

Abstract
Trimethoprim (TMP)-sulfamethoxazole (SMX) is a widely used synergistic antimicrobial combination to treat a variety of bacterial and certain fungal infections. These drugs act by targeting sequential steps in the biosynthetic pathway for tetrahydrofolate (THF), where SMX inhibits production of the THF precursor dihydropteroate, and TMP inhibits conversion of dihydrofolate (DHF) to THF. Consequently, SMX potentiates TMP by limiting de novo DHF production and this mono-potentiation mechanism is the current explanation for their synergistic action. Here, we demonstrate that this model is insufficient to explain the potent synergy of TMP-SMX. Using genetic and biochemical approaches, we characterize a metabolic feedback loop in which THF is critical for production of the folate precursor dihydropterin pyrophosphate (DHPPP). We reveal that TMP potentiates SMX activity through inhibition of DHPPP synthesis. Our study demonstrates that the TMP-SMX synergy is driven by mutual potentiation of the action of each drug on the other.
The antibiotics trimethoprim (TMP) and sulfamethoxazole (SMX) synergistically inhibit bacterial tetrahydrofolate biosynthesis, apparently because SMX potentiates TMP activity. Here, Minato et al. identify a metabolic feedback loop in this pathway, revealing that TMP also potentiates SMX activity.
Introduction
Some antimicrobial drug combinations show strongly synergistic effects, where the combined inhibitory activity is far greater than the sum of individual activities1. However, in most cases, it is not clear why combinations may act synergistically. A mixture of trimethoprim (TMP) and sulfamethoxazole (SMX), also known as Co-trimoxazole, is a widely used synergistic antimicrobial combination to treat a variety of bacterial infections2. TMP-SMX is also effective against certain fungal infections and is the major treatment choice for pneumocystis pneumonia, which is one of the most common opportunistic infections in people with HIV-AIDS3. In bacteria, SMX inhibits dihydropteroate (DHPte) production from the two folate precursors, p-aminobenzoic acid (PABA) and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (DHPPP) (Fig. 1a)4. TMP inhibits the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF)5. Although no conclusive evidence has been provided, it is generally accepted that the potent synergy between these drugs derives simply from the sequential inhibition of adjacent steps in microbial THF biosynthesis6. Mathematical modeling of this pathway suggests that this model is sufficient to explain how SMX action results in potentiation of TMP action, yet is inadequate to explain how TMP action potentiates SMX action7.
Fig. 1.
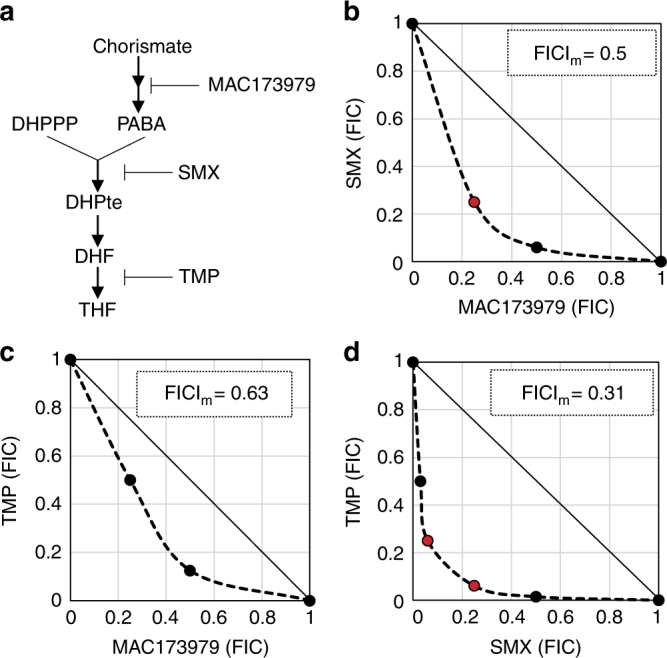
