

Abstract
Background and Purpose
Inflammatory bowel disease (IBD) is a chronic and relapsing inflammatory disorder of the gastrointestinal tract, and an impaired immune response plays a critical role in IBD. The current drugs and therapies for IBD treatment are of limited use, therefore, there is a need to find novel drugs or therapies for this disease. We investigated the effect of cambogin in a mouse model of dextran sulphate sodium (DSS)‐induced colitis and whether cambogin attenuates inflammation via a Treg‐cell‐mediated effect on the immune response.
Experimental Approach
Chronic colitis was established in mice using 2% DSS, and cambogin (10 mg·kg−1, p.o.) was administered for 10 days. Body weight, colon length and colon histology were assessed. Cytokine production was measured using elisa and quantitative real‐time PCR. To evaluate the mechanism of cambogin, human CD4+CD25hiCD127lo Treg cells were isolated from peripheral blood mononuclear cells. Major signalling profiles involved in Treg cell stability were measured.
Key Results
Cambogin attenuated diarrhoea, colon shortening and colon histological injury and IL‐6, IFN‐γ and TNF‐α production in DSS‐treated mice. Cambogin also up‐regulated Treg cell numbers in both the spleen and mesenteric lymph nodes. Furthermore, cambogin (10 μM) prevented Foxp3 loss in human primary Treg cells in vitro, and promoted USP7‐mediated Foxp3 deubiquitination and increased Foxp3 protein expression in LPS‐treated cells.
Conclusions and Implications
The effect of cambogin on DSS‐induced colitis is expedited by a Treg‐cell‐mediated modification of the immune response, suggesting that cambogin could be applied as a novel agent for treating colitis and other Treg cell‐related diseases.
Abbreviations
- CD25
cluster of differentiation 25
- CTLA4
cytotoxic T‐lymphocyte antigen 4
- DSS
dextran sulphate sodium
- DUB
deubiquitinating enzyme
- Foxp3
forkhead box P3
- Ni‐NTA
nickel–nitrilotriacetic acid
- PIM1
proto‐oncogene serine/threonine‐protein kinase Pim‐1
- Treg
regulatory T‐cell
- USP7
ubiquitin‐specific‐processing protease 7
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis and Crohn's disease, is a common intestinal disorder characterized by recurrent and serious inflammation of the gastrointestinal tract (Carpenter and Talley, 2000; Bouma and Strober, 2003). IBD generally originates from complicated interactions between various host genetics and environmental influences (Kaser et al., 2010). Although the precise aetiology of IBD remains uncertain, there is evidence indicating that an unregulated immune response in the intestine has a key pathogenic role in this disease. An aberrant infiltration of mononuclear phagocytes, neutrophils and inflammatory T‐cells, particularly CD4+ T lymphocytes, is observed in the colonic lamina propria of IBD patients and in animal models of IBD (Lohr et al., 2006). Regulatory T‐cells (Tregs) expressing the transcription factor forkhead box P3 (Foxp3) play a key role in the pathogenesis of IBD and other autoimmune disorders, and are crucial for intestinal immune homeostasis. Several studies have shown that patients with IBD display significantly reduced numbers and functionally defective peripheral Treg cells (Maul et al., 2005; Sakaguchi, 2005; Harrison and Powrie, 2013; Haribhai et al., 2016; Yamada et al., 2016). Therefore, it is likely that correcting the defects of Treg cells might be an effective treatment for IBD. However, until recently, few drug developments or studies have examined Treg cells or Foxp3 as targets for IBD treatment. The current therapies used for IBD treatment include aminosalicylates, corticosteroids and immunosuppressants (Faubion et al., 2001; Rosen et al., 2012); however, long‐term use of high‐dose corticosteroids may lead to harmful side effects, including hypertension and drug‐induced lupus (Guilbert et al., 2006; Haribhai et al., 2016).
For decades, natural plants have remained a major source for developing new drugs against a variety of diseases. In a previous study, we reported that the genus Garcinia Y.H. Li of the family Guttiferae exhibits various biological activities, including antibacterial, antifungal, anti‐inflammatory, antioxidant and cytotoxic effects (Lu et al., 2016). The novel compound cambogin, isolated from Garcinia esculenta, exhibits anticancer effects (Shen et al., 2015; 2016); however, to date, the anti‐inflammatory or immunoregulatory effect of cambogin has not been investigated. The purpose of the present study was to determine whether cambogin exerts an anti‐inflammatory effect by enhancing the Treg responses and suppressing immune responses in a dextran sulphate sodium (DSS)‐induced murine model of chronic colitis.
