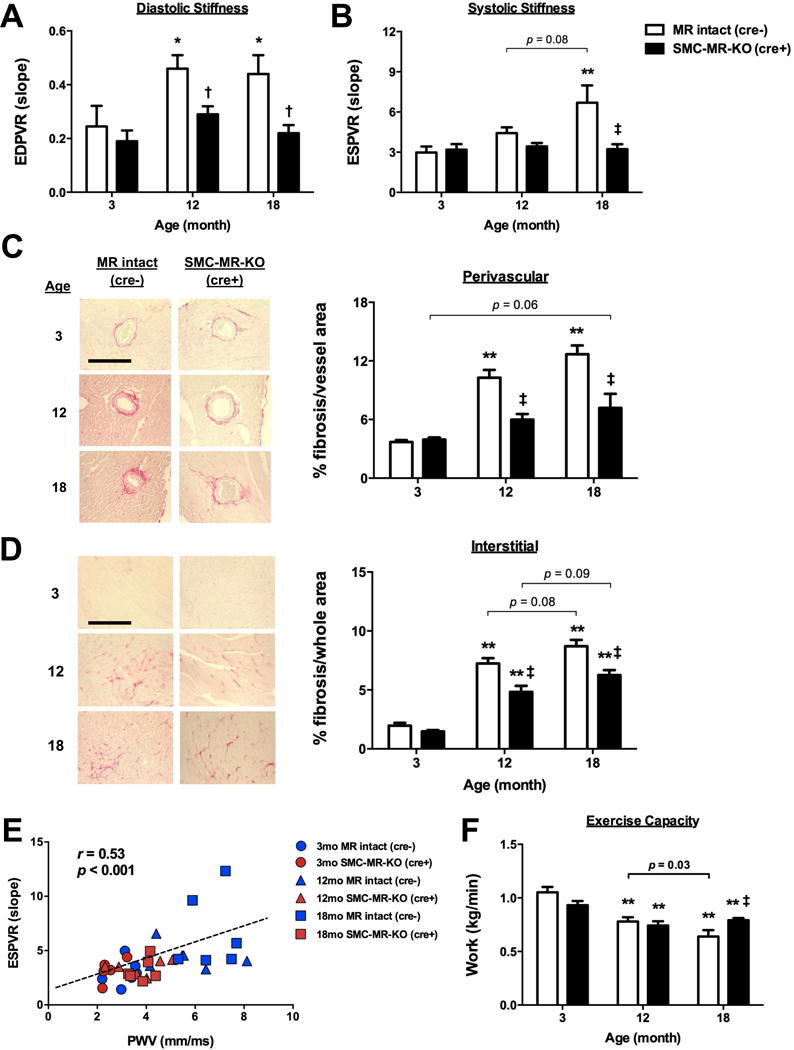
All measurements were made from 3-, 12- and 18-month old MR-intact and SMC-MR KO male littermates. (A) Left ventricular end-diastolic pressure volume relationship (EDPVR) and (B) end-systolic pressure volume relationship (ESPVR) were assessed by invasive hemodynamic measurements. (C) Perivascular and (D) interstitial fibrosis were quantified in left ventricle sections stained with Picrosirius Red. Scale bar=50 μm. (E) There is a significant correlation between cardiac stiffness (ESPVR) and vascular stiffness (PWV). (F) Endurance exercise capacity was determined my measuring run to exhaustion time normalized to body mass and vertical distance. n=5–6 mice per group. *p<0.05 and **p<0.01 compared to genotype-matched 3-month old mice; †p <0.05 and ‡p<0.01 compared to age-matched MR-intact mice.
We next assessed cardiac fibrosis histologically (Fig. 2C–D). Both perivascular and interstitial fibrosis increased with aging in MR-intact mice, while only interstitial fibrosis significantly increased with aging in SMC-MR-KO mice. Both perivascular and interstitial fibrosis were significantly attenuated in SMC-MR-KO at 12 and 18 months of age compared to age-matched MR-intact mice, consistent with reduced cardiac stiffening with aging. The degree of systolic stiffness as measured by ESPVR significantly correlated with aortic stiffness by PWV (Fig. 2E) as did cardiac diastolic stiffness (EDPVR), but to a less extent (r=0.46, p=0.003, data not shown). Changes in kidney hypertrophy and fibrosis also increased significantly with age but these renal changes were independent of the presence of SMC-MR (Fig. S1). Finally, while endurance exercise capacity decreased with aging in both MR-intact and SMC-MR-KO mice, the decline was attenuated such that 18-month old SMC-MR-KO had significantly improved endurance exercise capacity compared to age-matched MR-intact mice (Fig. 2F) with no difference in serum aldosterone, glucose or cholesterol in aged MR-intact compared to SMC-MR-KO mice (Figs. S2 and S3). Combined, these results demonstrate that SMC-MR plays a role in aging-associated cardiac fibrosis and stiffening, without influencing renal fibrosis, thus the mechanism of fibrosis in renal aging may differ from that in the heart and vessels and is not dependent on SMC-MR.
Distinct vascular aging transcriptomes in the presence or absence of SMC-MR
As MR is a transcriptional regulator, we next performed global aortic mRNA expression profiling to explore potential mechanisms by which SMC-MR deficiency protects from vascular aging. Microarray profiles from MR-intact and SMC-MR-KO littermates were compared in young 3-month old mice and aged 12-months old mice, the age at which SMC-MR dependent structural changes are first observed in Figure 1. Four differential gene expression comparisons were performed to assess the role of SMC-MR and aging and the numbers of up- or down-regulated genes for each comparison are shown in Figure 3A. The complete list of all regulated genes can be found in Tables S2–S5. Expression of MR gene (Nr3c2) in the aorta increased significantly with aging and was decreased in aortas from SMC-MR-KO compared to aortas from MR-intact mice at each age. Microarray data were validated for selected genes by independent RT-qPCR in aortic RNA derived from a second cohort of mice. Overall, 86% of tested gene expression changes from the microarray study were reproduced by PCR in biological replicates (Table S6). Compared to age-matched MR-intact mice, only 66 genes were differentially expressed in aortas from young SMC-MR-KO mice, while 579 genes were differentially expressed in aortas from aged SMC-MR-KO mice (Fig. 3A). This is consistent with the data in Fig. 1 revealing minimal effects of SMC-MR deletion on vascular structure and function in young mice, but substantial effects in aged mice.
Fig. 3. A distinct vascular aging transcriptome in MR-intact and SMC-MR-KO mice.