Synergistic activity of anti-folate combinations against E. coli and S. aureus. a Targets of anti-folate compounds. b–d E. coli BW25113 strain was grown overnight in LB medium. Cultures were washed twice and resuspended in M9-glucose, then inoculated into a 96-well round-bottom plate (Corning) containing the same medium with a range of concentrations of SMX, TMP, MAC173979, or combination of two compounds. Concentration ranges were as follows: SMX (0.024–25 µg ml−1), TMP (0.0078–1 µg ml−1), and MAC173979 (0.05–25 µg ml−1). MICs were determined by visible growth after 24 h incubation at 37 °C. Synergy was assessed by calculating FICI. FICIm, minimum value of FICI in the tested combinations is shown. Synergy (FICIm ≤ 0.5). No interaction (FICIm > 0.5). b–d Graphical representations of E. coli BW25113 checkerboard assays are shown. Representative data from at least three independent experiments are shown. b SMX and MAC173979. c TMP and MAC173979. d SMX and TMP
In this study, we take a systematic genetic approach, using Escherichia coli single-gene deletion mutants8, and discover that inhibition of DHPPP biosynthesis increases SMX activity. We also identify a functional metabolic feedback loop in the folate biosynthesis pathway by which TMP can also limit DHPPP biosynthesis. Collectively, our study indicates that TMP also potentiates SMX activity, and that the strong synergy between SMX and TMP is mediated by mutual potentiation. Our findings reveal a novel mechanism of drug synergism underlying the therapeutic efficacy of a widely established combination antimicrobial treatment and suggest that other metabolic pathways with functional feedback loops might be similarly susceptible to synergistic inhibitors.
Results
Inhibition of sequential steps in THF synthesis is not always synergistic
To test whether inhibition of other sequential steps in the THF pathway can produce synergistic activity we targeted synthesis of PABA, an essential precursor for DHPte, with the antimicrobial compound 3,3-dichloro-1-(3-nitrophenyl) prop-2-en-1-one (MAC173979), which has been shown to inhibit PABA synthesis in E. coli9 (Fig. 1a). As this biosynthetic module is located upstream of the SMX target, MAC173979 is expected to be synergistic with both SMX and TMP. We performed checkerboard assays to determine fractional inhibitory concentration indexes (FICIs) as a measure of interaction between antimicrobial agents10. We found that the combination of MAC173979 and SMX was only mildly synergistic (Fig. 1b and Supplementary Fig. 1a) and the combination of MAC173979 and TMP was merely additive and not synergistic (Fig. 1c and Supplementary Fig. 1b), while consistent with there being an unexplained aspect of synergy between TMP and SMX (Fig. 1d and Supplementary Fig. 1c). Further, although MAC173979 could potentiate susceptibility to SMX and, to a lesser degree, TMP, SMX and TMP did not potentiate susceptibility to MAC173979 (Fig. 1b,c). Thus, the interaction of the PABA biosynthesis inhibitor with both SMX and TMP is one of mono-potentiation. These FICI profiles show a stark contrast with the strong mutual potentiation observed between SMX and TMP (Fig. 1d and Supplementary Fig. 1c). Similar FICI profiles were observed for a clinical isolate of E. coli (strain B11)11 and methicillin-resistant Staphylococcus aureus (strain USA300)12, demonstrating that MAC173979 and TMP were not synergistic, whereas SMX and TMP were synergistic against these strains (Table 1). These results confirmed that a model based on sequential inhibition within the bacterial THF biosynthesis pathway is not adequate to explain the potent synergy between SMX and TMP.
Table 1.
