

Abstract
Background and Purpose
Adiponectin potently suppresses inflammatory mediator production. Autophagy is known to play a critical role in the modulation of inflammatory responses by adiponectin. However, the underlying mechanisms are not clearly understood. Interaction between Beclin‐1 and B‐cell lymphoma 2 (Bcl‐2) is a critical event in autophagy induction. We examined the effects of globular adiponectin (gAcrp) on the Beclin‐1/Bcl‐2 association and its underlying mechanisms.
Experimental Approach
The effect of gAcrp on the interaction between Beclin‐1 and Bcl‐2 was examined by immunoprecipitation followed by Western blotting. To elucidate the underlying mechanisms, we determined the effects of gAcrp on Beclin‐1 phosphorylation and Bcl‐2 mRNA stability, and investigated their role in the suppression of inflammatory mediators using pharmacological inhibitors and transient target gene knockdown.
Key Results
Globular adiponectin disrupted the association between Beclin‐1 and Bcl‐2 and increased Beclin‐1 phosphorylation at Thr119, critical residue for binding with Bcl‐2, via a death‐associated protein kinase‐1 (DAPK1)‐dependent mechanism. Moreover, gAcrp reduced Bcl‐2 expression via Bcl‐2 mRNA destabilization, without significantly affecting Bcl‐2 promoter activity and protein degradation, which was mediated by tristetraprolin (TTP) induction. Finally, DAPK1 and TTP were shown to play key roles in gAcrp‐induced autophagosome formation and suppression of LPS‐stimulated TNF‐α and IL‐1β expression.
Conclusion and Implications
Beclin‐1 phosphorylation and Bcl‐2 mRNA destabilization mediated by DAPK1 and TTP are crucial events leading to autophagy and the suppression of inflammatory cytokine production by gAcrp. These results provide novel mechanisms underlying adiponectin's modulation of inflammatory responses. DAPK and TTP are potential therapeutic targets for the management of inflammation.
Abbreviations
- 3′‐UTR
3′‐untranslated region
- Adipo receptors
adiponectin receptors
- AMPK
AMP‐activated protein kinase
- ARE
A (adenylate) + U (uridylate)‐rich elements
- ATG5
autophagy protein 5
- Bcl‐2
B‐cell lymphoma 2
- BH3
Bcl‐2 homology domain 3
- DAPK1
death‐associated protein kinase‐1
- EBSS
Earle's Balanced Salt Solution
- FoxO3A
forkhead box O3
- gAcrp
globular adiponectin
- NLRP3
NACHT domain‐, leucine‐rich repeat domain‐ and pyrin domain‐containing protein 3
- PI3P
phosphatidylinositol 3‐phosphate
- PP1/PP2A
protein phosphatase
- TTP
tristetraprolin
- VSP34
vacuolar sorting protein 34
Introduction
Adipokines, a group of cytokines produced by adipose tissue, are implicated in diverse physiological responses (Fasshauer and Bluher, 2015). Adiponectin, the most abundant adipokine, exerts a variety of biological functions, including insulin sensitization, fatty acid oxidation and glucose uptake (Bruce et al., 2005; Deepa and Dong, 2009) and plays a role in the maintenance of tissue homeostasis (Brochu‐Gaudreau et al., 2010). It has been well documented that adiponectin plays a beneficial role in the regulation of metabolic conditions (Kadowaki et al., 2006). In addition to its critical roles in metabolic diseases, adiponectin exhibits potent anti‐inflammatory properties via multiple mechanisms. For example, adiponectin suppresses the activity of pro‐inflammatory transcription factors, including NF‐κB (Thakur et al., 2006), and induces the expression of anti‐inflammatory genes, such as haem oxygenase‐1 (HO‐1) and IL‐10 (Mandal et al., 2010; Park et al., 2007). Moreover, recent studies have demonstrated that the induction of autophagy plays a critical role in the suppression of inflammatory mediators by adiponectin (Kim et al., 2017; Pun et al., 2015).
Autophagy, a highly conserved self‐digestive cellular process, removes dysfunctional intracellular components and misfolded proteins. This process involves the sequestration of these components into double‐membrane vesicles called autophagosomes followed by fusion with lysosomes (Parzych and Klionsky, 2014). Autophagy plays a crucial role in the maintenance of cellular homeostasis. For example, the breakdown of target proteins and intracellular organelles provides a free amino acid pool and contributes to new protein synthesis during starvation (Onodera and Ohsumi, 2005). The dysregulation of autophagy is closely associated with the development of various pathological conditions, including cancer and neurodegenerative disorders (Levine and Kroemer, 2008). Autophagy is a complicated biological process involving multiple steps, each of which is coordinated by specific genes, such as Beclin‐1, autophagy protein 5 (ATG5) and microtubule‐associated proteins light chain 3B (LC3). Of the proteins involved in autophagy induction, Beclin‐1 induces activation of vacuolar sorting protein 34 (VSP34), a class III PI3K‐III (McKnight and Zhenyu, 2013; Yue and Zhong, 2010), which catalyses the phosphorylation of phosphatidylinositol or phosphoinositides to produce phosphatidylinositol 3‐phosphate (PI3P). PI3P then elongates phagophores during autophagy (Simonsen and Tooze, 2009) and provides the platform for the recruitment of other ATGs for phagophore biogenesis and autophagosome maturation (Polson et al., 2010; Toton et al., 2014; Wirawan et al., 2012). Therefore, Beclin‐1 plays a key role in initiating autophagy.
The biological activity of Beclin‐1 in autophagy induction is regulated by binding with B‐cell lymphoma 2 (Bcl‐2). Interaction with Bcl‐2 through its Bcl‐2 homology domain 3 (BH3) inhibits the binding of Beclin‐1 with class III PI3K and prevents initiation of the autophagic process. Therefore, the modulation of the interaction between Beclin‐1 and Bcl‐2 is considered a critical step in the initiation of autophagy. The binding activity of Beclin‐1 with Bcl‐2 is determined by post‐translational modifications of Beclin‐1. In particular, the phosphorylation of Beclin‐1 causes the dissociation of Bcl‐2 (Zalckvar et al., 2009), which in turn leads to the formation of a Beclin‐1–class III PI3K complex. Recently, death‐associated protein kinase‐1 (DAPK1), a member of the death‐related kinase family, has been shown to phosphorylate Beclin‐1 (Zalckvar et al., 2009). DAPK1 was originally reported to be involved in apoptotic cell death (Gozuacik et al., 2008). Recent studies have further demonstrated that gene silencing of DAPK1 abolishes resveratrol‐ and IFN‐γ‐induced LC3II protein expression and autophagosome formation in human dermal fibroblasts and lymphocytic leukaemia, suggesting the involvement of DAPK1 signalling in autophagy (Choi et al., 2013; Gade et al., 2016). It has been reported that DAPK1 induces autophagy via the activation of PKD and further phosphorylation of VSP34 (Eisenberg‐Lerner and Kimchi, 2007; Eisenberg‐Lerner and Kimchi, 2012). The biological activity of DAPK1 is determined by phosphorylation. In particular, autophosphorylation at Ser308 in the kinase domain prevents DAPK1 from conformational change to the active state, and it remains inactive (Shiloh et al., 2014). Therefore, decreased levels of phosphorylation at Ser308 represent the active state of DAPK1, which leads to the phosphorylation of Beclin‐1 and further autophagy activation (Abrahamsen et al., 2012; Zalckvar et al., 2009). Although DAPK1 signalling is implicated in autophagy induction and adiponectin is well known to induce autophagy, the effect of adiponectin on the modulation of DAPK1 activity and its role in adiponectin‐induced autophagy have not been explored.
The interaction between Beclin‐1 and Bcl‐2 can also be determined by expression levels of Bcl‐2. Decreased Bcl‐2 expression results in an increase in the level of free Beclin‐1. The activity and expression levels of Bcl‐2 can be regulated at multiple stages. For example, the phosphorylation of Bcl‐2 by a JNK results in its dissociation from Beclin‐1 and subsequently induces autophagy (Abrahamsen et al., 2012; Wei et al., 2008). In addition, ubiquitination and degradation of Bcl‐2 by 26S proteasome reduces the level of cellular Bcl‐2, thereby inhibiting the association between Bcl‐2 and Beclin‐1 (Breitschopf et al., 2000). Furthermore, the expression of Bcl‐2 is determined by the stability of mRNA. Bcl‐2 mRNA contains A + U‐rich elements (AREs) in 3′‐untranslated region (3′‐UTR) that are involved in the regulation of mRNA stability (Bandyopadhyay et al., 2003). The stability of mRNA is regulated by various mRNA‐binding proteins, with different properties that regulate mRNA stability. For example, HuR and nucleolin bind to the ARE in Bcl‐2 mRNA and causes stabilization of mRNA by blocking exosome‐mediated degradation (Ishimaru et al., 2009; 2010), whereas tristetraprolin (TTP) mediates the destabilization of TNF‐α mRNA by globular adiponectin (gAcrp) in macrophage cell lines (Park et al., 2008).
In the present study, to better understand the molecular mechanisms underlying adiponectin‐induced autophagy activation, we examined the potential role of DAPK1 signalling in Beclin‐1/Bcl‐2 association and autophagosome formation by gAcrp. Herein, we found that gAcrp induced activation of DAPK1, which leads to Beclin‐1 phosphorylation, inhibition of Beclin‐1/Bcl‐2 association and ultimately induces autophagosome formation. We also demonstrated that gAcrp decreases Bcl‐2 expression through TTP‐mediated destabilization of Bcl‐2 mRNA, which also results in suppression of Beclin‐1 and Bcl‐2 complex formation, and autophagosome formation in macrophages. Finally, we showed that DAPK1 activation and TTP induction play crucial roles in the suppression of inflammatory cytokine expression by gAcrp in a gene‐selective manner in macrophages.
Methods
Cell culture of RAW 264.7 macrophages
An RAW 264.7 macrophage cell line was purchased from the Korean Cell Line Bank (Seoul, Korea) and maintained in DMEM supplemented with 10% (v .v‐1) FBS and 1% (v .v‐1) penicillin/streptomycin. The cells were routinely cultured in an incubator at 37°C in a humidified atmosphere of 5% CO2.
Isolation and culture of murine peritoneal macrophages
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All the animal experiments were conducted under the guidelines issued by the Yeungnam University Institutional Animal Care and Use Committee. The protocols for the experiments in this study were reviewed and approved by this Yeungnam University Research Committee (YU‐2016‐009). Murine peritoneal macrophages were isolated as described previously (Pun et al., 2015). In brief, 6‐ to 7‐week‐old male C57BL/6N mice (purchased from Orient Bio, South Korea) were injected i.p. with 1 mL of 4% (w.v‐1) thioglycollate medium (Difco, Detroit, MI, USA) to induce the accumulation of macrophages in the peritoneum region. After 3 days of thioglycollate injection, the macrophages from the peritoneum were extracted with ice‐cold HBSS (Sigma‐Aldrich, St Louis, MO, USA) and centrifuged at 300× g for 5 min. The cell pellet was suspended in red blood cell lysis buffer (BioLegend, San Diego, CA, USA) to remove contaminated red blood cells. After centrifugation, the remaining cell pellet was mixed with RPMI 1640 medium containing 10% FCS and 1% penicillin/streptomycin. The cells were then seeded onto a culture dish (35 mm) at a density of 2 × 106 cells per dish and incubated in an incubator (37°C) under a humidified atmosphere of 5% CO2 for further experiments.