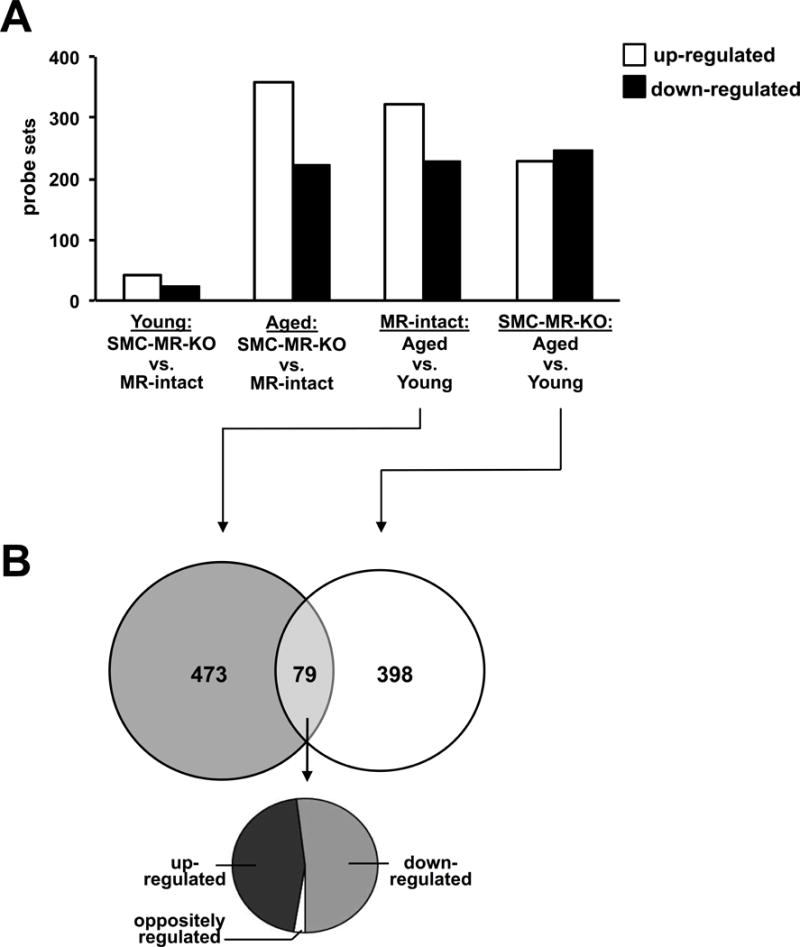
(A) Four comparisons were made using microarray gene expression profiling of aortic RNA from young (3-month old) and aged (12-month old) MR-intact and SMC-MR KO mice results (n=3–4 mice per age/genotype) and the number of probe sets differentially regulated by each comparison with a fold change >1.25 and p<0.01 is indicated. (B) Venn diagram depicts the degree of overlap between transcripts differentially regulated with aging in aortas from MR-intact and SMC-MR KO mice. The pie chart indicates the direction of regulation of the 79 transcripts regulated in both MR-intact and SMC-MR-KO mice with aging. Only 2 transcripts are oppositely regulated with aging between MR-intact and SMC-MR-KO mice.
Another approach to examining the role of SMC-MR in vascular aging is to contrast gene expression changes with aging in SMC-MR-KO with those with aging in MR-intact mice. Interestingly, although similar numbers of genes were differentially expressed with aging in MR-intact (552 genes) and SMC-MR-KO (477 genes) mice, only 79 genes changed significantly with aging in both MR-intact and SMC-MR-KO mice. Of these, only 2 genes, ubiquitin conjugating enzyme E2 Q family like 1 (ube2ql1) and small nucleolar RNA C/D box 34 (snord34), were regulated in opposite directions with aging in the two genotypes (Fig. 3B). These findings suggest that rather than merely preventing the expression of vascular aging-associated genes, the absence of SMC-MR results in recruitment of a distinct set of genes in the aging vasculature that are associated with protection from vascular stiffness and fibrosis.
Pathways associated with progression and protection from vascular aging
To systemically understand the genes driving the vascular aging phenotype in MR-intact mice compared to the protective gene program recruited with aging in SMC-MR-KO mice, Ingenuity Pathway Analysis (IPA) was performed. “Cardiovascular system development and function” was the top overrepresented biological function category within the genes differentially expressed with aging in MR-intact mice (Table S8). Within this category, there were 10 sub-categories significantly regulated with aging in both MR-intact and SMC-MR-KO mice. Comparing the z-score, a measure of the predicted direction of regulation of these sub-categories, revealed that 8 of those 10 sub-categories were predicted to be regulated in opposite directions with aging when comparing MR-intact and SMC-MR-KO mice (Fig. 4A). Notably, this opposing regulation was not driven by opposite regulation of the same genes. Rather, a distinct gene signature was regulated with aging in MR-intact versus SMC-MR-KO littermates that coordinately results in opposing effects on these cardiovascular pathways. For example, fibroblast growth factor 2 (FGF2) was up-regulated in MR-intact mice, but unchanged in SMC-MR-KO mice with aging. Whereas, connective tissue growth factor (CTGF) was unchanged in MR-intact mice and down-regulated in the aorta of SMC-MR-KO mice with aging. The gene signatures that contribute to opposing regulation of “Cardiovascular system development and function” are indicated in Fig. 4B. These results support the concept that the biological processes activated with aging in aortas from MR-intact mice are reversed in aortas from SMC-MR-KO mice by recruiting a distinct set of genes with opposing biological functions.
Fig. 4. The absence of SMC-MR recruits a unique anti-fibrotic gene signature in the aging vasculature.
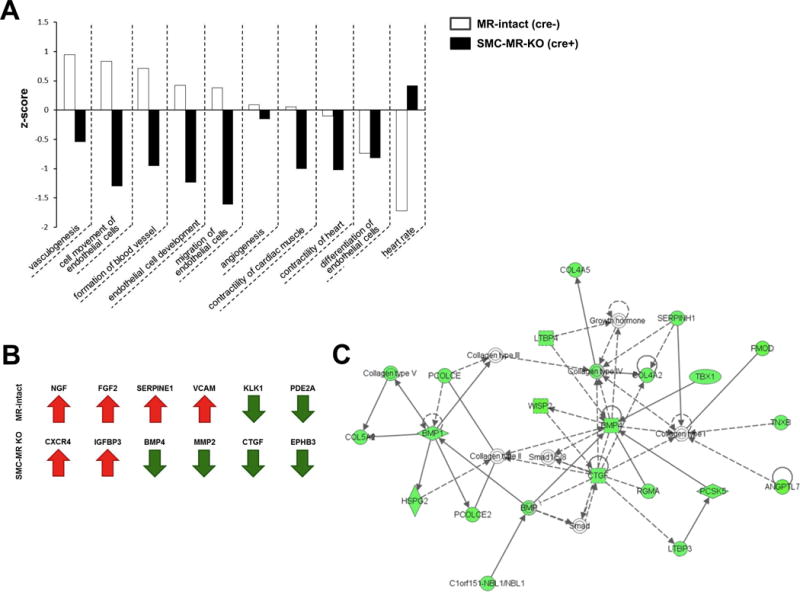
(A) Within the top biological function regulated in the aging vasculature, “Cardiovascular development and function”, there are 10 sub-categories regulated in both MR-intact and SMC-MR-KO mice. The Z-score, a predicted direction of the regulated pathway, is shown comparing MR-intact and SMC-MR-KO mice. (B) Determining a vascular aging gene signature (genes in at least 4 of the above sub-categories) in MR-intact and SMC-MR-KO mice. Red arrows indicate up-regulation and green arrows indicate down-regulation with aging. (C) Top network by IPA analysis of genes differentially expressed with aging in aortas from SMC-MR-KO mice. Green nodes indicate significant down-regulation with fold change >1.25 and p<0.01. This fibrosis-associated network is significantly and globally down-regulated with aging only in aortas from SMC-MR KO mice.