MICs of SMX, TMP, and MAC173979 and FICIm of SMX-TMP and MAC173979-TMP
| Bacterial strains | MIC (µg ml–1) | FICIm | |||
|---|---|---|---|---|---|
| SMX | TMP | MAC173979 | SMX-TMP | MAC173979-TMP | |
| E. coli BW25113 | 1.6 | 0.60 | 1.6 | 0.31 | 0.63 |
| E. coli B11 | 3.1 | 1.0 | 0.80 | 0.31 | 1.0–2.0 |
| S. aureus USA300 | 0.80 | 0.50 | 6.3 | 0.16 | 1.0 |
FICIm, minimum fractional inhibitory combination of antimicrobial agent pairs found to achieve growth inhibition; MIC, minimum concentration of antimicrobial agent required to inhibit at least 50% of growth relative to a no drug control after 24 h of incubation at 37 °C; SMX, sulfamethoxazole; TMP, trimethoprim
Folate precursors affect susceptibility to SMX and TMP
To further evaluate synergy through targeting of sequential steps in THF synthesis, we assessed the impact of genetic impairment of steps upstream of DHPte synthesis on potency of SMX and TMP (Fig. 2a). It was previously suggested13 and recently demonstrated that SMX acts by competing with PABA for ligation with DHPPP14. As a result, SMX forms dead-end complexes with DHPPP (dihydropterin-SMX)15 and inhibits DHPte production through metabolic wasting13, 16. Based on this model of metabolic wasting, we expected SMX activity would be influenced by the intracellular abundance of both PABA and DHPPP (Fig. 2a). We previously found that genetic disruption of the PABA biosynthesis pathway potentiates SMX activity against Mycobacterium tuberculosis17. Similar to M. tuberculosis, an E. coli mutant strain deleted for pabC, encoding the aminodeoxychorismate lyase involved in PABA biosynthesis, showed 4-fold enhanced susceptibility to SMX (Fig. 2b). In contrast, the E. coli pabC deletion mutant strain was only twofold more susceptible to TMP (Fig. 2c). This observation is consistent with the finding that the combination of MAC173979 and SMX is mildly synergistic (Fig. 1b and Supplementary Fig. 1a), whereas the combination of MAC173979 and TMP is merely additive (Fig. 1c and Supplementary Fig. 1b). To determine the impact of DHPPP levels on SMX and TMP susceptibility, we utilized an E. coli mutant strain deleted for nudB, which encodes dihydroneopterin triphosphate pyrophosphohydrolase18. Interestingly, this strain showed over 300-fold enhanced susceptibility to SMX and 60-fold enhanced susceptibility to TMP (Fig. 2b, c), demonstrating that DHPPP levels are far more impactful than PABA levels on SMX and TMP susceptibility. Of note, the ΔnudB strain showed the same susceptibility to ceftazidime and ciprofloxacin as the parent strain, indicating that the enhanced drug susceptibility phenotype of the ΔnudB strain was specific to anti-folate drugs (Supplementary Table 1).
Fig. 2.

Folate deficiency potentiates SMX activity. a Schematic of E. coli folate metabolism. Green characters indicate metabolites and enzymes that affect SMX susceptibility. Blue characters indicate enzymes that affect both SMX and TMP susceptibility. b MICs of SMX and TMP for E. coli folate pathway mutants were determined after 24 h of incubation at 37 °C in M9-glucose medium. The selected E. coli single-gene deletion mutants and their parental strain BW25113 (WT) from the Keio collection8 were used. c Effects of inosine (Ino) and methionine (Met) on SMX MIC against E. coli BW25113. Ino and Met were added to the medium at 10 µg ml−1. MICs of SMX were determined after 24 h incubation at 37 °C in M9-glucose medium. Representative data from at least three independent experiments are shown
Folate-dependent metabolites affect susceptibility to SMX but not TMP
As DHPPP is ultimately derived from the folate-dependent purine nucleotide guanosine-5’-triphosphate (GTP), we were curious to explore whether alterations in folate inter-conversion could impact on the susceptibility of E. coli to TMP and SMX. The genes glyA and gcvPHT code for serine hydroxymethyltransferase and the glycine cleavage system, respectively. These two independent steps yield the 10-formyl-THF precursor 5,10-methylene-THF from THF (Fig. 2a) and were previously associated with susceptibility to anti-folate drugs in a high throughput conditional gene essentiality study19, 20. We observed that the E. coli single-gene deletion mutants in these two steps had enhanced susceptibility to SMX (Fig. 2b) and complementation of individual mutants completely restored SMX susceptibility to the level of the parental strain (Supplementary Table 2). Interestingly, the glyA and gcv deletion mutant strains showed a level of TMP susceptibility that was comparable to that of the parental strain (Fig. 2c). These observations demonstrate that interference with 10-formyl-THF production mediates enhanced susceptibility to SMX and suggests that disruption of folate-dependent metabolism is the likely basis for potentiation of SMX activity by TMP.