To examine the effects of gAcrp on the expression of the genes of interest in in vivo conditions, 6‐ to 7‐week‐old male C57BL/6N mice were randomly divided into two groups (control and gAcrp‐treated groups, five mice for each group). On day 1, mice were injected i.p. with 1 mL of 4% thioglycollate solution. On day 2, mice were injected i.p. with adiponectin at a concentration of 1.5 μg·g−1 (mouse body weight). On day 3 (after 24 h treatment with gAcrp), peritoneal macrophages were isolated as described above and cultured for 2 h. Cells were then washed with cold PBS and used for the experiments.
Preparation of cellular extracts and Western blot analysis
The RAW 264.7 macrophages were seeded onto a 35 mm dish at a density of 1 × 106 cells per dish. After treatment as indicated, total cellular extracts were prepared using radio‐immunoprecipitation assay lysis buffer containing a Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL, USA). Total proteins were loaded onto an 8–15% SDS‐PAG, separated by electrophoresis and transferred to PVDF membranes. Possible non‐specific binding was blocked by incubation with 5% skimmed milk in PBS/Tween 20 for 1 h with shaking. The membranes were then incubated overnight at 4°C with the specific primary antibodies. After being washed with PBS/Tween three times, the membranes were incubated with the secondary antibodies conjugated with HRP for 1 h and again washed with PBS/Tween three times after a 10 min interval. Finally, images were captured using Fujifilm LAS‐4000 mini (Fujifilm, Tokyo, Japan). The membrane was stripped and reprobed with β‐actin antibody as a loading control.
Immunoprecipitation assay
The RAW 264.7 macrophages were seeded at a density of 5.5 × 106 cells per 100 mm dish. After overnight culture, the cells were transfected with DAPK1 siRNA or with scrambled siRNA for 24 h and stimulated with gAcrp for 24 h. Total proteins were extracted with immunoprecipitation lysis buffer (150 mM NaCl, 1 mM PMSF, 1% 4‐nonylphenyl‐PEG, 0.5 mM DTT, 50 mM HEPES and 5 mM EDTA) and incubated with 30 μL of Pierce Protein G Agarose (Thermo Scientific) for 1 h with gentle shaking and rocking at 4°C in a rotatory mixer. The reaction mixture was then centrifuged at 5000× g for 5 min. Protein from the supernatants (500 μg) was incubated overnight at 4°C with anti‐Beclin‐1 antibody (1:200 dilution) while being shaken and rocked to form the immune complexes. The immune complexes were pulled‐down by incubation with Protein G Agarose (30 μL) for 4 h at 4°C with gentle shaking and rocking, followed by centrifugation at 5000× g for 5 min at 4°C. The supernatant was removed, and the pellet containing the immune complexes was washed three times with immunoprecipitation lysis buffer. The final pellet was suspended in denaturing buffer and heated at 95°C for 10 min to completely denature the protein. The protein samples were stored until required for further immunoblot analysis.
RNA isolation, RT and quantitative PCR (qPCR)
To measure the expression levels of the mRNA of the target genes, total RNAs were extracted using Qiagen lysis buffer (Qiagen, MD, USA) according to the manufacturer's instructions. One microgram of the total extracted RNA was then reverse‐transcribed into cDNA using a GoScript Reverse Transcription system (Promega, Madison, WI, USA) following the manufacturer's instructions. The cDNA was amplified by qPCR using a Light Cycler 1.5 system (Roche Diagnostics, Mannheim, Germany) and an absolute QPCR SYBR green capillary mix (Thermo Scientific, UK) at 95°C for 15 min, 40 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 30 s. The relative mRNA expression levels of the target genes were determined using the comparative threshold (Ct) method, in which the Ct value of the target mRNA was normalized to that of GAPDH (ΔCt). The sequences of the primers used in the PCR amplification are shown in Table 1.
Table 1.
Sequences of the primers used for quantitative RT‐PCR and siRNA used in transient transfection
| Target gene | Purpose | Nucleotide sequence | |
|---|---|---|---|
| Bcl‐2 | PCR primer | F | 5′‐AGGAGCAGGTGCCTACAAGA‐3′ |
| R | 5′‐GCATTTTCCCACCACTGTCT‐3′ | ||
| TTP | PCR primer | F | 5′‐ATCCTGCCTTAGCCTTTTCC‐3′ |
| R | 5′‐GAGGGAAATTTGAGCACCAG‐3′ | ||
| TNF‐α | PCR primer | F | 5′‐CCCTCACACTCAGATCATCTTCT‐3′ |
| R | 5′‐GCTACGACGTGGGCTACAG‐3′ | ||
| IL‐1β | PCR primer | F | 5′‐GCCTCGTGCTGTCGGACCCATAT‐3′ |
| R | 5′‐TCCTTTGAGGCCCAAGGCCACA‐3′ | ||
| GAPDH | PCR primer | F | 5′‐ACCACAGTCCATGCCATCAC‐3′ |
| R | 5′‐TCCACCACCCTGTTGCTGTA‐3′ | ||
| DAPK | siRNA | F | 5′‐GUCAUGACGUCUACUCACA‐3′ |
| R | 5′‐UGUGAGUAGACGUCAUGAC‐3′ | ||
| TTP | siRNA | F | 5′‐GCUCCCACAAUACUAUCCU‐3′ |
| R | 5′‐AGGAUAGUAUUGUGGGAGC‐3′ | ||
| AMPKα1 | siRNA | F | 5′‐GCAGAAGUUUGUAGAGCAA‐3′ |
| R | 5′‐UCUUAUAGUUCAACCAUGA‐3′ | ||
| FoxO3A | siRNA | F | 5′‐GACGUCAUGAUGACCCAGU‐3′ |
| R | 5′‐ACUGGGUCAUCAUGACGUC‐3′ |
Transient transfection with siRNAs
The RAW 264.7 macrophages were seeded at a density of 7 × 105 cells per 35 mm dish. After overnight incubation, the cells were transfected for 24 h with siRNAs targeting specific genes or scrambled control siRNAs using HiPerFect Transfection Reagent (Qiagen, Hilden, Germany) according to the manufacturer's guidelines. Gene silencing efficiency was monitored by Western blot analysis. The siRNA duplexes used in this study were chemically synthesized by Bioneer (Daejeon, South Korea) or Dharmacon (Colorado, USA). The sequences of the siRNAs are listed in Table 1.
Confocal microscopic analysis
For the measurement of LC3 dots formation, RAW 26.7 macrophages were seeded at a density of 5 × 104 cells per well on an eight‐well glass chamber slide. After overnight culture, the cells were transfected either with DAPK1 siRNA or scrambled siRNA for 24 h using a HiPerFect Transfection Reagent according to the manufacturer's instructions and further co‐transfected with an eGFP‐tagged LC3 plasmid using FuGENE HD Transfection Reagent (Promega) for an additional 24 h. The cells were then stimulated with gAcrp for 24 h, washed twice with ice‐cold PBS and fixed in 4% paraformaldehyde solution for 10 min. The formation of LC3 green dots (indicating autophagosome formation) was captured using an A1 Confocal Laser Microscope System (Nikon Corp., Tokyo, Japan).
Luciferase assay for the measurement of Bcl‐2 promoter activity
Bcl‐2 promoter‐reporter constructs (pMAX‐Bcl‐2) containing a Bcl‐2 promoter region linked to the luciferase reporter gene were kindly provided by Prof. Kwon's laboratory. The RAW 264.7 macrophages were seeded onto a 24‐well plate at a density of 3 × 105 cells per well and incubated to reach 60–80% confluence. The cells were then transfected with a Bcl‐2/‐3254 plasmid using FuGENE HD Transfection Reagent (Promega). After incubation for 24 h, the cells were treated with gAcrp for the indicated time periods, washed twice with ice‐cold PBS and extracted with lysis buffer. After centrifugation, 20 μL of the supernatant was mixed with 100 μL of Luciferase Assay Reagent (Promega). Finally, luminescence was measured and values are presented relative to the basal value after normalizing the luminescence value with the respective protein concentrations.
elisa
To measure the level of TNF‐α secretion, RAW 264.7 macrophages were seeded onto 96‐well transparent plates at a density of 3 × 104 cells per well. After overnight incubation, the cells were transfected with scrambled control siRNA or siRNA targeting TTP (50 nM) or DAPK1 (25 nM) using HiPerfect Transfection Reagent. After incubation for 24 h, the cells were treated overnight with gAcrp. The media were replaced with new fresh media containing LPS (100 ng.mL−1) and further stimulated for 4 h. The cell culture media were collected and used for the measurement of TNF‐α secretion using TNF‐α elisa kits (BioLegend), according to the manufacturer's instructions.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Values are expressed as mean ± SEM from at least five independent experiments, and the data were analysed without the knowledge of treatment (blinding assessment). The data were analysed by one‐way ANOVA combined with the Bonferroni post hoc multiple comparison tests to find out the significant difference between groups. Graph Pad Prism software version 7.0 (La Jolla, CA, USA) was used to analyse the data. A P value <0.05 (*P < 0.05) denotes that the difference between groups is significant, and n used in figure legends denotes the number of independent sets of experiments.
Materials
All the cell culture reagents were obtained from HyClone Laboratories (South Logan, UT, USA). Recombinant human gAcrp was acquired from PeproTech Inc. (Rocky Hill, NJ, USA). MG‐132 and actinomycin D were purchased from Sigma‐Aldrich. Okadaic acid and anti‐TTP antibody were obtained from Santa Cruz (Delaware, CA, USA). Compound C, a pharmacological inhibitor of AMP‐activated protein kinase (AMPK), was obtained from Tocris Bioscience (Tocris House, IO Centre, Bristol, UK). The primary antibodies against Beclin‐1, LC3, total DAPK1, phospho‐AMPKα, total AMPKα and Bcl‐2 were procured from Cell Signaling Technology Inc. (Beverly, MA, USA). Antibodies against phospho‐specific Beclin‐1, β‐actin and phospho‐DAPK1 were from EMD Millipore (Denmark, Germany), Thermo Scientific Inc. and GeneTex Inc. (North America) respectively. The secondary antibody conjugated with HRP (goat anti‐rabbit IgG) was obtained from Pierce Biotechnology (Rockford, IL, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Globular adiponectin induces Beclin‐1 phosphorylation in macrophages
Phosphorylation of Beclin‐1 at threonine 119 (Thr119), a critical residue for binding with Bcl‐2, inhibits the association between Beclin‐1 and Bcl‐2, which leads to the initiation of autophagy (Zalckvar et al., 2009). To investigate the effect of gAcrp on Beclin‐1/Bcl‐2 association in macrophages, we first examined the effect of gAcrp on Beclin‐1 phosphorylation. As shown in Figure 1A, gAcrp treatment significantly increased Beclin‐1 phosphorylation at Thr119 in a dose‐dependent manner in RAW 264.7 macrophages. We also found that gAcrp treatment enhanced Beclin‐1 phosphorylation at 24 h treatment, whereas no significant effect was observed up to 8 h (Figure 1B). The effect of gAcrp on Beclin‐1 phosphorylation was further confirmed in primary peritoneal macrophages (Figure 1C, D). To confirm these effects in vivo, we treated the mice with gAcrp and measured phosphorylated level of Beclin‐1 in peritoneal macrophages. As indicated in Figure 1E, gAcrp treatment enhanced Beclin‐1 phosphorylation without a significant effect on total Beclin‐1 expression, essentially similar to in vitro experiments. Collectively, these data suggest that gAcrp induces Beclin‐1 phosphorylation in macrophages.