The top five IPA gene networks differentially regulated with aging in MR-intact and SMC-MR-KO littermates are summarized in Table S9. The top regulated gene network in SMC-MR-KO mice is made up of genes involved in “connective tissue development and function” (Fig. 4C). Of 29 genes in this network, 23 genes were down-regulated with aging. The remaining 6 were unchanged with aging. On the other hand, all 29 genes in this network were unchanged with aging in MR-intact mice (data not shown). This network is known to play a critical role in promoting tissue fibrosis, thus the profound down-regulation of this pathway in aged vessels lacking SMC-MR supports recruitment of an anti-fibrotic gene program as a possible mechanism for the protection from vascular fibrosis with aging in SMC-MR-KO mice.
Upstream regulator analysis was employed to identify regulators predicted to coordinately control the gene expression changes observed (Table S10). Interestingly, the MR antagonist eplerenone was predicted to be inhibited with aging in MR-intact mice, while the MR agonist aldosterone was predicted to be inhibited with aging in SMC-MR-KO mice, supporting a critical mechanistic role for MR signaling in driving global vascular gene expression changes with aging. The upstream regulator most substantially inhibited with aging in SMC-MR-KO mice is transforming growth factor beta 1 (TGFβ-1) which has a well-established role in promoting vascular fibrosis.
MR antagonism in aged mice attenuates vascular stiffness and fibrosis and recruits the anti-fibrotic vascular gene program associated with SMC-MR deficiency
We next tested whether systemic MR inhibition with spironolactone could attenuate the vascular stiffness and fibrosis associated with aging via similar anti-fibrotic mechanisms. Twelve month-old male wild type (WT) mice with established vascular stiffness and fibrosis (Fig. 1) were randomized to 4 months of treatment with placebo or a dose of spironolactone previously shown to have no depressor effect on blood pressure.21 PWV significantly increased from 12 to 16 months of age in placebo-treated mice, while this was prevented in spironolactone-treated mice, resulting in significantly lower PWV in 16-month old spironolactone-treated mice (Fig. 5A). As PWV was measured every 2 months in the same mice, we examined the change in PWV from the 12-month old baseline in each mouse and determined that spironolactone treatment prevented the progression of vascular stiffness after 2 and 4 months of treatment (Fig. 5B). The degree of aortic, carotid, and renal fibrosis was also significantly attenuated by spironolactone compared to placebo (Figs. 5C and D). As expected, serum aldosterone levels increased with spironolactone treatment (Table S11), confirming effective MR blockade, and BP measured by tail-cuff method was not affected at this dose of spironolactone (Fig. 5E). Next, aortic mRNA expression was quantified for the protective gene expression signature previously identified in SMC-MR-KO mice. C-X-C chemokine receptor type 4 (CXCR4) was significantly up-regulated and bone morphogenetic protein 4 (BMP4) and TGFβ were significantly down-regulated in the aortas from spironolactone- compared to placebo-treated mice (Fig. 5F). As in the SMC-MR-KO mice (Fig. 4C), collagen type 1 and 3 (Col1 and Col3, respectively) mRNA expression levels were not significantly changed after 4-month spironolactone treatment. While not universal for all genes, overall spironolactone treatment induced a similar protective vascular gene expression signature as that observed with aging in SMC-MR-KO mice (compare 4B to 5F). As expected, aortic MR gene (Nr3C2) expression was unchanged with spironolactone treatment. Overall, systemic MR inhibition prevented aging-associated increases in vascular stiffness and fibrosis in aged mice while inducing an anti-fibrotic vascular gene expression signature similar to that induced with aging in SMC-MR-KO mice.
Fig. 5. MR antagonism attenuates vascular stiffness and vascular and renal fibrosis in aged mice while promoting the anti-fibrotic vascular gene signature.
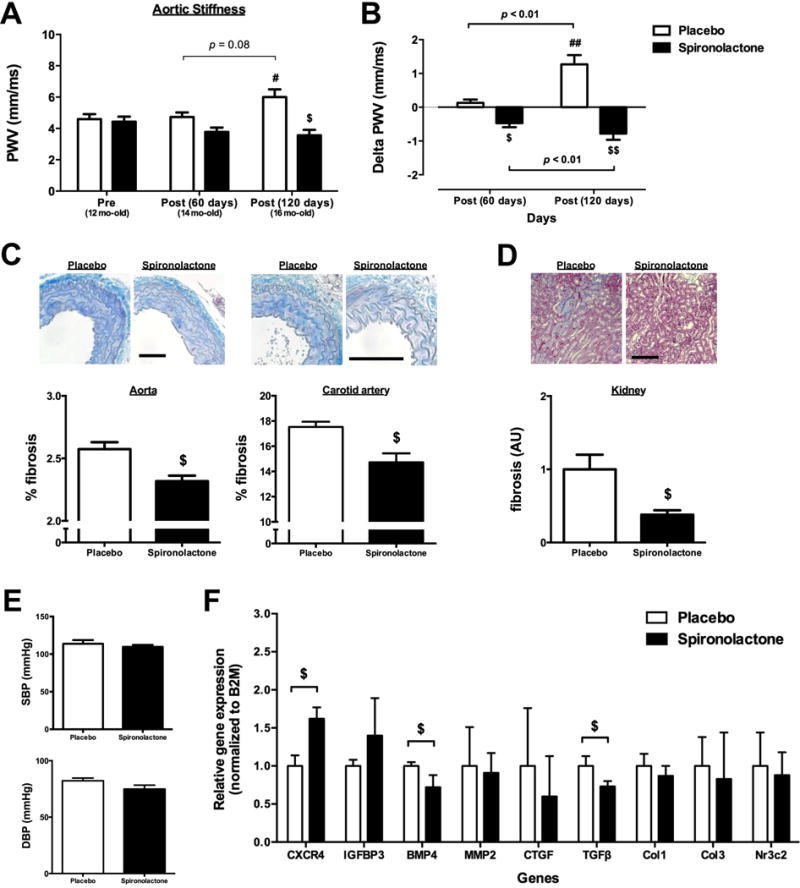
(A) Abdominal aortic PWV was measured in 12-month old male WT mice pre-randomization and again after 2-month (14-month old) and 4-month (16-month old) treatment with placebo or spironolactone (n=5–7 mice/group). (B) The change in vascular stiffness (Delta PWV) in each individual mouse was compared between placebo- and spironolactone-treated mice. Fibrosis was quantified in (C) aortic and carotid sections and (D) left kidney sections stained with Masson’s Trichrome from mice treated with placebo or spironolactone for 4 months. (E) Blood pressure was recorded after 4 months treatment with placebo or spironolactone by tail cuff plethysmography. (F) Expression of the aging gene signature identified in SMC-MR-KO mice was analyzed by RT-qPCR in aortas after 4 months of treatment. n=4–5 mice/group. Scale bar=25 μm. #p<0.05 and ##p<0.01 compared to pre-pellet implantation; $p<0.05 and $$p<0.01 compared to age-matched mice implanted with placebo.