We next investigated how disruption of 10-formyl-THF synthesis impacts SMX susceptibility. It has long been known that the combination of two THF-dependent metabolites, inosine and methionine, antagonizes SMX activity by an, as yet, uncharacterized mechanism (Fig. 2b)21. We found that these metabolites also antagonized SMX susceptibility in the E. coli glyA and gcv deletion mutant strains. Since these deletion mutants showed the same level of SMX susceptibility as the parental strain in the presence of inosine and methionine, they are able to overcome the limited availability of 10-formyl-THF by supplementation with these two THF-dependent metabolites. Of note, inosine and methionine had no effect on TMP susceptibility in E. coli (Supplementary Fig. 2). Given that 10-formyl-THF is the essential cofactor for the de novo purine biosynthesis pathway and biosynthesis of the folate precursor DHPPP starts from GTP4, we hypothesized that limitation of 10-formyl-THF ultimately potentiates SMX susceptibility by impairing the DHPPP biosynthesis pathway. Consistent with this hypothesis, we observed that inosine and methionine did not antagonize SMX activity in the ΔnudB strain, indicating that these metabolites interfere with SMX action by increasing metabolic flux towards DHPPP biosynthesis (Fig. 2b). Taken together, our findings suggest the existence of a previously unrecognized functional metabolic connection between downstream and upstream components of the bacterial folate biosynthetic pathway that dramatically impacts on SMX susceptibility.
TMP inhibits DHPPP biosynthesis
As decreased DHPPP biosynthesis enhances SMX susceptibility, it is likely to be that TMP potentiates SMX activity by negatively impacting the DHPPP biosynthesis pathway (Fig. 3a). To test this hypothesis, we developed liquid chromatography-tandem mass spectrometry (LC-MS/MS) analytical methods using authentic standards to quantify two of the DHPPP precursors, 7,8-dihydroneopterin (DHN) and 6-hydroxymethyl-7,8-dihydropterin (DHPt). We verified that when treated with TMP, E. coli BW25113 strain showed significantly lower intracellular levels of both DHN and DHPt, relative to untreated controls (Fig. 3b, c). TMP treatment did not affect intracellular level of PABA, suggesting that the TMP treatment only affected intracellular levels of a specific set of metabolites (Fig. 3d). TMP treatment also negatively affected DHPt levels in E. coli B11 and S. aureus USA300 strains (Fig. 3e,f). Therefore, inhibition of THF biosynthesis by TMP directly impairs DHPPP biosynthesis.
Fig. 3.

TMP inhibits DHPPP biosynthesis pathway. The impact of TMP on intracellular levels of a 7,8-dihydroneopterin (DHN) and b, e, f 6-hydroxymethyl-7,8-dihydropterin (DHPt), c PABA were determined by using LC-MS/MS. The results represent the mean and standard deviation of three biological replicates. **p < 0.05. NS indicates no significant difference (p > 0.05). p-values of pairwise comparisons were calculated by using the Student’s t-test. d Effects of TMP on DHPPP biosynthesis pathway. Red characters are metabolites that decrease intracellular amount in response to TMP treatment (THF23, GTP29, and ATP29 were reported previously)
If impairment of DHPPP synthesis is the mechanistic basis for TMP-mediated potentiation of SMX activity, it follows that TMP-SMX synergy should be abolished in the E. coli ΔnudB strain. To evaluate TMP-SMX synergy we performed growth assays with wild-type E. coli BW25113 and the ΔnudB strain in the presence of each drug at a concentration that by itself led to mild growth impairment (Fig. 4a, b). As anticipated when TMP-SMX were combined, their synergistic action resulted in full growth inhibition of the wild-type strain (Fig. 4a). In striking contrast, the combination of TMP-SMX showed an additive inhibitory effect on growth of the E. coli ΔnudB strain (Fig. 4b). Furthermore, through the use of checkerboard assays we found that TMP-SMX showed additive effects against the ΔnudB strain (Fig. 4c). Thus, synergy between TMP and SMX is contingent upon the ability of TMP to disrupt synthesis of DHPPP (Fig. 4d).
Fig. 4.