Figure 1.
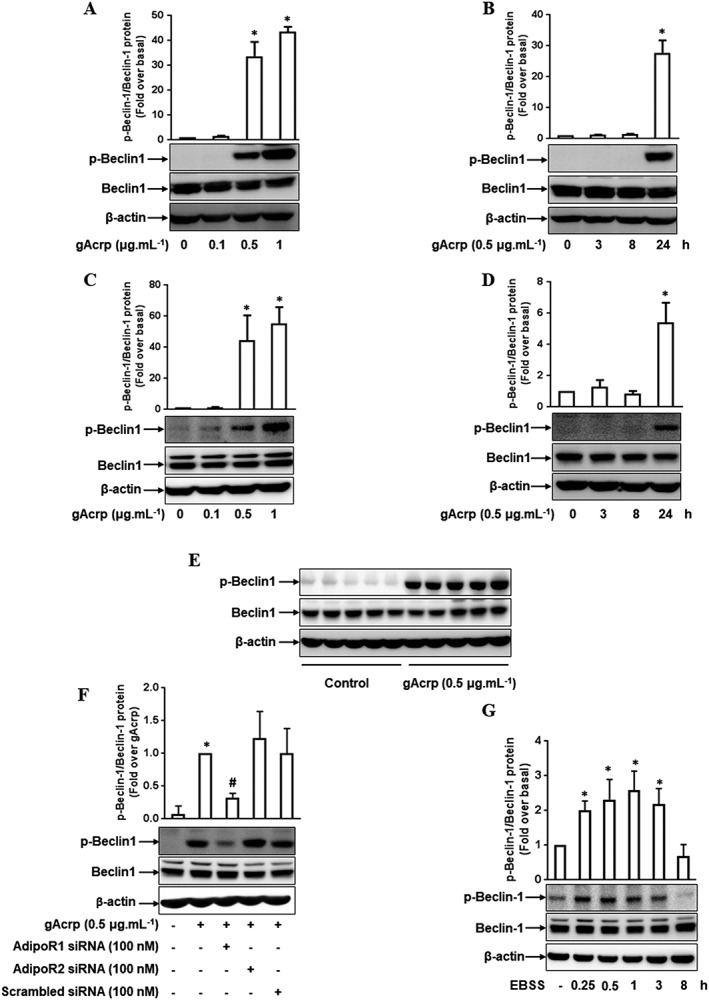
Effect of gAcrp on Beclin‐1 phosphorylation at Thr119 in macrophages. (A, B) RAW 264.7 macrophages were stimulated with the indicated concentrations of gAcrp for 24 h (A) or with 0.5 μg·mL−1 of gAcrp for different periods (B). Total and phosphorylated levels of Beclin‐1 were measured by Western blot analysis. (C, D) Macrophages isolated from murine peritoneum were stimulated with different concentrations of gAcrp for 24 h (C) or with gAcrp (0.5 μg·mL−1) for different durations (D). Total and phosphorylated Beclin‐1 levels were detected by Western blot analysis. (E) Mice were injected i.p. with gAcrp. After 24 h, peritoneal macrophages were isolated, and total and phosphorylated levels of Beclin‐1 were measured by Western blot analysis. (F) RAW 264.7 macrophages were transfected either with Adipo1, Adipo2 receptors or scrambled siRNA. After 24 h, the cells were treated with gAcrp for an additional 24 h. Expression of phosphorylated Beclin‐1 protein was measured by Western blot analysis. (G) RAW 264.7 macrophages were incubated in starvation EBSS media for the indicated times, and the expression levels of total and phosphorylated Beclin‐1 were determined by Western blot analysis. Throughout the Western blot analysis, the phosphorylated level of Beclin‐1 was quantified by densitometric analysis. The expression level of phospho‐Beclin‐1 was normalized to the level of total Beclin‐1 (used as an internal loading control) and shown in the upper panel of each Western blot image. Values represent the fold change relative to control (fold over basal) and are presented as mean ± SEM (n = 5). *P < 0.05 as compared to control cells and # P < 0.05 as compared to gAcrp treatment.
Diverse physiological responses induced by adiponectin are initiated by its binding to adiponectin receptors, either the Adipo1 or Adipo2 receptor. In the following experiments to identify the specific receptor mediating Beclin‐1 phosphorylation, gene silencing of the Adipo1 receptor significantly inhibited gAcrp‐induced beclin‐1 phosphorylation but gene silencing of the Adipo2 receptor had no significant effects (Figure 1F), suggesting that the Adipo1 receptor is crucial for Beclin‐1 phosphorylation by gAcrp. To further investigate the characteristics of gAcrp‐induced Beclin‐1 phosphorylation, we examined starvation‐induced Beclin‐1 phosphorylation using Earle's Balanced Salt Solution (EBSS) media, which is a typical condition for autophagy induction, and compared it with the effects of gAcrp treatment. EBSS treatment rapidly induced Beclin‐1 phosphorylation at Thr119 starting from 15 min, and it returned to the basal level at 8 h treatment (Figure 1G), which is a different pattern from gAcrp treatment. These results indicate that both starvation and gAcrp induce Beclin‐1 phosphorylation, which mediates the induction of autophagy in macrophages; however, the signalling events required for gAcrp‐induced and starvation‐stimulated Beclin‐1 phosphorylation are different from each other.
DAPK1 signalling is involved in Beclin‐1 phosphorylation by globular adiponectin in macrophages
We next examined the molecular mechanisms underlying gAcrp‐induced Beclin‐1 phosphorylation. For Beclin‐1 phosphorylation, it has been reported that DAPK1 phosphorylates Beclin‐1 at Thr119, which leads to autophagy activation (Zalckvar et al., 2009). We therefore hypothesized that DAPK1 might play a role in gAcrp‐induced Beclin‐1 phosphorylation in macrophages. To test this, we first investigated the effect of gAcrp on DAPK1 (de)phosphorylation. As depicted in Figure 2, gAcrp prominently suppressed the phosphorylation of DAPK1 at Ser308 in a dose‐ and time‐dependent manner in RAW 264.7 macrophages without a significant effect on total DAPK1 expression level (Figure 2A, B). Essentially similar effects were also observed in primary peritoneal macrophages (Figure 2C, D). It was further confirmed in the in vivo model (Figure 2E), collectively implying that gAcrp activates DAPK1 in macrophages. In addition, (de)phosphorylation of DAPK1 by gAcrp was restored by transfection with Adipo1 receptor siRNA without significant effects of Adipo2 receptor siRNA (Figure 2F), similar to the modulation of Beclin‐1 phosphorylation, suggesting that DAPK1 dephosphorylation is mainly mediated by Adipo1 receptor signalling. Moreover, gene silencing of DAPK1 almost completely inhibited gAcrp‐induced Beclin‐1 phosphorylation (Figure 2G), indicating the critical role of DAPK1 signalling in gAcrp‐induced Beclin‐1 phosphorylation. In the following experiments, pretreatment with okadaic acid, an inhibitor of serine/Thr‐specific protein phosphatase 1 (PP1) and 2A (PP2A), inhibited the gAcrp‐induced dephosphorylation of DAPK1 (Figure 2H), raising the possibility of the involvement of PP in the (de)phosphorylation of DAPK1 by gAcrp. Taken together, these results suggest that gAcrp induces Beclin‐1 phosphorylation through DAPK1 activation.
Figure 2.
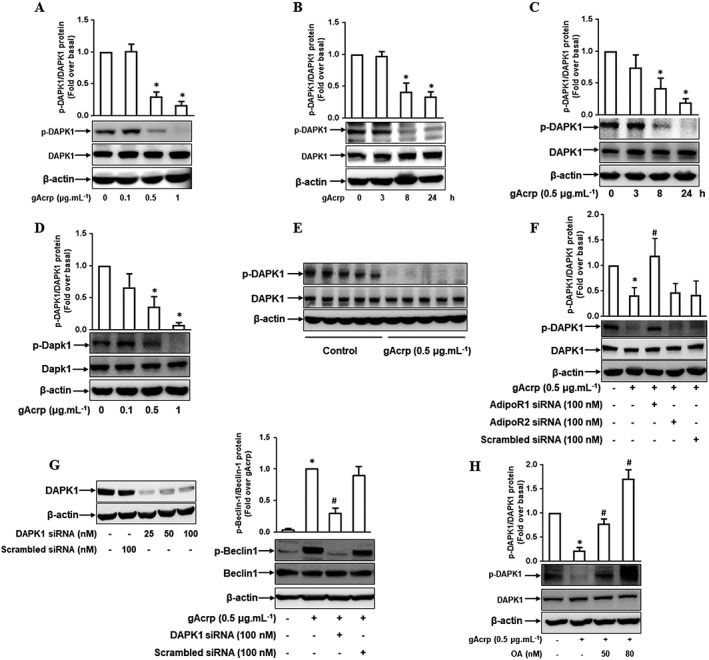
Effect of gAcrp on DAPK1 activation in macrophages and its role in Beclin‐1 phosphorylation in macrophages. (A, B) RAW 264.7 macrophages were stimulated with different doses of gAcrp for 24 h (A) or with 0.5 μg·mL−1 of gAcrp for the indicated periods (B). Protein expression levels of total and phosphorylated DAPK1 were determined by Western blot analysis. (C, D) Murine peritoneal macrophages were stimulated with the indicated doses of gAcrp for 24 h (C) or with 0.5 μg·mL−1 of gAcrp for the designated times (D). Total and phosphorylated DAPK1 levels were measured by Western blot analysis. (E) After 24 h injection with gAcrp, macrophages were isolated from murine peritoneum. Total and phosphorylated levels of DAPK1 were measured by Western blot analysis. (F) RAW 264.7 macrophages were transfected with siRNA targeting Adipo1, Adipo2 receptors or scrambled siRNA. After 24 h incubation, the cells were stimulated with gAcrp for 24 h. Total and phosphorylated levels of DAPK1 were measured by Western blot analysis. (G) RAW 264.7 macrophages were transfected either with DAPK1 siRNA or control scrambled siRNA for 24 h, followed by stimulation with gAcrp for 24 h. The gene silencing efficiency of DAPK1 siRNA was monitored by Western blot analysis (left panel). Total and phosphorylated Beclin‐1 levels were detected by Western blot analysis (right panel). (H) RAW 264.7 macrophages were initially stimulated with the indicated concentrations of okadaic acid (OA) for 1 h, followed by treatment with gAcrp for an additional 24 h. Total and phosphorylated DAPK1 levels were determined by Western blot analysis. Representative images from at least five sets of independent experiments are shown for all the Western blot analyses. Phosphorylated DAPK1 and Beclin‐1 expression levels were quantified by densitometric analysis and are shown above each Western blot image. Values are presented as the fold change relative to control group (fold over basal) and are expressed as mean ± SEM (n = 5). *P < 0.05 as compared with control cells. # P < 0.05 as compared with the cells treated with gAcrp.