MR antagonism in aged humans induces changes in circulating biomarkers that correlate with the anti-fibrotic vascular gene signature
As SMC-MR deletion or systemic MR inhibition induced a protective vascular gene expression program in aged mouse vessels, we aimed to test whether MR inhibition could induce similar effects in humans. As human vascular tissue cannot be routinely obtained, we assessed circulating levels of the secreted protein products of the vascular protective SMC-MR gene signature as well as other known fibrotic biomarkers. These biomarkers were measured in serum collected from a small study of 11 healthy male subjects average age 63.6±2.1 years treated for one month with eplerenone and one month with placebo in a randomized crossover study design, as previously described.31
In general, after one month of MR antagonism, circulating levels of secreted products of genes from the SMC-MR vascular protective signature were reduced in older male subjects (Table 1). Specifically, after eplerenone treatment, the serum matrix metallopeptidase 2 (MMP2) level decreased significantly, there was a strong trend towards decreased serum BMP4 (50% decrease, p=0.068) and CTGF also tended to decrease, although this was not statistically significant in this small cohort of patients. This pattern matched the protective gene signature identified with aging in SMC-MR-KO mice and was also similar to the aortic gene expression pattern after 4 months of spironolactone treatment in aged WT mice (Figs. 4B and 5F). Circulating levels of procollagen type I C-terminal peptide (PIP) and procollagen type III N-terminal propeptide (P3NP), biomarkers respectively of collagen type 1 and 3 production, were not significantly altered by eplerenone treatment. This is consistent with results from mouse vessels in which vascular Col1 and Col3 gene expression were unchanged with aging in SMC-MR-KO mice and with MR antagonism in aged WT mice (Table S5 and Fig. 5F). The circulating level of C-telopeptide of type I collagen (ICTP), a biomarker of collagen degradation, was significantly decreased in humans after MR antagonist treatment. This is consistent with the eplerenone-induced decrease in circulating MMP2, a matrix protease that degrades collagen. As in the mice, the aldosterone level was increased after eplerenone treatment, confirming the effectiveness of MR blockade in these patients. This short duration of eplerenone treatment did not affect vascular stiffness as previously reported by measuring PWV in this cohort.31 Similarly, the changes in circulating levels of biomarkers between eplerenone and placebo treatment did not significantly correlate with the minimal changes in PWV (Table S12). While this study is very small, the data support that short term systemic inhibition of MR produces an anti-fibrotic biomarker signature that correlates with the protective gene expression program induced with aging in SMC-MR-KO mice and with spironolactone treatment in aged mice.
Table 1.
Fibrotic biomarkers in serum from elderly males administered with placebo or eplerenone for 1 month
| Circulating level in human serum
|
Gene expression change in mouse aorta
|
||||
|---|---|---|---|---|---|
| Biomarker | Placebo | Eplerenone | p value | Aged vs. young SMC-MR-KO (Table S5) | Spironolactone vs. placebo in aged WT(Figure 5F) |
| BMP4 (pg/mL) | 51.7 ± 10.9 | 26.3 ± 6.3 | 0.068 | ↓† | ↓* |
| MMP2 (ng/mL) | 224.2 ± 7.8 | 196.5 ± 8.7 | 0.035* | ↓† | no change |
| CTGF (pg/mL) | 287.0 ± 65.1 | 181.1 ± 20.1 | 0.154 | ↓† | ↓ |
| PIP (ng/mL) | 454.2 ± 29.2 | 478.6 ± 31.0 | 0.591 | (Col1) no change | (Col1) no change |
| P3NP (ng/mL) | 4.2 ± 0.8 | 2.9 ± 0.3 | 0.194 | (Col3) no change | (Col3) no change |
| ICTP (ng/mL) | 1115.9 ± 50.7 | 946.2 ± 46.2 | 0.029* | N.D. | N.D. |
| PIP:ICTP (ng/mL) | 0.4 ± 0.0 | 0.5 ± 0.0 | 0.123 | N.D. | N.D. |
| Aldosterone (pg/mL) | 49.9 ± 5.9 | 86.1 ± 9.8 | 0.007† | N.D. | ↑ |
Average age = 63.6 ± 2.1; n=11/group; BMP4, bone morphogenetic protein 4; MMP2, matrix metallopeptidase 2; CTGF, connective tissue growth factor; PIP, procollagen type I C-terminal peptide (biomarker of Type 1 collagen (Col1) production); P3NP, procollagen type III N-terminal propeptide (biomarker of Type 3 collagen (Col3) production); ICTP, C-telopeptide of type I collagen (biomarker of collagen breakdown); N.D., not determined; Arrows indicate the direction of the change in each gene in aorta with aging from SMC-MR-KO mice or with spironolactone treatment in aged WT mice;
p < 0.05;
p < 0.01.
Discussion
By comparing aging MR-intact mice to SMC-MR-KO littermates, we identified a critical role for SMC-MR in cardiovascular aging. Specifically, we found that: (i) Increased vascular stiffness and fibrosis with aging are mitigated when MR is specifically deleted from SMC in mice; (ii) Aging-associated increases in cardiac stiffness and fibrosis and the decline in exercise capacity are also mitigated in the absence of SMC-MR; (iii) Gene expression profiling in mouse aortas revealed that vascular aging is associated with induction of gene pathways associated with cardiovascular development while when SMC-MR is deleted, aging induces a distinct anti-fibrotic gene signature; (iv) In aged mice, systemic inhibition of MR prevents the progression of vascular stiffness and fibrosis with induction of a similar anti-fibrotic pattern of aortic gene expression to that seen with SMC-MR deletion; (v) Systemic inhibition of MR, but not SMC-MR deletion, prevented renal fibrosis suggesting a role for non-SMC MR in renal fibrosis with aging; and finally (vi) Short term MR antagonist treatment in a small human cohort of older males produces an analogous anti-fibrotic serum biomarker signature. Overall, this study identifies MR in SMC as a mediator of cardiovascular aging and as a potential target to prevent the adverse consequences of vascular stiffness with aging. These data support the initiation of larger trials to test whether longer-term treatment with MR antagonists in older humans could prevent the progression of vascular fibrosis and stiffness and potentially improve cardiovascular outcomes with aging.