A metabolic feedback loop amplifies SMX-TMP activity. a, b SMX-TMP synergy was assessed by growth kinetics in the presence of a sub-inhibitory concentration of each drug and in combination. The results represent the mean and SD of three biological replicates. a Wild type (E. coli BW25113 strain) was grown in the presence of SMX (0.06 µg ml−1), TMP (0.125 µg ml−1), or a combination of SMX (0.06 µg ml−1) and TMP (0.125 µg ml−1). b ΔnudB (E. coli BW25113 ΔnudB) was grown in the presence of SMX (0.003 µg ml−1), TMP (0.0125 µg ml−1), or a combination of SMX (0.003 µg ml−1) and TMP (0.0125 µg ml−1). c SMX-TMP synergy against ΔnudB (E. coli BW25113 ΔnudB) was assessed by FICI. FICIm, minimum value of FICI for the tested combinations is shown. Representative data from at least three independent experiments are shown. d Relative metabolite abundance and metabolic flux are shown. No drug treatment (i), sub-inhibitory concentrations of each drug (ii), and combination of sub-inhibitory concentrations of each drug (iii). SMX treatment inhibits the accumulation of DHF by TMP that results in potentiation of TMP activity, and TMP treatment decreases DHPPP that potentiates SMX activity. Cyclic mutual potentiation results in amplified depletion of the essential cofactor THF
Discussion
Over the past 50 years, the mechanistic basis for the potently synergistic action between TMP and SMX has been explained by an overly simplistic model. Hitchings first proposed that sulfa drugs such as SMX potentiate the action of TMP through inhibition of DHF accumulation, which enhances the interaction of TMP with its target DHF reductase22. Several studies have confirmed key elements of this model23, 24. However, this model does not account for the ability of TMP to enhance microbial susceptibility to SMX. Our findings reveal that TMP potentiates SMX activity through the disruption of a previously unrecognized metabolic feedback loop and the cyclic mutual potentiation of these disruptions results in amplified depletion of the essential cofactor THF (Fig. 4d). These findings highlight the importance of metabolic pathway structure in understanding antimicrobial drug interaction and will enable the identification of additional pathways that can be explored for potently synergistic antimicrobial based targeting.
Methods
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplementary Table 3. Bacterial strains were grown in Lysogeny Broth (LB, Difco) or M9 minimal medium supplemented with 0.2% (vol:vol) glucose (M9-glucose) at 37 °C. 0.5% Casamino acids, pantothenate (f.c. 2 µg ml−1), nicotinamide (f.c. 2 µg ml−1), thiamin (f.c. 2 µg ml−1), and biotin (f.c. 100 ng ml−1) were supplemented to M9-glucose for growing S. aureus USA300 strain. When required, media were amended with penicillin G (Sigma-Aldrich) 150 µg ml−1 or kanamycin (Teknova) 50 µg ml−1.
Strains construction
For the complementation analyses, we cloned the nudB, gcvP, gcvH, and gcvT genes as follows. The DNA fragment, which contained the respective open reading frame, was amplified by PCR using chromosomal DNA of E. coli BW25113 as a template. Primers used for these constructs included SacI and BamHI restriction sites, respectively, and are listed in Supplementary Table 4. The obtained PCR products were digested with SacI and BamHI, gel-purified and then ligated into SacI–BamHI digested pUC19. The resultant plasmid constructs were verified by DNA sequencing.
Antimicrobial agent susceptibility testing
SMX and TMP were purchased from Sigma-Aldrich. MAC173979 was synthesized as previously described17. The minimum inhibitory concentrations (MICs) of SMX, TMP, and MAC173979 were determined by using the microdilution method.
Twofold dilution series of each antimicrobial agent in M9-glucose was prepared in 96-well round-bottom plates (Corning). Bacterial strains were grown overnight in LB medium, sub-cultured into fresh LB medium, and grown to mid-log phase. The cultures were washed twice and resuspended in M9-glucose, then inoculated into each well containing M9-glucose to OD600 0.001. MICs were determined by visible growth after 24 h incubation at 37 °C. For Supplementary Figure 1, the same drug susceptibility testing was performed in 96 well flat bottom plate (Corning) and MICs were read spectrophotometrically (OD600) to determine the minimum amount of antimicrobial agent required to inhibit at least 50% of growth relative to a no drug control after 24 h incubation at 37 °C. Interaction between antimicrobial agents was assessed by using a checkerboard assay format to determine FICI of each agent in the presence of sub-inhibitory concentrations of another. FICI of antimicrobial agent combinations were calculated using the following equation, FICI = ([MIC drug A in presence of Drug B] [MIC of drug A]−1) + ([MIC of drug B in the presence of drug A] [MIC of drug B]−1). Minimum FICI (FICIm) represents the lowest fractional combination of antimicrobial agent pairs to achieve growth inhibition. FICIm ≤ 0.5 is regarded as synergistic.