DAPK1 is involved in globular adiponectin‐induced autophagy activation via inhibition of Beclin‐1 and Bcl‐2 protein interaction in macrophages
Because Beclin‐1 phosphorylation is a critical event in the modulation of Beclin‐1/Bcl‐2 association, we next determined if gAcrp affects the interaction between Beclin‐1 and Bcl‐2 by immunoprecipitation and Western blotting. As shown in Figure 3A, gAcrp stimulation significantly reduced the interaction between Beclin‐1 and Bcl‐2 protein. Interestingly, the decreased level of Beclin‐1/Bcl‐2 interaction was restored by DAPK1 gene silencing (Figure 3B), suggesting that DAPK1 signalling‐mediated Beclin‐1 phosphorylation plays a crucial role in the modulation of Beclin‐1 and Bcl‐2 interaction by gAcrp. As mentioned earlier, gAcrp has been shown to increase the expression of autophagy‐related genes and autophagy flux (Nepal and Park, 2013; Pun et al., 2015). Because the association between Beclin‐1 and Bcl2 is a key regulatory step in autophagy induction, we next determined if DAPK1 signalling is implicated in gAcrp‐induced autophagy activation. As indicated in Figure 3C, treatment with gAcrp enhanced autophagosome formation (indicated by LC3 green dots) consistent with previous reports, but it was almost completely abolished by DAPK1 gene silencing. These data collectively indicate that DAPK1 signalling plays an important role in gAcrp‐induced autophagy activation via Beclin‐1 phosphorylation and modulation of Beclin‐1/Bcl‐2 association in macrophages.
Figure 3.
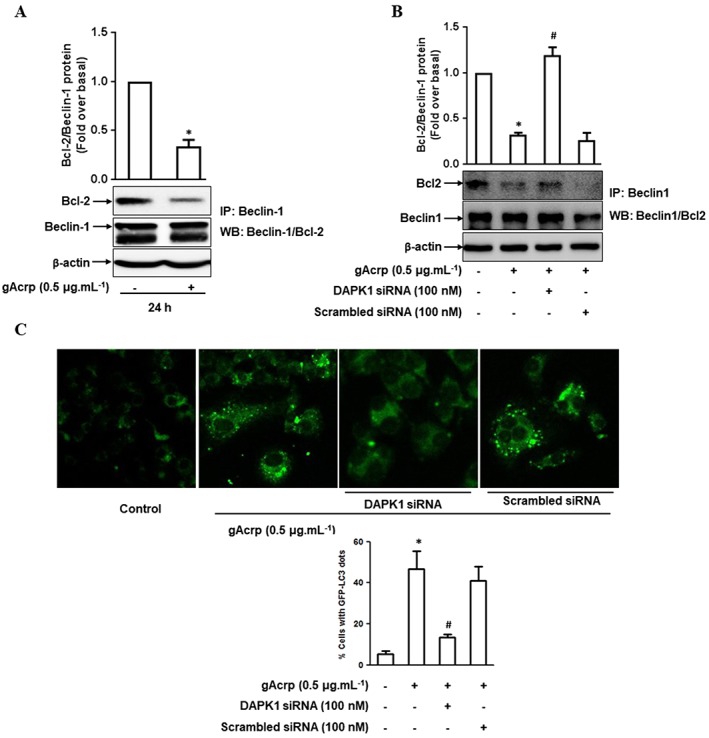
Role of DAPK1 signalling in the modulation of Beclin‐1 and Bcl‐2 protein interaction and autophagosome formation by gAcrp in macrophages. (A, B) RAW 264.7 macrophages were treated with gAcrp (0.5 μg·mL−1) for 24 h (A), or cells were transfected with DAPK1 siRNA or scrambled control siRNA for 24 h, followed by stimulation with gAcrp for an additional 24 h (B). The physical interaction between Beclin‐1 and Bcl‐2 was measured by immunoprecipitation using anti‐Beclin‐1 antibody and further immuno‐blotting with anti‐Bcl‐2 or anti‐Beclin‐1 antibody, as described in the Methods section. The representative images from independent experiments are shown. Data from densitometric analysis for the measurement of Bcl‐2 protein are presented above each Western blot image. (C) After transfection with DAPK1 siRNA or with scrambled control siRNA for 24 h, RAW 264.7 macrophages were further co‐transfected with eGFP‐tagged LC3 plasmids (eGFP‐LC3) followed by treatment with gAcrp for an additional 24 h. The cells were then fixed with paraformaldehyde solution and the formation of autophagosomes was indicated by the presence of LC3 dots (green dots) in the cytosol, as captured using an A1 Confocal Laser Microscope. Representative images from five sets of independent experiments are shown along with the quantification of LC3 dots in the lower panel. In (A), (B) and (C), the values are shown as the fold increases relative to the control (fold over basal) and are indicated as mean ± SEM (n = 5). *P < 0.05 as compared to control and # P < 0.05 as compared to the cells treated with gAcrp.
Globular adiponectin decreases Bcl‐2 expression via mRNA destabilization in macrophages
Changes in the expression of Bcl‐2 also affect the Beclin‐1/Bcl‐2 interaction. Therefore, we next examined the effect of gAcrp on Bcl‐2 expression. gAcrp substantially reduced Bcl‐2 expression at both mRNA (Figure 4A, B) and protein levels (Figure 4C, D) in a dose‐ and time‐dependent manner in RAW 264.7 macrophages. Essentially similar effects on Bcl‐2 expression were observed in primary peritoneal macrophages (Figure 4E, F). Treatment with gAcrp generated the consistent effects in the in vivo model (Figure 4G). Furthermore, gene silencing of the Adipo1 receptor prominently restored the suppression of Bcl‐2 expression by gAcrp, whereas no significant effect was observed with Adipo2 receptor siRNA (4H).
Figure 4.
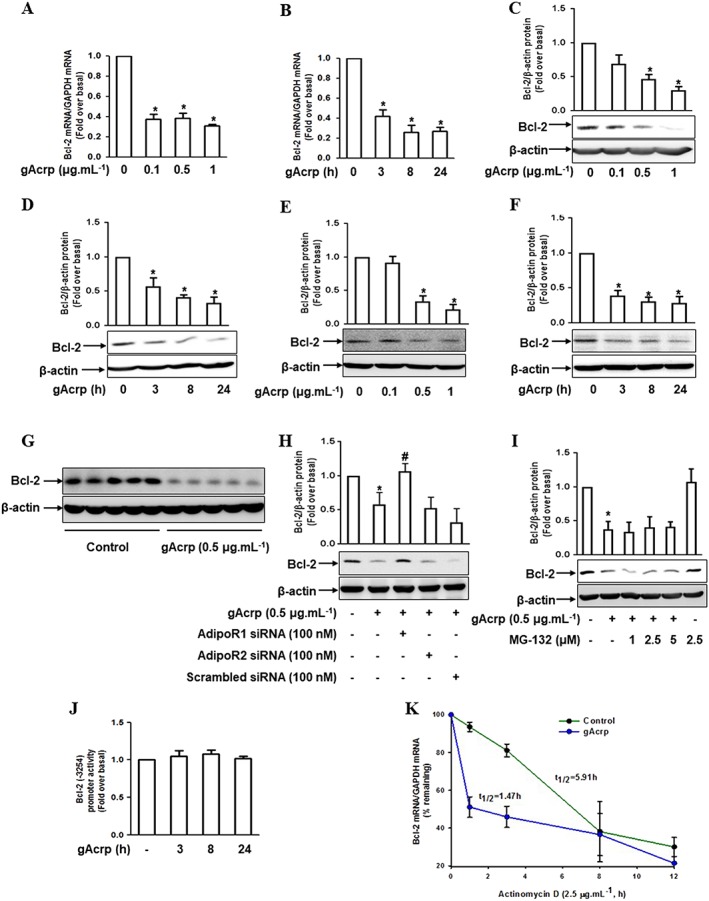
Effect of gAcrp on Bcl‐2 mRNA stability in macrophages. (A, B) RAW 264.7 macrophages were stimulated with different doses of gAcrp for 24 h (A) or with 0.5 μg·mL−1 of gAcrp for designated periods (B). The expression levels of Bcl‐2 mRNA were measured by qRT‐PCR analysis as indicated in the Methods section. (C, D) Cells were stimulated with different concentrations of gAcrp for 24 h (C) or with 0.5 μg·mL−1 of gAcrp for the indicated periods (D). The expression levels of Bcl‐2 protein were determined by Western blot analysis. (E, F) Macrophages isolated from murine peritoneum were stimulated with different concentrations of gAcrp for 24 h (E) or with 0.5 μg·mL−1 of gAcrp for the indicated times (F). The expression levels of Bcl‐2 protein were measured by Western blot analysis. (G) Mice were injected i.p. with gAcrp. After 24 h, peritoneal macrophages were isolated, and Bcl2 expression levels were measured by Western blot analysis. (H) RAW 264.7 macrophages were transfected with siRNA targeting Adipo1 or Adipo2 receptors, or scrambled siRNA. Cells were then further treated with gAcrp for 24 h and Bcl‐2 protein expression was measured by Western blot analysis. (I) RAW 264.7 macrophages were pretreated with the indicated concentrations of MG‐132 for 1 h, followed by gAcrp (0.5 μg·mL−1) for an additional 24 h. Bcl‐2 protein levels were measured by Western blot analysis. Expression levels of Bcl‐2 were quantified by densitometric analysis and the data are presented above each Western blot image. (J) RAW 264.7 macrophages were transfected with Bcl‐2 promoter plasmids tagged with luciferase using FuGENE HD transfection reagent, and the cells were stimulated with gAcrp for different time points. The luciferase activity (indicative of Bcl‐2 promoter activity) was measured by a luciferase assay. (K) Cells were pre‐stimulated with gAcrp for 24 h and then further treated with actinomycin D (2.5 μg·mL−1) for the indicated time periods. The levels of Bcl‐2 mRNA were measured by qPCR, and the half‐life (t 1/2) of Bcl‐2 mRNA was calculated as the percentage remaining by measuring the levels of Bcl‐2 mRNA. Values represent fold change relative to the control cells and are presented as mean ± SEM (n = 5). *P < 0.05 as compared with control cells and # P < 0.05 as compared to cells treated with gAcrp.
To further elucidate the mechanisms underlying the suppression of Bcl‐2 expression, treatment with MG‐132, a proteasome inhibitor, was shown not to significantly affect the suppression of Bcl‐2 protein levels by gAcrp (Figure 4I), suggesting that the ubiquitin‐proteasomal system is not involved in the suppression of Bcl‐2 expression by gAcrp. We also observed that gAcrp did not significantly affect Bcl‐2 promoter activity, as determined by a luciferase reporter assay (Figure 4J), indicating that gAcrp does not regulate Bcl‐2 expression at the transcriptional level. Finally, we speculated that gAcrp has an effect on Bcl‐2 mRNA stabilization. We measured the half‐life of Bcl‐2 mRNA following treatment with actinomycin D, an inhibitor of de novo mRNA synthesis. As indicated in Figure 4K, treatment with gAcrp significantly reduced the half‐life of Bcl‐2 mRNA (control group 5.91 h versus gAcrp treatment 1.47 h). Taken together, these results imply that gAcrp suppresses Bcl‐2 expression via mRNA destabilization in macrophages.