Conduit vessel stiffness significantly increases with aging in humans and in rodents thereby promoting resistance vessel dysfunction and contributing to the development of hypertension and to impaired regional blood flow.7,9 As a consequence, vascular stiffness is a significant independent risk factor for MI, stroke, and CVD death.6,11 The RAAS contributes to vascular fibrosis and stiffness with aging,2,12 but the underlying mechanisms have not been completely elucidated. Here, we demonstrate for the first time that SMC-MR contributes to aging-associated changes in vascular fibrosis and stiffness and that MR deletion in vascular smooth muscle is sufficient to inhibit vascular fibrosis related to aging. Prior studies have also implicated MR in myeloid cells in vascular dysfunction and fibrosis,32 although this has not been explored with aging. EC stiffness has also been shown to increase with aging29 and EC-MR has been implicated in vascular stiffness in response to obesity19 by a mechanism implicating up-regulation of epithelial sodium channels.33,34 Thus, MR in other cell types likely also contribute to vascular aging.
A role for SMC-MR was previously demonstrated in arterial stiffening and fibrotic remodeling in young mice in response to hypertension or to direct vascular injury.20,26 Notably, in both models, vascular structure was unchanged by deletion of MR in the absence of a vascular injury stimulus. Indeed, in uninjured carotid arteries, MR activation by aldosterone infusion did not affect vascular structure.20 Combined with the current study, the data support the overall concept that SMC-MR does not modulate vascular structure in the healthy state but rather SMC-MR is a critical mediator of the vascular fibrotic response in the setting of vascular damage induced by direct injury, hypertension and now also aging. Interestingly, the serum aldosterone level was significantly decreased in 18-month compared to 12-month old mice regardless of the presence of SMC-MR. Prior studies show that MR can be activated in a ligand-independent manner by Angiotensin II (Ang II)25 and ras-related C3 botulinum toxin substrate 1 (Rac1).35 Ang II signaling is augmented with aging13 and Rac1 is activated by oxidative stress,36 which also increases with aging. This raises the possibility that other hormones or hormone-independent mechanism might activate vascular MR with aging.
It is well known that cardiac structure also changes with age resulting in wall thickening, fibrosis and stiffness that contributes to heart failure with preserved ejection fraction and in some cases, ultimately leads to cardiac dilation and systolic dysfunction.37 Ample data show that MR antagonist treatment reduces cardiac remodeling and mortality associated with heart failure and hypertension in animals and in humans.16,17,21,22 These benefits are generally independent of changes in BP and kidney function yet the specific cell type in which MR inhibition is cardio-protective is only beginning to be elucidated.15 Deletion of MR in cardiomyocytes (but not fibroblasts) in mice prevented cardiac dysfunction and remodeling in response to pressure overload or MI, and MR deletion in macrophages prevented cardiac fibrosis in response to deoxycorticosterone/salt hypertension.15 In the current study, the aging-associated increase in cardiac fibrosis is attenuated by SMC-MR deletion. Several potential mechanisms could contribute to the role of SMC-MR in aging-induced cardiac fibrosis. First, increased aortic stiffness exposes to the heart to higher pressures and this can lead to left ventricular hypertrophy, fibrosis and systolic stiffening.9,10 Indeed, the degree of cardiac systolic stiffness correlated with the degree of vascular stiffness so perhaps the cardiac benefits in SMC-MR-KO mice are secondary to the prevention of vascular stiffening. Another potential mechanism could involve a role for SMC-MR in leukocyte extravasation into the heart. Extracellular matrix stiffening has been shown to enhance leukocyte extravasation38 and secreted factors induced by MR in SMC have been shown to recruit leukocytes in vitro.39 MR in EC has also been shown to play a role in leukocyte recruitment, adhesion and transmigration through the endothelium.40 However, specific deletion of MR from EC in a mouse model protected from cardiac dysfunction in response to pressure overload without affecting cardiac leukocyte infiltration.41 Indeed, it was previously demonstrated that MR inhibition prevents AngII/salt-induced cardiac perivascular inflammation and fibrosis in rats, although the cell specific role of MR was not addressed.42 SMC-MR in coronary arteries may contribute to leukocyte infiltration that drives perivascular fibrosis, limiting oxygen delivery into interstitial spaces, thereby contributing to interstitial fibrosis. Alternatively, or in addition, SMC-MR may contribute to vasoconstriction in coronary vessels and in this way lead to cardiac ischemia and fibrosis with aging. A prior study demonstrated a role for SMC-MR in coronary vasoconstriction in response to ischemia-reperfusion in the heart.43 Overall, the current study demonstrates a role for SMC-MR in cardiac remodeling with aging and further studies are warranted to differentiate between these potential mechanisms.
Exercise capacity decreases with aging in humans, is a powerful predictor of mortality, and contributes to declining quality of life.44,45 In the current study, we confirmed that exercise capacity decreases with aging in mice and this decline is mitigated in SMC-MR-KO mice. There are many physiological factors involved in determining exercise capacity, including pulmonary diffusion, cardiac output, vascular function, oxygen carrying in the blood, skeletal muscle blood flow, and energy metabolism, all factors that are compromised with aging.44 Some studies suggest that MR may regulate energy metabolism, potentially via MR in adipocytes46, but there is no evidence that this is affected by SMC-MR as growth and glucose were unchanged in knockout mice, although metabolic function was not formally tested. Given the notion that a critical limiting factor for exercise capacity is oxygen delivery to exercising muscles in response to increasing physiologic demands,45 reduced fibrosis and stiffness in the heart and vessels of old SMC-MR-KO mice could contribute to increased oxygen delivery to exercising muscles. Further studies are needed to directly test the role of SMC-MR in regulating skeletal muscle blood flow in response to exercise and how this might change with aging.
As MR is a hormone-activated transcription factor, we compared the vascular aging transcriptomes in the presence and absence of SMC-MR in 12-month old mice to identify early mechanisms driving the protection from vascular aging. As the mouse aorta is largely composed of SMC,47 gene expression profiles from aortas predominantly represents SMC gene expression. Rammos et al. previously reported that 561 genes were differentially regulated with aging in aortas from C57BL/6 mice.48 These genes were enriched in signaling pathways including extracellular matrix integrity, chemokine signaling, cell adhesion as well as the RAAS, thus our findings are consistent with prior studies. Importantly, rather than merely shutting down the genes recruited with aging in MR-intact vessels, SMC-MR deletion produced a distinct gene expression pattern with aging. MR deletion resulted in profound down-regulation of BMP4, CTGF, and TGFβ, well characterized pro-fibrotic regulators that contribute to vascular fibrosis with aging.9 These genes were unchanged with aging in MR-intact mice supporting that MR activation in SMC is driving the consistent expression of these pro-fibrotic genes in the vasculature over time. Several mechanisms have been identified by which vascular MR regulates gene expression including direct binding to MR-responsive elements in gene promoter,49 regulation of other transcriptional factors such as activator protein 1 (AP-1) and nuclear factor-kB (NF-kB) in the heart,50 and epigenetic mechanisms including suppression of miR-155 expression in SMC.28 As these mechanisms are cell-, tissue- or gene-specific, further studies are required to unravel the mechanisms by which SMC-MR globally regulates fibrotic vascular gene expression with aging.