Growth assays
Bacterial strains were grown overnight in LB medium. The cultures were washed twice and resuspended in M9-glucose then inoculated into each well of a 96-well flat-bottom plate (Corning) containing M9-glucose containing different concentrations of SMX, TMP, or combination of SMX and TMP. Growth was monitored in a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek) with the following settings (temperature 37 °C, continuous orbital shaking at 282 cpm (3 mm), read every hour at OD600).
Synthesis of DHPt
Synthesis of DHPt was accomplished as shown in Supplementary Fig. 3. The synthesis started with the degradation of commercially available folic acid. A solution of folic acid in 40% aqueous hydrogen bromide and excess bromine was heated using a previously described method to obtain 6-formylpterin in 52% yield25. Sodium borohydride-mediated reduction of 6-formylpterin in 0.1 N NaOH solution provided 6-hydroxymethylpterin in 95% yield. A well-established method for the selective reduction of 7,8 double bond of folate species was employed for the conversion of 6-hydroxymethylpterin to DHPt. This transformation was accomplished by using sodium dithionite as a reducing agent in the presence of ascorbic acid at pH 6.526. Synthesized DHPt was fully characterized using 1H, 13C NMR and high-resolution mass spectrometry.
Liquid chromatography-tandem mass spectrometry
Sample preparation
bacterial strains were grown until early log growth phase (OD600 0.2–0.3). The cells were then diluted to OD600 0.2 and were treated with 4 µg ml−1 TMP. After 30 min, and 60 min with or without TMP treatment, 40 ml of each culture was collected by centrifugation. Metabolites were extracted by using 500 µl of acetonitrile:methanol:water (40:40:20) extraction buffer as previously described27. One hundred and fifty microliters of each sample was evaporated using a SpeedVac Concentrator (Savant SC210A, Thermo Scientific) and reconstituted with 50 µL of 10 mM ammonium acetate (95:5 H2O:MeCN, pH 9.0) solution containing 200 nM internal standard (hydroxypteroic acid) for DHPt and DHN quantification. Similarly, for PABA quantification, 150 µL of each sample was evaporated and reconstituted with 50 µL of 10 mM aqueous ammonium acetate (pH 4.0) solution containing 100 nM internal standard (d4-PABA).
DHPt and DHN quantification
LC-MS/MS experiments were performed with Shimadzu UFLC-XR (LC) and AB Sciex QTRAP 5500 (MS) instruments. Synthetic DHPt and DHN (Santa Cruz Biotechnology) were used as authentic standards. Hydroxypteroic acid was used as an internal standard. Reverse-phase LC was performed on an Eclipse XDB-C8 column (4.6 × 150 mm, 5 µm particle size; Agilent, Santa Clara, CA). Mobile phase A was 10 mM ammonium acetate in H2O (pH 9.0), whereas mobile phase B was 10 mM ammonium acetate in 5:95 H2O:MeCN (pH 9.0). Initial conditions were 5% B from 0 to 1.0 min, after which the %B was increased to 70% from 1.0 to 5.3 min, then %B was increased to 90% from 5.3 to 5.7 min. The column was washed with 90% B from 5.7 to 6.5 min, returned to 5% B at 7.0 min, and allowed to re-equilibrate for 4 min in 5% B, to provide a total run time of 11 min. The flow rate was 0.5 mL min−1 and the column oven was maintained at 35 °C. The injection volume was 10 µL. All analytes were analyzed by MS in positive ionization mode by Multiple Reaction Monitoring (MRM). To determine the optimum MRM settings (Supplementary Table 3), each analyte was infused at a concentration of 10 µM in 50:50 H2O:MeCN containing 10 mM ammonium acetate at pH 9.0 onto the MS by a syringe pump at a flow of 10 µL min−1. During direct infusion, we also observed the presence of oxidized forms of both the analytes, i.e., the presence of neopterin and 6-hydroxymethylpterin. Such auto air oxidation of reduced form of pterin species has been known; thus, we decided to measure both the oxidized and reduced forms of these species (Supplementary Table 3). Peak areas of both species were combined for the analytes before converting to the concentration. Analyte and internal standard peak areas were calculated (MultiQuant, version 2.0.2). Analyte peak areas were normalized to internal standard peak areas and the concentrations of DHN and DHPt were determined using appropriate standard curves.