Tristetraprolin (TTP) is involved in Bcl‐2 mRNA destabilization by globular adiponectin
To further elucidate the mechanisms underlying Bcl‐2 mRNA destabilization by gAcrp, we examined the involvement of TTP, an mRNA‐destabilizing protein. We first analysed the effect of gAcrp on TTP and showed that gAcrp treatment led to a significant increase in TTP expression at both mRNA (Figure 5A, B) and protein (Figure 5C, D) levels in RAW 264.7 macrophages, primary peritoneal macrophages (Figure 5E, F) and the in vivo model (Figure 5G). In addition, the increase in TTP induced by gAcrp was returned to the normal level by gene silencing of the Adipo1 receptor, but not the Adipo2 receptor (Figure 5H), consistent with the modulation of Bcl‐2. In the ensuing experiments to verify the functional role of TTP in the modulation of Bcl‐2 mRNA stability, we found that gene silencing of TTP reversed the decrease in Bcl‐2 mRNA half‐life by gAcrp to almost normal levels (1.16 h gAcrp treatment versus 4.5 h gAcrp treatment plus TTP siRNA transfection) (Figure 5I). Finally, gene silencing of TTP restored the expression of Bcl‐2 both at mRNA (Figure 5J) and protein (Figure 5K) levels. In summary, these results suggest that TTP plays a pivotal role in the suppression of Bcl‐2 expression by gAcrp through Bcl‐2 mRNA destabilization in macrophages.
Figure 5.
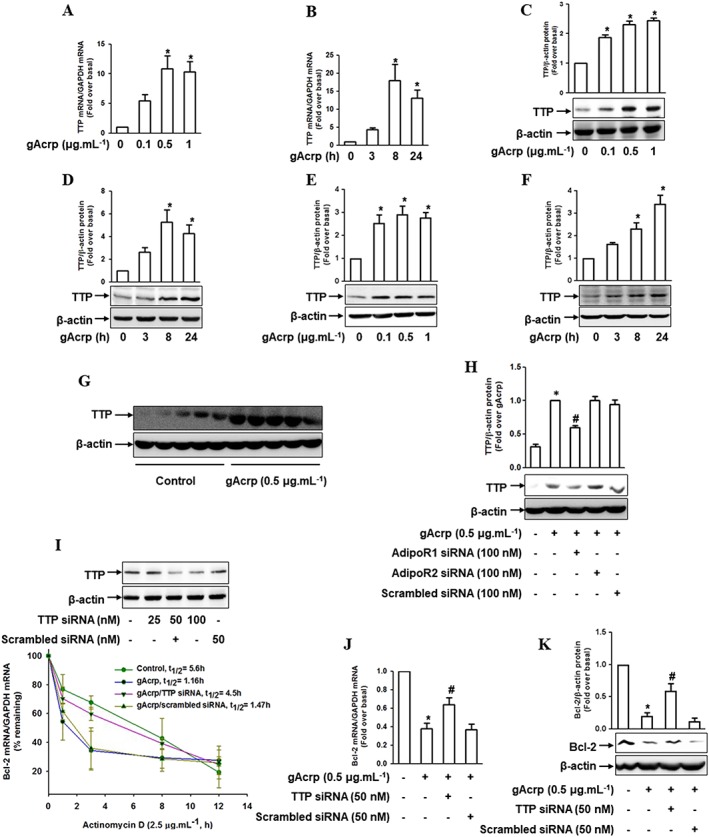
Effect of gAcrp on TTP induction and the role of TTP in Bcl‐2‐mRNA destabilization by gAcrp in macrophages. (A, B) RAW 264.7 macrophages were stimulated with the indicated concentrations of gAcrp for 24 h (A) or with 0.5 μg·mL−1 of gAcrp for the indicated time points (B). The TTP mRNA levels were measured by qRT‐PCR analysis. (C, D) Cells were stimulated with different concentrations of gAcrp for 24 h (C) or with gAcrp (0.5 μg·mL−1) for the indicated periods (D). TTP protein levels were determined by Western blot analysis along with β‐actin as an internal loading control. (E, F) Murine peritoneal macrophages were stimulated with different concentrations of gAcrp for 24 h (E) or with 0.5 μg·mL−1 of gAcrp for the indicated times (F). TTP protein expression levels were measured by Western blot analysis along with β‐actin as an internal loading control. (G) Peritoneal macrophages were isolated from mice injected with gAcrp (1.5 μg.g−1 wt. of mouse), and TTP protein expression was measured by Western blot analysis. (H) RAW 264.7 macrophages were transfected with Adipo1, Adipo2 receptors’ or scrambled siRNA. After 24 h of transfection, cells were treated with gAcrp for an additional 24 h. TTP protein expression was measured by Western blot analyses. (I) RAW 264.7 macrophages were transfected with siRNA targeting TTP or with scrambled control siRNA for 24 h. The gene silencing efficiency of TTP was monitored by Western blot analysis (upper panel). After transfection with TTP siRNA, the cells were pretreated with gAcrp for 24 h and then further treated with actinomycin D (2.5 μg·mL−1) for up to 12 h. Bcl‐2 mRNA levels were measured by qPCR, and the half‐life was calculated as the percentage remaining by measuring the levels of Bcl‐2 mRNA (lower panel). (J, K) RAW 264.7 macrophages were transfected with TTP siRNA or with scrambled control siRNA for 24 h. The cells were then treated with gAcrp for 24 h. Bcl‐2 mRNA (J) and protein (K) levels were determined by qRT‐PCR and Western blot analysis respectively. In all the Western blot analyses, the representative images from at least five independent experiments are shown. Quantitative analyses of proteins were performed by densitometric analysis and the data are shown above the images. The values are expressed as mean ± SEM (n = 5). *P < 0.05 as compared with the control cells and # P < 0.05 as compared with the cells treated with gAcrp.
The AMPKα1/forkhead box O3 (FoxO3A) axis is implicated in globular adiponectin‐induced increase in TTP in macrophages
As indicated in Figures 4 and 5, gAcrp decreased Bcl‐2 expression via TTP‐mediated Bcl‐2 mRNA destabilization. Since TTP plays a critical role in the suppression of Bcl‐2 expression, we investigated the upstream signalling involved in TTP induction. It has been shown that FoxO3A is involved in the induction of autophagy (Nepal et al., 2015) and it is also known to suppress Bcl‐2 expression in cancer cells (Park et al., 2014; Zhang et al., 2013). Thus, we hypothesized that FoxO3A could play a role in TTP induction by gAcrp. As indicated in Figure 6A, stimulation with gAcrp rapidly increased FoxO3A protein expression in RAW 264.7 macrophages. In addition, gene silencing of FoxO3A markedly inhibited gAcrp‐induced TTP expression at both mRNA (Figure 6B) and protein levels (Figure 6C), indicating the crucial role of FoxO3A signalling in TTP induction by gAcrp. Furthermore, the suppression of Bcl‐2 mRNA (Figure 6D) and protein (Figure 6E) expression by gAcrp was significantly restored by transfection with FoxO3A siRNA, consistent with the results showing TTP regulates Bcl‐2 expression (shown in Figure 5J, K). These results collectively imply that FoxO3A mediates the transcription of TTP gene, which in turn leads to the suppression of Bcl‐2 expression.
Figure 6.
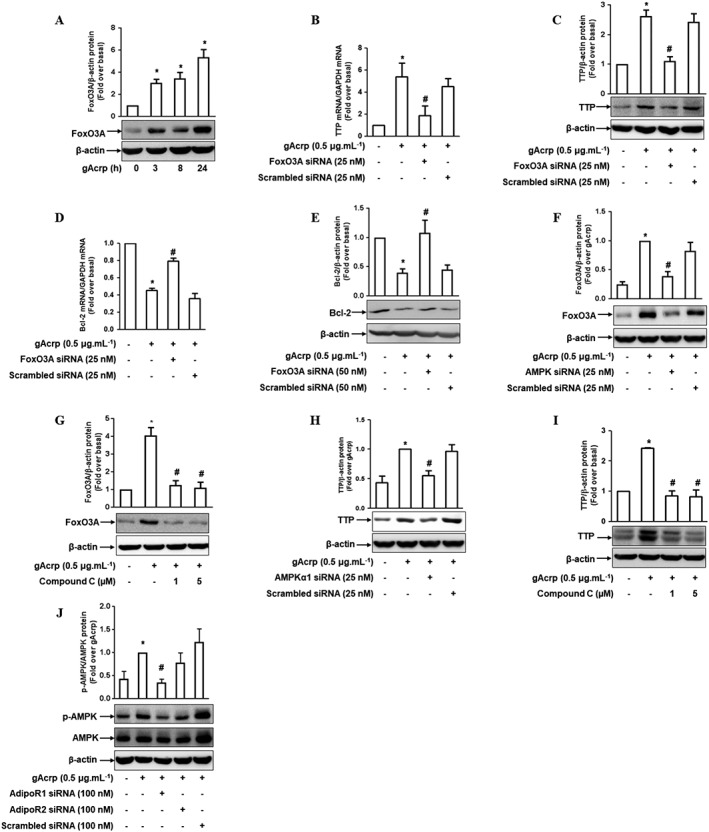
The crucial role of AMPK and FoxO3A in TTP induction by gAcrp in RAW 264.7 macrophages. (A) The cells were treated with gAcrp (0.5 μg·mL−1) for the indicated periods and the total FoxO3A protein level was determined by Western blot analysis along with β‐actin as an internal loading control. (B, C) The cells were transfected with siRNA targeting FoxO3A gene and further stimulated with gAcrp for an additional 6 h. The levels of TTP mRNA (B) and protein (C) were determined by qRT‐PCR and Western blot analysis respectively. (D, E) After transfection with FoxO3A siRNA, the cells were treated with gAcrp for 24 h. The expression levels of Bcl‐2 mRNA (D) and protein (E) were measured by qRT‐PCR and Western blot analysis respectively. (F) Cells were transfected with siRNA targeting AMPKα1, followed by treatment with gAcrp for an additional 6 h. The total FoxO3A protein level was determined by Western blot analysis. (G) The cells were pretreated with the indicated concentrations of compound C for 2 h, followed by stimulation with gAcrp for an additional 6 h. The FoxO3A protein expression level was determined by Western blot analysis. (H) After transfection with siRNA targeting AMPKα1, the cells were treated with gAcrp for 6 h. The TTP protein expression level was determined by Western blot analysis. (I) Cells were pretreated with compound C for 2 h, followed by further stimulation with gAcrp for an additional 6 h. TTP protein expression was measured by Western blot analysis. (J) RAW 264.7 macrophages were transfected with Adipo1, Adipo2 receptors’ or scrambled siRNA. Cells were then further treated with gAcrp for 24 h. Phospho‐ and total AMPK protein expression were measured by Western blot analysis. The representative images from five independent sets of experiments are shown in all Western blots. Quantitative analyses of FoxO3A, TTP, Bcl‐2 and p‐AMPKα1 protein expression bands were performed by densitometric analysis, and the results are presented above each Western blotting image. Values are presented as mean ± SEM (n = 5). *P < 0.05 compared with the control cells and # P < 0.05 compared with the cells treated with gAcrp.