MR antagonism was previously shown to prevent the development of vascular stiffness and fibrosis induced by hypertension or aging in animal models,21,22 so we explored the therapeutic potential of systemic MR-inhibition to induce a similar anti-fibrotic phenotype in aged mice using a low dose of spironolactone that did not reduce BP in a prior study.21 A direct effect of spironolactone on the vasculature is supported by demonstration that MR antagonism induced similar aortic gene expression changes as those in aging SMC-MR-KO mice. Specifically, TGFβ and BMP4 were significantly down-regulated by spironolactone treatment and these genes are known to play a role in SMC proliferation, hypertrophy, calcification, and matrix remodeling, which together contribute to fibrosis.9 In addition, CTGF is a critical mediator of fibrosis and a known vascular MR target gene.51 Together, these findings support the idea that increased SMC-MR expression and signaling with aging alters vascular gene expression and directly contributes to vascular structural changes and that MR inhibition prevents this process and the associated cardiovascular dysfunction. Interestingly, renal fibrosis is also inhibited by global MR inhibition but not by SMC-MR deletion suggesting that MR in non-SMC mediated the protective effect of spironolactone on kidney fibrosis with aging.
To explore the translational implications of these preclinical studies, we measured serum circulating levels of SMC-MR target genes and known fibrosis biomarkers in a small cohort of elderly males after one month of eplerenone treatment. Biomarkers were measured as surrogates, since vascular gene expression could not be directly assessed in human vessels. Eplerenone reduced circulating levels of MMP2 and ICTP with a trend towards a decrease in BMP4 and CTGF (49% decrease, p=0.068 and 37% decrease, p=0.15, respectively). This overall pattern of circulating biomarkers in response to eplerenone mirrors the aortic gene expression signature in aging SMC-MR-KO and spironolactone-treated aged WT mice. Changes in collagen turnover biomarkers have been associated with adverse CVD outcomes including heart failure and atherosclerosis.52,53 Since ICTP is an MMP-dependent degradation product of type I collagen,53 the decreased level of MMP2 with eplerenone treatment could contribute to the decrease in ICTP level induced by MR inhibition. A previous clinical study showed that the serum ICTP levels positively correlate with arterial stiffness.54 While there was no change in PWV with MR inhibition in our clinical study, this might be expected due to the short-term (one month) treatment. Indeed, this is consistent with our mouse study in which 2-month spironolactone treatment in aged mice did not significantly affect vascular stiffness, yet vascular stiffness was significantly decreased after 4 months of treatment. It remains possible that the observed changes in serum fibrosis biomarkers in our small human study reflect early changes in vascular gene expression that could lead to improvements in vascular function and structure with longer duration of treatment. These results support the need for larger and longer clinical studies to clarify the potential benefits of MR antagonism to treat vascular stiffness and associated morbidity with aging.
Some limitations and future directions should be acknowledged. (1) Spironolactone or eplerenone globally antagonizes MR thus the changes observed with MR antagonist treatment may be driven by inhibition of SMC-MR as well as MR in other cell types. A recent study reported that EC-specific deletion of MR in female mice prevented vascular remodeling and stiffness induced by obesity.19 As SMC and EC function in concert to regulate vascular function and structure,55 globally blocking both SMC- and EC-MR in the vasculature by MR antagonist might contribute concomitantly to the observed changes in vascular fibrosis and stiffness. Accordingly, future studies are needed to explore the specific role of EC-MR in vascular aging. As MR antagonists have a small but concerning risk of hyperkalemia through MR inhibition in the kidney,17 this study supports further development of vascular-specific drug delivery platforms. (2) BP was measured by the less sensitive method of tail cuff plethysmography in the spironolactone study so as to not disturb aortic structure with a telemetry catheter to allow for accurate PWV and fibrosis measurements. As this method would not be sensitive to very small changes in BP, we cannot rule out the possibility that spironolactone resulted in a small difference in BP that is below the level of detection of this otherwise well validated tail cuff method. (3) Previous studies have shown that SMC-MR contributes to vascular calcification56 and to oxidative stress with aging,18,28 however, there are other important processes that contribute to vascular aging including mitochondrial dysfunction, autophagy, DNA-damage, and cell senescence.57 While beyond the scope of this study, it would be interesting to determine whether SMC-MR contributes to these additional aging mechanisms in the vasculature. (4) Finally, for practical reasons only male mice were used in this long-term aging study. Substantial evidence indicates sex differences in vascular aging58 and recent studies suggest a potential sex-specific contribution of MR to other aspects of vascular function.19,59 Accordingly, future studies are warranted to examine sex difference in the role of SMC-MR in vascular aging.
Supplementary Material
Perspectives.
This study reveals that MR in smooth muscle cells plays a critical role in vascular stiffness and fibrosis with aging. Moreover, deletion of MR specifically from SMC attenuates cardiac stiffening and fibrosis and recruits a potent anti-fibrotic vascular gene expression program with aging. Results from both aged mice and humans treated with an MR antagonist suggest that MR antagonism may prevent the progression of aging-associated vascular stiffness by inhibiting SMC-MR driven fibrosis pathways and supports the possibility of using fibrosis biomarkers to monitor therapy. These data support the initiation of large long-term clinical studies of MR antagonists to determine potential vascular benefits that could mitigate cardiovascular aging and the associated cardiovascular morbidity and mortality that is so common in the elderly.
What is New?
This study demonstrates for the first time that mineralocorticoid receptor (MR) deletion restricted to smooth muscle cells (SMC) protects from vascular and cardiac stiffening and fibrosis with aging.
Transcriptomics analysis reveals that aging in the absence of SMC-MR recruits a potent anti-fibrotic gene expression program in the vasculature.
MR antagonist treatment prevents vascular stiffness and fibrosis and induces a similar anti-fibrotic vascular gene signature in aged mice and an analogous anti-fibrotic serum biomarker signature in elderly men.
What is Relevant?
These findings suggest the potential for MR to be a pharmacological target to prevent the progression of vascular fibrosis and stiffness with aging.
As vascular stiffness increases with aging in humans and predicts adverse cardiovascular outcomes, the potential to prevent vascular stiffness to improve functional outcomes and decrease mortality is highly clinically relevant.
Summary.
Deletion of mineralocorticoid receptors specifically from smooth muscle cells protects from aging-associated vascular and cardiac stiffness and fibrosis and improves exercise capacity in male mice. This correlates with recruitment of an anti-fibrotic vascular gene signature. Pharmacological mineralocorticoid receptors blockade in aged mice reproduces the vascular protection and gene signature. In a small study in aged men, mineralocorticoid receptors antagonism induced a similar anti-fibrotic biomarker signature. These findings support a potential therapeutic strategy that could be immediately tested in large clinical trials using a new biomarker panel to monitor therapy.
Acknowledgments
We thank Nathan. Li for assistance with sectioning and staining mouse tissues; Jeung-ki. Yoo, PhD for assistance with handling human serum. We acknowledge Adam Gower and the Boston University Microarray and Sequencing Resource Core Facility that is supported by CTSA grant UL1-TR000157.