PABA quantification
PABA was quantified using a previously developed LC-MS/MS method28. d4-PABA was used as an internal standard. Reverse-phase LC was performed on a Kinetix C18 column (50 × 2.1 mm, 2.6 μm particle size; Phenomenex, Torrance, CA). Mobile phase A was 10 mM ammonium acetate in H2O (pH 4.0), whereas mobile phase B was 0.1% formic acid in 5:95 H2O:MeOH. Initial conditions were 5% B, after which the proportion of B was increased to 10% in 1.0 min, 15% in 4.0 min, 55% in 4.5 min, 92% in 4.75 min, returned to initial composition of eluent (5% B) in 5.0 min, and then held for 3.0 min in order to re-equilibrate the column, which provided a total run time of 8 min. The flow rate was 0.5 mL min−1 and the column oven was maintained at 35 °C. The injection volume was 10 µL. Both analyte and internal standard were analyzed by electrospray ionization MS in positive ionization mode by MRM. The transformations m/z 138.05 → 94.07 for PABA and 142.08 → 98.09 for d4-PABA were used for MRM (Supplementary Table 5).
Data availability
All relevant data are available in this article and its Supplementary Information files, or from the corresponding authors upon request.
Electronic supplementary material
Acknowledgements
We thank Peter Southern for critical reading of the manuscript and helpful comments, Bruce Witthuhn for expert technical assistance with LC-MS/MS analysis, Arkady B. Khodursky for providing us the E. coli single-gene deletion mutants, Ryan C. Hunter for providing S. aureus USA300, and Betsy Hirsch for providing E. coli B11. We also thank Allison Bauman for her excellent technical assistance. This work was supported by a grant from the University of Minnesota Academic Health Center Faculty Research Development Program to A.D.B. and C.C.A., and by startup funds from the University of Minnesota to A.D.B. and C.C.A.
Author contribution
Y.M., A.B., S.L.K., and S.D. performed experiments. S.D. synthesized custom reagents. Y.M., C.C.A., and A.D.B. conceived the work. Y.M., S.D., and A.D.B. wrote the manuscript. All authors contributed to analyzing data and editing of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Surendra Dawadi, Shannon L. Kordus and Abiram Sivanandam.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-03447-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yusuke Minato, Email: yminato@umn.edu.
Anthony D. Baughn, Email: abaughn@umn.edu
References
- 1.Yeh P, Tschumi AI, Kishony R. Functional classification of drugs by properties of their pairwise interactions. Nat. Genet. 2006;38:489–494. doi: 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- 2.Smilack JD. Trimethoprim-sulfamethoxazole. Mayo Clin. Proc. 1999;74:730–734. doi: 10.4065/74.7.730. [DOI] [PubMed] [Google Scholar]
- 3.Morris A, et al. Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 2004;10:1713–1720. doi: 10.3201/eid1010.030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green, J. M. & Matthews, R. G. Folate biosynthesis, reduction, and polyglutamylation and the interconversion of folate derivatives. EcoSal Plus2,doi:10.1128/ecosalplus.3.6.3.6 (2007). [DOI] [PubMed]
- 5.Poe M, Breeze AS, Wu JK, Short CR, Hoogsteen K. Dihydrofolate reductase from trimethoprim-resistant Escherichia coli MB 3746 and MB 3747. Purification, amino acid composition, and some kinetic properties. J. Biol. Chem. 1979;254:1799–1805. [PubMed] [Google Scholar]
- 6.Bushby SR. Synergy of trimethoprim-sulfamethoxazole. Can. Med Assoc. J. 1975;112:63–66. [PMC free article] [PubMed] [Google Scholar]
- 7.Harvey RJ. Interaction of two inhibitors which act on different enzymes of a metabolic pathway. J. Theor. Biol. 1978;74:411–437. doi: 10.1016/0022-5193(78)90223-0. [DOI] [PubMed] [Google Scholar]