We next further elucidated the upstream signalling molecule mediating FoxO3A expression. Activation of AMPKα1 and treatment with AICAR (a pharmacological activator of AMPK) lead to nuclear translocation and induction of FoxO3A (Nakashima and Yakabe, 2007; Nepal and Park, 2013; Pun et al., 2015). Moreover, AMPK signalling is well known to mediate various physiological responses by gAcrp. Thus, we hypothesized that AMPKα1 signalling is involved in the induction of FoxO3A by gAcrp. As expected, we observed that silencing of AMPKα1 gene or treatment with an inhibitor of AMPK (Compound C) blocked gAcrp‐induced FoxO3A protein expression (Figure 6F, G), indicating the critical role of AMPKα1 signalling in gAcrp‐induced FoxO3A expression. Moreover, similar effects of AMPKα1 siRNA or compound C were observed on the induction of TTP by gAcrp (Figure 6H, I). Taken together, these results imply that the AMPKα1/FoxO3A axis plays an important role in TTP induction and Bcl‐2 suppression by gAcrp in macrophages. In addition, gene silencing of the Adipo1 recepor significantly suppressed AMPKα1 phosphorylation by gAcrp with a slight, but not significant, effect seen with Adipo2 receptor siRNA (Figure 6J), consistent with the results from Bcl‐2 and TTP. Collectively, these findings indicate that Adipo1 receptor signalling plays a predominant role in the regulation of the AMPK/FoxO3A/TTP/Bcl2 axis.
DAPK1 activation and TTP mediate the suppression of LPS‐stimulated inflammatory mediator expression by globular adiponectin in macrophages
Autophagy induction plays a critical role in the suppression of inflammatory cytokine expression by gAcrp (Pun et al., 2015). Because Beclin‐1 phosphorylation, mediated by DAPK1 activation, and Bcl‐2 suppression, mediated by and increase in TTP, are critical steps in the initiation of autophagy, we next confirmed the role of DAPK1 signalling and TTP induction in the suppression of LPS‐induced inflammatory cytokine expression by gAcrp. As indicated in Figure 7, DAPK1 gene silencing resulted in restoration of gAcrp‐induced suppression of LPS‐induced TNF‐α expression at both the mRNA (Figure 7A) and protein levels (Figure 7B). DAPK1 knockdown also restored the suppression of LPS‐stimulated IL‐1β mRNA expression (Figure 7C), suggesting the crucial role of DAPK1 signalling in the suppression of LPS‐stimulated inflammatory cytokine expression by gAcrp. Essentially similar effects on TNF‐α and IL‐1β expression were observed following transfection with TTP siRNA (Figure 7D–F). Finally, the suppression of LPS‐induced IL‐1β expression by gAcrp was restored to the normal level by LC3B gene silencing (Figure 7G), consistent with the results obtained from DAPK and TTP gene silencing (Figure 7C, F). Taken together, these results suggest that both DAPK1 activation and TTP induction play crucial roles in the suppression of LPS‐induced inflammatory cytokine expression by gAcrp via modulation of autophagy induction.
Figure 7.
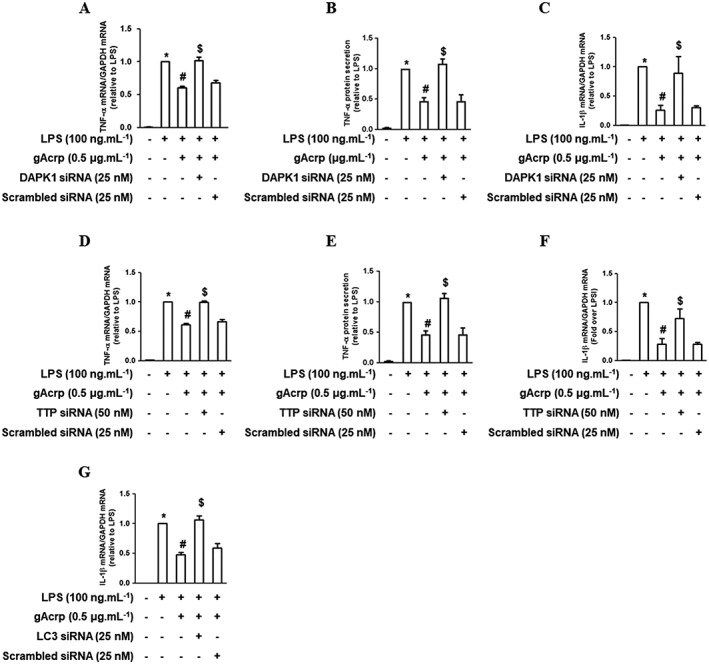
Role of DAPK1 and TTP signalling in the suppression of LPS‐induced cytokine expression by gAcrp in macrophages. (A, B) After transfection with siRNA targeting DAPK1, cells were treated with gAcrp for 24 h, followed by LPS treatment for an additional 2 h (A) or 4 h (B). (A) TNF‐α mRNA levels were determined by qRT‐PCR. (B) Cell culture media were collected and the secreted amount of TNF‐α was measured by elisa. (C) After DAPK1 gene silencing and gAcrp treatment as indicated, the cells were further stimulated with LPS for an additional 6 h. The IL‐1β mRNA expression level was determined by qRT‐PCR. (D–F) After transfection with siRNA targeting TTP, the cells were treated with gAcrp for 24 h and LPS for 2 h (D), 4 h (E) or 6 h (F). (D) The TNF‐α mRNA level was determined by qRT‐PCR. (E) The secretion of TNF‐α protein was measured by elisa. (F) The IL‐β mRNA level was determined by qRT‐PCR. (G) Cells were transfected either with LC3B siRNA or scrambled siRNA. The cells were then treated with gAcrp for 18 h, followed by incubation with LPS for an additional 6 h. The expression of IL‐1β mRNA was detected by qRT‐PCR. Values are expressed as mean ± SEM (n = 5). *P < 0.05 compared with the control cells. # P < 0.05 compared with the cells treated with LPS. $ P < 0.05 compared with the cells treated with LPS and gAcrp together.
Discussion and conclusions
Beclin‐1, which is encoded by the first identified autophagy‐related gene (Liang et al., 1999), plays a key role in the initiation of autophagy via complex formation with VSP34. Binding with Bcl‐2 hinders the communication between Beclin‐1 and VSP34, inhibits the overall kinetic function of the class III PI3K and finally inhibits the initiation of autophagy (Pattingre et al., 2005). Therefore, the interaction between Beclin‐1 and Bcl‐2 is considered a critical step in the regulation of autophagy induction, and inhibition of such an interaction is a plausible mechanism for the initiation of autophagy. Emerging evidence suggests that autophagy induction plays a crucial role in mediating many physiological responses induced by adiponectin (Nepal and Park, 2015). However, its underlying molecular mechanisms are not clearly understood. In the present study, we have demonstrated that adiponectin‐induced autophagy is mediated by disruption of Beclin‐1/Bcl‐2 association. Mechanistically, this process is mediated by Beclin‐1 phosphorylation through DAPK1 activation and decreased Bcl‐2 mRNA stability through TTP induction. Furthermore, we have shown that Beclin‐1 phosphorylation and Bcl‐2 mRNA destabilization are critical for the suppression of inflammatory mediators by gAcrp in macrophages.
Since the interaction between Beclin‐1 and Bcl‐2 is a key regulatory event in autophagy induction, the molecular mechanisms involved in the regulation of Beclin‐1/Bcl‐2 association have been extensively studied. In this study, we observed that gAcrp treatment induced Beclin‐1 phosphorylation at Thr119 without a significant effect on Beclin‐1 expression (Figure 1) and inhibited Beclin‐1/Bcl‐2 interaction (Figure 3A), supporting the hypothesis that Beclin‐1 phosphorylation might be involved in adiponectin‐induced autophagy activation. Beclin‐1 phosphorylation could be mediated by a number of kinases. For example, DAPK3 and unc‐51 like autophagy activating kinase (ULK) phosphorylate Beclin‐1 at Ser90 and Ser14, respectively (Fujiwara et al., 2016; Russell et al., 2013), thereby enhancing the kinase activity of VSP34. In addition, DAPK1 phosphorylates Beclin‐1 at Thr119 and inhibits the association between Beclin‐1 and Bcl‐2 (Zalckvar et al., 2009). DAPK1 activity is regulated by autophosphorylation at Ser308. In the basal state, in which substrate access to the kinase domain of DAPK1 is inhibited, Ser308 within the Ca2+/CaM autoregulatory domain is autophosphorylated. Therefore, a reduced level of phosphorylation at Ser308 represents the active state of DAPK1 (Shiloh et al., 2014). Herein, we found that gAcrp markedly reduced phosphorylation of DAPK1 at residue Ser308 (Figure 2). Interestingly, inhibition of DAPK1 phosphorylation by gAcrp was restored by pretreatment with okadaic acid, an inhibitor of PP1 and PP2A (Figure 2H), indicating the role of PP in the suppression of DAPK1 phosphorylation by adiponectin. Adiponectin has been shown to enhance the activity of PP2A (Kim et al., 2009) and protein tyrosine phosphatase 1B in breast cancer cells (Taliaferro‐Smith et al., 2013). Therefore, it is possible that the maintenance of a low level of DAPK1 phosphorylation by adiponectin is mediated by the actions of PP. At this stage, we did not thoroughly investigate the effect of adiponectin on the expression/activity of various types of PP or identify the specific type of PP involved in the regulation of DAPK1 phosphorylation. Further studies are needed to elucidate the role of PP in adiponectin‐induced DAPK1 dephosphorylation and autophagy induction.
In addition to the modulation of Beclin‐1, Bcl‐2 regulation is also critical to the interaction between Beclin‐1 and Bcl‐2. Bcl‐2 activity is regulated by direct phosphorylation. For example, phosphorylation of Bcl‐2 at Thr69, Ser70 and Ser87 by JNK1 causes the dissociation of Bcl‐2 from Beclin‐1 under starvation conditions (Wei et al., 2008). Moreover, Bcl‐2 protein expression is down‐regulated by ubiquitination and proteasomal degradation (Azad et al., 2008; Breitschopf et al., 2000), but proteasomal degradation was not involved in the decrease in Bcl‐2 induced by gAcrp in our study (Figure 4I), suggesting that the suppression of Bcl‐2 expression by gAcrp is not mediated by proteasomal degradation. Moreover, adiponectin did not significantly affect Bcl‐2 promoter activity (Figure 4J). Based on this result, we also excluded the possibility of the transcriptional regulation of Bcl‐2 by gAcrp and determined whether suppression of Bcl‐2 expression by gAcrp is mediated via a change in mRNA stability. Intracellular mRNA stability can be regulated by a number of proteins that bind to the AREs of the mRNA (Guhaniyogi and Brewer, 2001). Bcl‐2 mRNA contains four potential AU‐rich elements in the 3′‐UTRs (Bandyopadhyay et al., 2003). The results of the present study indicate the pivotal role of TTP induction in Bcl‐2 mRNA destabilization by gAcrp (Figure 5). To the best of our knowledge, this is the first report to demonstrate that adiponectin regulates Bcl‐2 expression via mRNA destabilization. In addition to TTP, many mRNA‐binding proteins regulate Bcl‐2 expression. For example, AU‐binding factor 1 (AUF1) and zinc finger protein 36 like‐1 (ZFP36LI) bind to the 3′‐UTR region of Bcl‐2 mRNA and induce its degradation (Ishimaru et al., 2010; Zekavati et al., 2014), raising the possibility that adiponectin regulates Bcl‐2 mRNA stability through the concerted action of various mRNA‐binding molecules. Further studies to investigate the mechanisms underlying the regulation of Bcl‐2 mRNA stability are required to gain a better insight into autophagy induction by adiponectin and to provide potential molecular targets for the regulation of inflammatory responses. Apart from the modulation of inflammatory responses, adiponectin is widely known to induce apoptosis of cancer cells via suppression of Bcl‐2 expression (Dos Santos et al., 2008; Konturek et al., 2008). Given that suppressing the expression of Bcl‐2 is critical for the suppression of cancer cell growth, these results would provide a novel insight into the suppression of tumour growth by adiponectin, as well as anti‐inflammatory responses.