Sources of Funding: This work was supported by grants from the National Institutes of Health: HL095590 and HL119290 to IZJ, F30HL137255 to MEM and AG032067 to DDC, and the American Heart Association: EIA18290005 to IZJ, 15POST21300000 to JJD, 17PRE32910003 to MEM, and 0865117F to DDC.
Footnotes
Disclosures: None.
References
- 1.Dhingra R, Vasan RS. Age as a risk factor. Med Clin North Am. 2012;96(1):87–91. doi: 10.1016/j.mcna.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med. 2010;65(10):1028–1041. doi: 10.1093/gerona/glq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulton RL, McMurdo MET, Hill A, Abboud RJ, Arnold GP, Struthers AD, Khan F, Vermeer C, Knapen MHJ, Drummen NEA, Witham MD. Effect of Vitamin K on Vascular Health and Physical Function in Older People with Vascular Disease–A Randomised Controlled Trial. J Nutr Health Aging. 2016;20(3):325–333. doi: 10.1007/s12603-015-0619-4. [DOI] [PubMed] [Google Scholar]
- 4.Snyder PJ, Bhasin S, Cunningham GR, et al. Effects of Testosterone Treatment in Older Men. N Engl J Med. 2016;374(7):611–624. doi: 10.1056/NEJMoa1506119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308(9):875–881. doi: 10.1001/2012.jama.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Rourke MF, Nichols WW. Aortic Diameter, Aortic Stiffness, and Wave Reflection Increase With Age and Isolated Systolic Hypertension. Hypertension. 2005;45(4):652–658. doi: 10.1161/01.HYP.0000153793.84859.b8. [DOI] [PubMed] [Google Scholar]
- 7.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107(1):139–146. doi: 10.1161/01.CIR.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 8.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am. 2009;93(3):583–604. doi: 10.1016/j.mcna.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can J Cardiol. 2016;32(5):659–668. doi: 10.1016/j.cjca.2016.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duprez DA. Arterial stiffness/elasticity in the contribution to progression of heart failure. Heart Fail Clin. 2012;8(1):135–141. doi: 10.1016/j.hfc.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial Stiffness and Cardiovascular Events: The Framingham Heart Study. Circulation. 2010;121(4):505–511. doi: 10.1161/CIRCULATIONAHA.109.886655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duprez DA. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens. 2006;24(6):983–991. doi: 10.1097/01.hjh.0000226182.60321.69. [DOI] [PubMed] [Google Scholar]
- 13.Yoon HE, Kim EN, Kim MY, Lim JH, Jang I, Ban TH, Shin SJ, Park CW, Chang YS, Choi BS. Age-Associated Changes in the Vascular Renin-Angiotensin System in Mice. Oxid Med Cell Longev. 2016;2016:6731093. doi: 10.1155/2016/6731093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossier BC, Pradervand S, Schild L, Hummler E. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol. 2002;64:877–897. doi: 10.1146/annurev.physiol.64.082101.143243. [DOI] [PubMed] [Google Scholar]
- 15.Cole TJ, Young MJ. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Mineralocorticoid receptor null mice: informing cell-type-specific roles. J Endocrinol. 2017;234(1):T83–T92. doi: 10.1530/JOE-17-0155. [DOI] [PubMed] [Google Scholar]
- 16.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 17.P Pitt B, Reichek N, Willenbrock R, Zannad F, Phillips RA, Roniker B, Kleiman J, Krause S, Burns D, Williams GH. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation. 2003;108(15):1831–1838. doi: 10.1161/01.CIR.0000091405.00772.6E. [DOI] [PubMed] [Google Scholar]
- 18.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18(9):1429–1433. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ Res. 2016;118(6):935–943. doi: 10.1161/CIRCRESAHA.115.308269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, Jaffe IZ. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscler Thromb Vasc Biol. 2014;34(2):355–364. doi: 10.1161/ATVBAHA.113.302854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacolley P, Safar ME, Lucet B, Ledudal K, Labat C, Benetos A. Prevention of aortic and cardiac fibrosis by spironolactone in old normotensive rats. J Am Coll Cardiol. 2001;37(2):662–667. doi: 10.1016/s0735-1097(00)01129-3. [DOI] [PubMed] [Google Scholar]
- 22.Benetos A, Lacolley P, Safar ME. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 1997;17(6):1152–1156. doi: 10.1161/01.atv.17.6.1152. [DOI] [PubMed] [Google Scholar]
- 23.Bernini G, Galetta F, Franzoni F, Bardini M, Taurino C, Bernardini M, Ghiadoni L, Bernini M, Santoro G, Salvetti A. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J Hypertens. 2008;26(12):2399–2405. doi: 10.1097/HJH.0b013e32831286fd. [DOI] [PubMed] [Google Scholar]
- 24.Savoia C, Touyz RM, Amiri F, Schiffrin EL. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension. 2008;51(2):432–439. doi: 10.1161/HYPERTENSIONAHA.107.103267. [DOI] [PubMed] [Google Scholar]
- 25.Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res. 2005;96(6):643–650. doi: 10.1161/01.RES.0000159937.05502.d1. [DOI] [PubMed] [Google Scholar]
- 26.Galmiche G, Pizard A, Gueret A, Moghrabi El S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension. 2014;63(3):520–526. doi: 10.1161/HYPERTENSIONAHA.113.01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krug AW, Allenhöfer L, Monticone R, Spinetti G, Gekle M, Wang M, Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55(6):1476–1483. doi: 10.1161/HYPERTENSIONAHA.109.148783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo J-K, Aronovitz M, Baur WE, Christou DD, Hill MA, Jaffe IZ. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight. 2016;1(14):e88942. doi: 10.1172/jci.insight.88942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paar M, Pavenstädt H, Kusche-Vihrog K, Drüppel V, Oberleithner H, Kliche K. Endothelial sodium channels trigger endothelial salt sensitivity with aging. Hypertension. 2014;64(2):391–396. doi: 10.1161/HYPERTENSIONAHA.114.03348. [DOI] [PubMed] [Google Scholar]
- 30.Hemmeryckx B, Hoylaerts MF, Deloose E, et al. Age-associated pro-inflammatory adaptations of the mouse thoracic aorta. Thromb Haemost. 2013;110(4):785–794. doi: 10.1160/TH13-01-0022. [DOI] [PubMed] [Google Scholar]
- 31.Hwang M-H, Yoo J-K, Luttrell M, Kim H-K, Meade TH, English M, Nichols WW, Christou DD. Role of mineralocorticoid receptors in arterial stiffness in human aging. Exp Gerontol. 2013;48(8):701–704. doi: 10.1016/j.exger.2013.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Usher MG, Duan S-Z, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120(9):3350–3364. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kusche-Vihrog K, Sobczak K, Bangel N, Wilhelmi M, Nechyporuk-Zloy V, Schwab A, Schillers H, Oberleithner H. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Archiv. 2008;455(5):849–857. doi: 10.1007/s00424-007-0341-0. [DOI] [PubMed] [Google Scholar]