- 8.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zlitni S, Ferruccio LF, Brown ED. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat. Chem. Biol. 2013;9:796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odds FC. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003;52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch EB, et al. Activity of fosfomycin and comparison of several susceptibility testing methods against contemporary urine isolates. Int J. Antimicrob. Agents. 2015;46:642–647. doi: 10.1016/j.ijantimicag.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy AD, et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc. Natl Acad. Sci. USA. 2008;105:1327–1332. doi: 10.1073/pnas.0710217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown GM. The biosynthesis of folic acid. II. Inhibition by sulfonamides. J. Biol. Chem. 1962;237:536–540. [PubMed] [Google Scholar]
- 14.Yun MK, et al. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science. 2012;335:1110–1114. doi: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bock L, Miller GH, Schaper KJ, Seydel JK. Sulfonamide structure-activity relationships in a cell-free system. 2. Proof for the formation of a sulfonamide-containing folate analog. J. Med Chem. 1974;17:23–28. doi: 10.1021/jm00247a006. [DOI] [PubMed] [Google Scholar]
- 16.Palmer AC, Kishony R. Opposing effects of target overexpression reveal drug mechanisms. Nat. Commun. 2014;5:4296. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiede JM, et al. Targeting intracellular p-aminobenzoic acid production potentiates the anti-tubercular action of antifolates. Sci. Rep. 2016;6:38083. doi: 10.1038/srep38083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabelli SB, et al. Structure and function of the E. coli dihydroneopterin triphosphate pyrophosphatase: a Nudix enzyme involved in folate biosynthesis. Structure. 2007;15:1014–1022. doi: 10.1016/j.str.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Ogwang S, et al. Bacterial conversion of folinic acid is required for antifolate resistance. J. Biol. Chem. 2011;286:15377–15390. doi: 10.1074/jbc.M111.231076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nichols RJ, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry RJ. The mode of action of sulfonamides. Bacteriol. Rev. 1943;7:175–262. doi: 10.1128/br.7.4.175-262.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hitchings GH. Folate antagonists as antibacterial and antiprotozoal agents. Ann. N. Y. Acad. Sci. 1971;186:444–451. doi: 10.1111/j.1749-6632.1971.tb31171.x. [DOI] [PubMed] [Google Scholar]
- 23.Quinlivan EP, McPartlin J, Weir DG, Scott J. Mechanism of the antimicrobial drug trimethoprim revisited. FASEB J. 2000;14:2519–2524. doi: 10.1096/fj.99-1037com. [DOI] [PubMed] [Google Scholar]
- 24.Kwon YK, et al. A domino effect in antifolate drug action in Escherichia coli. Nat. Chem. Biol. 2008;4:602–608. doi: 10.1038/nchembio.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thijssen HH. A simple method for preparing 2-amino-4-hydroxy-6-formylpteridine, a precursor of the pteridine substrate of dihydropteroate biosynthesis. Anal. Biochem. 1973;54:609–611. doi: 10.1016/0003-2697(73)90394-1. [DOI] [PubMed] [Google Scholar]
- 26.Furrer H-J, Bieri JH, Viscontini M. Conformational analysis of 5,6,7,8-tetrahydropteroic acid and 5,6,7,8-tetrahydro-L-folic acid. Helvetica Chimica Acta. 1978;61:2744–2751. doi: 10.1002/hlca.19780610743. [DOI] [Google Scholar]
- 27.Rabinowitz JD, Kimball E. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 2007;79:6167–6173. doi: 10.1021/ac070470c. [DOI] [PubMed] [Google Scholar]
- 28.Dhananjeyan MR, et al. Simultaneous determination of procaine and para-aminobenzoic acid by LC-MS/MS method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007;847:224–230. doi: 10.1016/j.jchromb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Kwon YK, Higgins MB, Rabinowitz JD. Antifolate-induced depletion of intracellular glycine and purines inhibits thymineless death in E. coli. ACS Chem. Biol. 2010;5:787–795. doi: 10.1021/cb100096f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available in this article and its Supplementary Information files, or from the corresponding authors upon request.