There is a growing appreciation that autophagy induction is critical for the control of inflammatory responses (Levine et al., 2011). For example, adiponectin suppresses endotoxin‐stimulated inflammatory mediator production via autophagy induction in macrophages (Pun et al., 2015). Although it is well established that autophagy induction contributes to the modulation of inflammatory responses by adiponectin, the underlying molecular mechanisms are not clearly understood. Herein, we demonstrated that adiponectin induces autophagy by suppressing the association between Beclin‐1 and Bcl‐2. DAPK1 and TTP play key roles in this process via Beclin‐1 phosphorylation and suppression of Bcl‐2 expression respectively. We further confirmed that DAPK1 and TTP are also critical to the suppression of inflammatory mediator expression by gAcrp (Figure 7). To the best of our knowledge, this is the first report demonstrating the involvement of DAPK1 signalling in biological responses to adiponectin. In this study, we observed that DAPK1 and TTP were crucial for the suppression of LPS‐stimulated TNF‐α and IL‐1β expression by gAcrp (Figure 7). However, these molecules were not involved in the suppression of IFN‐β expression (Supporting Information Figure S1). Although we do not understand the detailed mechanisms for the differential regulation of the inflammatory cytokines, these results suggest that DAPK1 and TTP modulate inflammatory responses in a gene‐selective manner.
Previous studies have demonstrated that DAPK1 knockout mice exhibit an increased accumulation of nuclear NF‐κB and sensitivity towards LPS‐induced cytokine expression (Nakav et al., 2012). Furthermore, DAPK1 induction is involved in the suppression of NF‐κB activity in cancer cells stimulated with TNF‐α and IFN‐γ (Yoo et al., 2012), demonstrating that DAPK1 regulates inflammatory responses, which is consistent with the results from the present study. However, DAPK1 has been also shown to induce IL‐1β and IL‐18 expression via inflammasomes activation in human monocytes (Chuang et al., 2011). In the latter study, DAPK1 was shown to directly bind to the leucine‐rich repeat motif of NLRP3 to facilitate apoptosis‐associated speck‐like protein containing a CARD, caspase‐1 and NLRP3 assembly formation, indicating that DAPK1 signalling can act as a pro‐inflammatory mediator, particularly associated with abnormal production of IL‐1β. These contradictory results suggest that DAPK1 signalling regulates inflammatory responses in a complicated manner, and a pro‐ or anti‐inflammatory effect of DAPK1 depends on the experimental conditions, including cell type, cellular environment and stimulus used (Figure 8).
Figure 8.
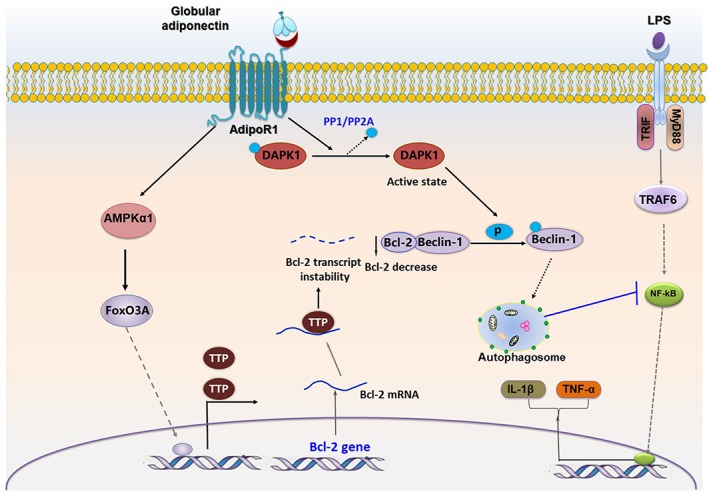
Proposed model for the role of DAPK1‐mediated Beclin‐1 phosphorylation and TTP‐mediated Bcl‐2 mRNA destabilization in autophagy leading to the suppression of inflammatory responses by gAcrp in macrophages. Bcl‐2 protein binds to Beclin‐1 through its BH3 domain. The activation of DAPK1 by gAcrp facilitates Beclin‐1 phosphorylation at Thr119 and inhibits binding to its inhibitory protein Bcl‐2, which leads to autophagosome formation. For the other way, the level of Bcl‐2 protein itself is reduced by gAcrp through TTP‐mediated mRNA instability. AMPKα1/FoxO3A axis signalling acts as an upstream pathway that leads to the induction of TTP protein. The Adipo1 receptor (AdipoR1), rather than the Adipo2 receptor, plays a predominant role in the modulation of DAPK/Beclin‐1 signalling and AMPK/FoxO3A/TTP/Bcl2 axis by gAcrp in macrophages. Both DAPK1 activation and TTP induction are involved in autophagy activation via the modulation of Beclin‐1 and Bcl‐2 protein interaction, which inhibits LPS‐primed inflammatory cytokines expression
The interaction between Beclin‐1 and Bcl‐2 could be determined by the status of both Beclin‐1 and Bcl‐2. In the present study, we observed that gAcrp did not affect Beclin‐1 expression but enhanced phosphorylation level (Figure 1), which suppresses its association with Bcl‐2 and subsequently induces autophagy. In addition, treatment with gAcrp suppressed Bcl‐2 expression (Figure 4), which also inhibits the Beclin‐1/Bcl‐2 interaction, indicating that both Beclin‐1 phosphorylation and Bcl‐2 suppression are involved in the suppression of association. But it is worth mentioning that there are differences in the time periods required for these events. A decrease in Bcl‐2 expression occurs at early time points, starting from 3 h, while Beclin‐1 phosphorylation was delayed and required prolonged stimulation (not observed until 8 h). Therefore, we assume that the modulation of the Beclin‐1/Bcl‐2 association would be caused by suppression of Bcl‐2 expression at an early stage, but both events (Beclin‐1 phosphorylation and Bcl‐2 suppression) would be implicated in the later stage.
There is a growing appreciation that AMPKα1 acts as a critical signalling molecule in mediating diverse physiological responses by adiponectin. For example, AMPKα1 mediates fatty acid oxidation and insulin sensitivity in muscle cells (Tomas et al., 2002) and stimulates endothelial NO production leading to vasodilatation and increased blood flow by gAcrp (Chen et al., 2003). AMPKα1 is rapidly phosphorylated by gAcrp, and its underlying mechanisms have been extensively studied. Among the diverse signalling mechanisms activated by Adipo receptors, recent studies have demonstrated that liver kinase B1 plays a crucial role in mediating AMPK activation by gAcrp in various experimental conditions (Deepa et al., 2011; Kim et al., 2014). While both Adipo1 and Adipo2 receptors can mediate gAcrp‐induced AMPKα1 phosphorylation depending on the experimental condition, the Adipo1 receptor possesses higher binding affinity with gAcrp (Achari and Jain, 2017). In accord with previous reports, herein, we also observed that Adipo1 receptor signalling is predominantly involved in AMPKα1 phosphorylation (Figure 6J) and the modulation of subsequent signalling molecules, including TTP and Bcl‐2 induction, and further phosphorylation of DAPK1 and Beclin‐1 (Figures 1F, 2F, 4H and 5H).
It has been recently shown that adiponectin induces autophagy in macrophages. However, the underlying molecular mechanisms are still largely unknown. We have previously reported that AMPK‐FoxO3A axis is involved in the induction of autophagy‐related genes by gAcrp in cancer cells (Nepal and Park, 2013), suggesting that the AMPK‐FoxO3A axis plays a key role in autophagy induction by gAcrp. In the present study, we have further demonstrated the role of the AMPK‐FoxO3A axis in the suppression of Bcl‐2 expression through TTP induction and its involvement in regulating the association between Beclin‐1 and Bcl‐2 in macrophages. While adiponectin has been shown to modulate Bcl‐2 expression, the detailed mechanisms are not clearly understood. Herein, we have found that TTP mediates gAcrp‐suppression of Bcl‐2 expression by inducing Bcl‐2 mRNA destabilization. In addition, DAPK1 is known to phosphorylate Beclin‐1 on Thr119. Beclin‐1 acts as a core protein in autophagy machinery, and Beclin‐1 phosphorylation leads to autophagy induction, collectively suggesting that DAPK1 plays a crucial role in the phosphorylation‐based autophagy induction. Although it has been shown that (de)phosphorylation of DAPK1 contributes to autophagy activation, the effect of adiponectin on the modulation of DAPK1 activity and, further, its role in adiponectin‐induced autophagy have not been explored. To the best of our knowledge, this is the first report to demonstrate the role of DAPK1 in adiponectin‐induced autophagy.
In conclusion, the present study has demonstrated that suppression of LPS‐stimulated inflammatory mediator production by gAcrp is mediated by inhibiting the formation of a Beclin‐1 and Bcl‐2 complexes in macrophages. Inhibition of the Beclin‐1/Bcl‐2 interaction is mediated by two parallel mechanisms: phosphorylation of Beclin‐1 in the BH3 domain by DAPK1 and destabilization of Bcl‐2 mRNA by TTP induction. In addition, TTP induction was mediated by, at least in part, by AMPKα1/FoxO3A axis‐dependent mechanisms. Furthermore, DAPK1 and TTP play crucial roles in autophagy induction and the regulation of inflammatory mediator production. Based on these results, DAPK1 and TTP are promising therapeutic targets for the treatment of diseases associated with inflammatory disorders.
Author contributions
P.H.P. and N.T.P. designed the study. N.T.P. performed the experiments. P.H.P. and N.T.P. analysed the data. P.H.P. and N.T.P. wrote the manuscript. P.H.P. contributed funding to the purchasing of materials and reagents.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Role of DAPK1 and tristetraprolin (TTP) signalling in the suppression of LPS‐induced IFN‐β mRNA expression by globular adiponectin (gAcrp) in RAW 264.7 macrophages.
Acknowledgements
This work was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF‐2015R1D1A1A01058203).
Tilija Pun, N. , and Park, P.‐H. (2018) Adiponectin inhibits inflammatory cytokines production by Beclin‐1 phosphorylation and B‐cell lymphoma 2 mRNA destabilization: role for autophagy induction. British Journal of Pharmacology, 175: 1066–1084. doi: 10.1111/bph.14144.