- 34.Drüppel V, Kusche-Vihrog K, Grossmann C, Gekle M, Kasprzak B, Brand E, Pavenstadt H, Oberleithner H, Kliche K. Long-term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J. 2013;27(9):3652–3659. doi: 10.1096/fj.13-228312. [DOI] [PubMed] [Google Scholar]
- 35.Nagase M, Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol. 2013;9(2):86–98. doi: 10.1038/nrneph.2012.282. [DOI] [PubMed] [Google Scholar]
- 36.Kawarazaki W, Nagase M, Yoshida S, Takeuchi M, Ishizawa K, Ayuzawa N, Ueda K, Fujita T. Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J Am Soc Nephrol. 2012;23(6):997–1007. doi: 10.1681/ASN.2011070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai D-F, Chen T, Johnson SC, Szeto H, Rabinovitch PS. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012;16(12):1492–1526. doi: 10.1089/ars.2011.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB, Reinhart-King CA. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med. 2011;3(112):112ra122. doi: 10.1126/scitranslmed.3002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGraw AP, Bagley J, Chen W-S, Galayda C, Nickerson H, Armani A, Caprio M, Carmeliet P, Jaffe IZ. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J Am Heart Assoc. 2013;2(1):e000018–e000018. doi: 10.1161/JAHA.112.000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young MJ. Mechanisms of mineralocorticoid receptor-mediated cardiac fibrosis and vascular inflammation. Curr Opin Nephrol Hypertens. 2008;17(2):174–180. doi: 10.1097/MNH.0b013e3282f56854. [DOI] [PubMed] [Google Scholar]
- 41.Salvador AM, Moss ME, Aronovitz M, Mueller KB, Blanton RM, Jaffe IZ, Alcaide P. Endothelial mineralocorticoid receptor contributes to systolic dysfunction induced by pressure overload without modulating cardiac hypertrophy or inflammation. Physiol Rep. 2017;5(12):e13313. doi: 10.14814/phy2.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rocha R, Martin-Berger CL, Yang P, Scherrer R, Delyani J, McMahon E. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart. Endocrinology. 2002;143(12):4828–4836. doi: 10.1210/en.2002-220120. [DOI] [PubMed] [Google Scholar]
- 43.Gueret A, Harouki N, Favre J, Galmiche G, Nicol L, Henry J-P, Besnier M, Thuillez C, Richard V, Kolkhof P, Mulder P, Jaisser F, Ouvrard-Pascaud A. Vascular Smooth Muscle Mineralocorticoid Receptor Contributes to Coronary and Left Ventricular Dysfunction After Myocardial Infarction. Hypertension. 2016;67(4):717–723. doi: 10.1161/HYPERTENSIONAHA.115.06709. [DOI] [PubMed] [Google Scholar]
- 44.Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346(11):793–801. doi: 10.1056/NEJMoa011858. [DOI] [PubMed] [Google Scholar]
- 45.Bassett DR, Howley ET. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med Sci Sports Exer. 2000;32(1):70–84. doi: 10.1097/00005768-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Armani A, Marzolla V, Fabbri A, Caprio M. Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J Mol Endocrinol. 2015;55(2):R1–R10. doi: 10.1530/JME-15-0122. [DOI] [PubMed] [Google Scholar]
- 47.Gao YZ, Saphirstein RJ, Yamin R, Suki B, Morgan KG. Aging impairs smooth muscle-mediated regulation of aortic stiffness: a defect in shock absorption function? Am J Physiol Heart Cir Physiol. 2014;307(8):H1252–H1261. doi: 10.1152/ajpheart.00392.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rammos C, Hendgen-Cotta UB, Deenen R, Pohl J, Stock P, Hinzmann C, Kelm M, Rassaf T. Age-related vascular gene expression profiling in mice. Mech Ageing Dev. 2014;135:15–23. doi: 10.1016/j.mad.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Marzolla V, Armani A, Mammi C, Moss ME, Pagliarini V, Pontecorvo L, Antelmi A, Fabbri A, Rosano G, Jaffe IZ, Caprio M. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int J Cardiol. 2017;232:233–242. doi: 10.1016/j.ijcard.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiebeler A, Schmidt F, Müller DN, Park J-K, Dechend R, Bieringer M, Shagdarsuren E, Breu V, Haller H, Luft FC. Mineralocorticoid Receptor Affects AP-1 and Nuclear Factor-κB Activation in Angiotensin II–Induced Cardiac Injury. Hypertension. 2001;37(2):787–793. doi: 10.1161/01.HYP.37.2.787. [DOI] [PubMed] [Google Scholar]
- 51.Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, Huang PL, Mendelsohn ME, Jaffe IZ. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol. 2011;31(8):1871–1880. doi: 10.1161/ATVBAHA.111.229070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.López B, González A, Díez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation. 2010;121(14):1645–1654. doi: 10.1161/CIRCULATIONAHA.109.912774. [DOI] [PubMed] [Google Scholar]
- 53.Kato S, Endo I, Fujimura M, Kuriwaka-Kido R, Fujinaka Y, Aihara K-I, Iwase T, Inoue D, Akaike M, Abe M, Matsumoto T. Serum carboxy-terminal telopeptide of type I collagen (ICTP) as a surrogate marker for vulnerable plaques in atherosclerotic patients: A pilot study. Atherosclerosis. 2013;229(1):182–185. doi: 10.1016/j.atherosclerosis.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 54.McNulty M, Mahmud A, Spiers P, Feely J. Collagen type-I degradation is related to arterial stiffness in hypertensive and normotensive subjects. J Hum Hypertens. 2006;20(11):867–873. doi: 10.1038/sj.jhh.1002015. [DOI] [PubMed] [Google Scholar]
- 55.Segal SS. Regulation of blood flow in the microcirculation. Microcirculation. 2005;12(1):33–45. doi: 10.1080/10739680590895028. [DOI] [PubMed] [Google Scholar]
- 56.Jaffe IZ, Tintut Y, Newfell BG, Demer LL, Mendelsohn ME. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler Thromb Vasc Biol. 2007;27(4):799–805. doi: 10.1161/01.ATV.0000258414.59393.89. [DOI] [PubMed] [Google Scholar]
- 57.Dai D-F, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110(8):1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santos dos RL, da Silva FB, Ribeiro RF, Stefanon I. Sex hormones in the cardiovascular system. Horm Mol Biol Clin Investig. 2014;18(2):89–103. doi: 10.1515/hmbci-2013-0048. [DOI] [PubMed] [Google Scholar]
- 59.DeMarco VG, Habibi J, Jia G, Aroor AR, Ramirez-Perez FI, Martinez-Lemus LA, Bender SB, Garro M, Hayden MR, Sun Z, Meininger GA. Low-Dose Mineralocorticoid Receptor Blockade Prevents Western Diet-Induced Arterial Stiffening in Female Mice. Hypertension. 2015;66(1):99–107. doi: 10.1161/HYPERTENSIONAHA.115.05674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.