References
- Abrahamsen H, Stenmark H, Platta HW (2012). Ubiquitination and phosphorylation of Beclin 1 and its binding partners: tuning class III phosphatidylinositol 3‐kinase activity and tumor suppression. FEBS Lett 586: 1584–1591. [DOI] [PubMed] [Google Scholar]
- Achari AE, Jain SK (2017). Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci 18: 1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y (2008). Superoxide‐mediated proteasomal degradation of Bcl‐2 determines cell susceptibility to Cr(VI)‐induced apoptosis. Carcinogenesis 29: 1538–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Sengupta TK, Fernandes DJ, Spicer EK (2003). Taxol‐ and okadaic acid‐induced destabilization of bcl‐2 mRNA is associated with decreased binding of proteins to a bcl‐2 instability element. Biochem Pharmacol 66: 1151–1162. [DOI] [PubMed] [Google Scholar]
- Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmeler S (2000). Posttranslational modification of Bcl‐2 facilitates its proteasome‐dependent degradation: molecular characterization of the involved signaling pathway. Mol Cell Biol 20: 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochu‐Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, Palin MF (2010). Adiponectin action from head to toe. Endocrine 37: 11–32. [DOI] [PubMed] [Google Scholar]
- Bruce CR, Mertz VA, Heigenhauser GJ, Dyck DJ (2005). The stimulatory effect of globular adiponectin on insulin‐stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes 54: 3154–3160. [DOI] [PubMed] [Google Scholar]
- Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ (2003). Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 278: 45021–45026. [DOI] [PubMed] [Google Scholar]
- Choi MS, Kim Y, Jung JY, Yang SH, Lee TR, Shin DW (2013). Resveratrol induces autophagy through death‐associated protein kinase 1 (DAPK1) in human dermal fibroblasts under normal culture conditions. Exp Dermatol 22: 491–494. [DOI] [PubMed] [Google Scholar]
- Chuang YT, Lin YC, Lin KH, Chou TF, Kuo WC, Yang KT et al (2011). Tumor suppressor death‐associated protein kinase is required for full IL‐1beta production. Blood 117: 960–970. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deepa SS, Dong LQ (2009). APPL1: role in adiponectin signaling and beyond. Am J Physiol Endocrinol Metab 296: E22–E36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deepa SS, Zhou L, Ryu J, Wang C, Mao X, Li C et al (2011). APPL1 mediates adiponectin‐induced LKB1 cytosolic localization through the PP2A‐PKCzeta signaling pathway. Molecular endocrinology (Baltimore, Md.) 25: 1773–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos E, Benaitreau D, Dieudonne MN, Leneveu MC, Serazin V, Giudicelli Y et al (2008). Adiponectin mediates an antiproliferative response in human MDA‐MB 231 breast cancer cells. Oncol Rep 20: 971–977. [PubMed] [Google Scholar]
- Eisenberg‐Lerner A, Kimchi A (2007). DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ 14: 1908–1915. [DOI] [PubMed] [Google Scholar]
- Eisenberg‐Lerner A, Kimchi A (2012). PKD is a kinase of Vps34 that mediates ROS‐induced autophagy downstream of DAPk. Cell Death Differ 19: 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer M, Bluher M (2015). Adipokines in health and disease. Trends Pharmacol Sci 36: 461–470. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Usui T, Ohama T, Sato K (2016). Regulation of beclin 1 protein phosphorylation and autophagy by protein phosphatase 2a (PP2A) and Death‐associated Protein Kinase 3 (DAPK3). J Biol Chem 291: 10858–10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gade P, Kimball AS, DiNardo AC, Gangwal P, Ross DD, Boswell HS et al (2016). Death‐associated protein kinase‐1 expression and autophagy in chronic lymphocytic leukemia are dependent on activating transcription factor‐6 and CCAAT/enhancer‐binding protein‐beta. J Biol Chem 291: 22030–22042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H et al (2008). DAP‐kinase is a mediator of endoplasmic reticulum stress‐induced caspase activation and autophagic cell death. Cell Death Differ 15: 1875–1886. [DOI] [PubMed] [Google Scholar]
- Guhaniyogi J, Brewer G (2001). Regulation of mRNA stability in mammalian cells. Gene 265: 11–23. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru D, Ramalingam S, Sengupta TK, Bandyopadhyay S, Dellis S, Tholanikunnel BG et al (2009). Regulation of Bcl‐2 expression by HuR in HL60 leukemia cells and A431 carcinoma cells. Mol Cancer Res 7: 1354–1366. [DOI] [PubMed] [Google Scholar]
- Ishimaru D, Zuraw L, Ramalingam S, Sengupta TK, Bandyopadhyay S, Reuben A et al (2010). Mechanism of regulation of bcl‐2 mRNA by nucleolin and A+U‐rich element‐binding factor 1 (AUF1). J Biol Chem 285: 27182–27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116: 1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Baek A, Hwang JE, Choi YA, Jeong J, Lee MS et al (2009). Adiponectin‐activated AMPK stimulates dephosphorylation of AKT through protein phosphatase 2A activation. Cancer Res 69: 4018–4026. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Kim EH, Pun NT, Chang JH, Kim JA, Jeong JH et al (2017). Globular adiponectin inhibits lipopolysaccharide‐primed inflammasomes activation in macrophages via autophagy induction: the critical role of AMPK signaling. Int J Mol Sci 18: 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Nagy LE, Park PH (2014). Globular adiponectin inhibits ethanol‐induced reactive oxygen species production through modulation of NADPH oxidase in macrophages: involvement of liver kinase B1/AMP‐activated protein kinase pathway. Mol Pharmacol 86: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konturek PC, Burnat G, Rau T, Hahn EG, Konturek S (2008). Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig Dis Sci 53: 597–605. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2008). Autophagy in the pathogenesis of disease. Cell 132: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW (2011). Autophagy in immunity and inflammation. Nature 469: 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676. [DOI] [PubMed] [Google Scholar]
- Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE (2010). The anti‐inflammatory effects of adiponectin are mediated via a heme oxygenase‐1‐dependent pathway in rat Kupffer cells. Hepatology (Baltimore, Md.) 51: 1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight NC, Zhenyu Y (2013). Beclin 1, an essential component and master regulator of PI3K‐III in health and disease. Curr Pathobiol Rep 1: 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Yakabe Y (2007). AMPK activation stimulates myofibrillar protein degradation and expression of atrophy‐related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem 71: 1650–1656. [DOI] [PubMed] [Google Scholar]
- Nakav S, Cohen S, Feigelson SW, Bialik S, Shoseyov D, Kimchi A et al (2012). Tumor suppressor death‐associated protein kinase attenuates inflammatory responses in the lung. Am J Respir Cell Mol Biol 46: 313–322. [DOI] [PubMed] [Google Scholar]
- Nepal S, Kim MJ, Hong JT, Kim SH, Sohn DH, Lee SH et al (2015). Autophagy induction by leptin contributes to suppression of apoptosis in cancer cells and xenograft model: involvement of p53/FoxO3A axis. Oncotarget 6: 7166–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal S, Park PH (2013). Activation of autophagy by globular adiponectin attenuates ethanol‐induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis. Biochim Biophys Acta 1833: 2111–2125. [DOI] [PubMed] [Google Scholar]
- Nepal S, Park PH (2015). Modulation of cell death and survival by adipokines in the liver. Biol Pharm Bull 38: 961–965. [DOI] [PubMed] [Google Scholar]
- Onodera J, Ohsumi Y (2005). Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem 280: 31582–31586. [DOI] [PubMed] [Google Scholar]
- Park PH, Huang H, McMullen MR, Mandal P, Sun L, Nagy LE (2008). Suppression of lipopolysaccharide‐stimulated tumor necrosis factor‐alpha production by adiponectin is mediated by transcriptional and post‐transcriptional mechanisms. J Biol Chem 283: 26850–26858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PH, McMullen MR, Huang H, Thakur V, Nagy LE (2007). Short‐term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor‐alpha (TNF‐alpha) expression via ERK1/2 activation and Egr‐1 expression: role of TNF‐alpha in adiponectin‐stimulated interleukin‐10 production. J Biol Chem 282: 21695–21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Lee JH, Berek JS, Hu MC (2014). Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. Int J Oncol 45: 1691–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzych KR, Klionsky DJ (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N et al (2005). Bcl‐2 antiapoptotic proteins inhibit Beclin 1‐dependent autophagy. Cell 122: 927–939. [DOI] [PubMed] [Google Scholar]
- Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ et al (2010). Mammalian Atg18 (WIPI2) localizes to omegasome‐anchored phagophores and positively regulates LC3 lipidation. Autophagy 6: 506–522. [DOI] [PubMed] [Google Scholar]
- Pun NT, Subedi A, Kim MJ, Park PH (2015). Globular adiponectin causes tolerance to LPS‐induced TNF‐alpha expression via autophagy induction in RAW 264.7 macrophages: involvement of SIRT1/FoxO3A axis. PloS one 10: e0124636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J et al (2013). ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh R, Bialik S, Kimchi A (2014). The DAPK family: a structure‐function analysis. Apoptosis 19: 286–297. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Tooze SA (2009). Coordination of membrane events during autophagy by multiple class III PI3‐kinase complexes. J Cell Biol 186: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro‐Smith L, Nagalingam A, Knight BB, Oberlick E, Saxena NK, Sharma D (2013). Integral role of PTP1B in adiponectin‐mediated inhibition of oncogenic actions of leptin in breast carcinogenesis. Neoplasia (New York, N.Y.) 15: 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur V, Pritchard MT, McMullen MR, Nagy LE (2006). Adiponectin normalizes LPS‐stimulated TNF‐alpha production by rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol 290: G998–G1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang Cc C, Itani SI et al (2002). Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl‐CoA carboxylase inhibition and AMP‐activated protein kinase activation. Proc Natl Acad Sci U S A 99: 16309–16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toton E, Lisiak N, Sawicka P, Rybczynska M (2014). Beclin‐1 and its role as a target for anticancer therapy. J Physiol Pharmacol 65: 459–467. [PubMed] [Google Scholar]
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B (2008). JNK1‐mediated phosphorylation of Bcl‐2 regulates starvation‐induced autophagy. Mol Cell 30: 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M et al (2012). Beclin1: a role in membrane dynamics and beyond. Autophagy 8: 6–17. [DOI] [PubMed] [Google Scholar]
- Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY, Rho SB (2012). DAPk1 inhibits NF‐kappaB activation through TNF‐alpha and INF‐gamma‐induced apoptosis. Cell Signal 24: 1471–1477. [DOI] [PubMed] [Google Scholar]
- Yue Z, Zhong Y (2010). From a global view to focused examination: understanding cellular function of lipid kinase VPS34‐Beclin 1 complex in autophagy. J Mol Cell Biol 2: 305–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M et al (2009). DAP‐kinase‐mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl‐XL and induction of autophagy. EMBO Rep 10: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekavati A, Nasir A, Alcaraz A, Aldrovandi M, Marsh P, Norton JD et al (2014). Post‐transcriptional regulation of BCL2 mRNA by the RNA‐binding protein ZFP36L1 in malignant B cells. PloS one 9: e102625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhao Y, Xu M, Yu L, Chen J, Yuan Y et al (2013). FoxO3a modulates hypoxia stress induced oxidative stress and apoptosis in cardiac microvascular endothelial cells. PloS one 8: e80342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Role of DAPK1 and tristetraprolin (TTP) signalling in the suppression of LPS‐induced IFN‐β mRNA expression by globular adiponectin (gAcrp) in RAW 264.7 macrophages.