Methods
Animals
All the animal care and experimental studies were approved by and conducted in accordance with the guidelines of the Animal Ethical Committee of Shanghai University of Traditional Chinese Medicine. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Since male mice are likely to be more irritable and fight than female mice, we used female Balb/c mice (20 ± 2 g) between 6 and 8 weeks of age, which were purchased from the Slac Animal Laboratory (Shanghai, China). All the mice were fed standard food (purchased from the Slac Animal Laboratory) and water ad libitum and kept in a constant environment, 20 ± 2°C and 12 h light/dark cycle, which they were acclimatized to for a week before the experiments.
Experimental colitis was induced by the p.o. administration of DSS as previously described (Wirtz et al., 2007). The mice were divided into three groups: normal control group (healthy mice given only water to drink), DSS model group (mice administered DSS in their drinking water) and DSS plus cambogin group (DSS mice also administered cambogin p.o.). In our preliminary experiments, we found that the toxicity of cambogin was negligible at 20 μM (in vitro) and 20 mg·kg−1 (in vivo). Hence, we chose to study the effects of cambogin 10 μM (in vitro) and 10 mg·kg−1 (in vivo). We also tested lower concentrations of cambogin, 1 μM in vitro and 1 and 5 mg·kg−1 in vivo, but they had no obvious effects on Treg cells or the colitis model. In the two DSS groups, mice were fed 2% (w.v‐1) DSS in drinking water for 7 days followed by plain water for 14 days, and DSS for another 7 days. For the cambogin group: cambogin (10 mg·kg−1) was dissolved in 2% carboxymethyl cellulose (CMC) for intragastric administration, and it was administered each day from days 18 to 27; the normal control group and DSS model group were administered 2% CMC solution daily by p.o. gavage on those days. At day 28, the mice were killed via a rising concentration of CO2 and subsequent cervical dislocation, and the colon, mesenteric lymph nodes (MLNs) and spleen were harvested for subsequent use in various assays as indicated. The severity of colitis was evaluated by monitoring body weight changes, disease activity and colon length, and histological changes were analysed. Sample sizes were n = 7 per group for all studies.
Disease activity evaluation
During the study, the body weight, stool consistency and occult blood in the stool were recorded to determine the disease activity index (DAI). The DAI was calculated as previously described (Liu et al., 2013). Generally, (i) diarrhoea (0 point = normal, 2 points = loose stool and 4 points = watery diarrhoea) and (ii) haematochezia (0 point = no bleeding, 2 points = slight bleeding and 4 points = gross bleeding); the DAI scores were the summation of (i) and (ii).
Flow cytometry analysis
The murine spleen cells and lymph nodes cells were stimulated using PMA, ionomycin and protein transport inhibitor (BD GolgiStop) for 4 h. At the end of stimulation, the cells were permeabilized using IC fixation buffer and 1× permeabilization buffer (eBioscience, San Diego, CA, USA). The following antibodies were used for flow cytometry: anti‐IFN‐γ‐APC (177311, eBioscience, San Diego, CA, USA), anti‐IL‐17A‐PE (130103015, Miltenyi, Auburn, CA, USA), anti‐Tbet‐APC (644813, Biolegend, San Diego, CA, USA), anti‐Foxp3‐FITC (eBio7979; 115773, eBioscience, San Diego, CA, USA), anti‐RORγt‐PE (562607, BD Pharmingen, San Diego, CA, USA), anti‐CTLA4‐PE (121522, eBioscience, San Diego, CA, USA) and anti‐CD4‐VioBlue (130102456, Miltenyi, Auburn, CA, USA). Single‐cell suspensions were examined on a FACS Fortessa (BD Immunocytometry Systems, San Jose, CA, USA), and the data were analysed using FlowJo software. The numbers in the corners of the FACS dot plots represent the percentage of each cell population within that quadrant as a fraction of the total cell population.
Isolation and expansion of human Treg cells
Human PBMCs were isolated from the buffy coat of healthy donors (Shanghai Blood Centre, Shanghai, China). All studies were approved by the Institutional Ethics Committee. Human CD4+CD25loCD127hi naive T‐cells and CD4+CD25hiCD127lo Treg cells were isolated from PBMC using FACS on a BD FACS ARIA II sorter. Treg cells were expanded in the presence of rhIL‐2 (500 U·mL−1, R&D, Minneapolis, MN, USA), rapamycin (100 nM) and anti‐CD3/CD28 DynaBeads (Invitrogen, Carlsbad, CA, USA) for 10 days followed by a quiescent period in a lower concentration of rIL‐2 (100 U·mL−1).
Transfection, immunoprecipitation and Western blotting
HEK293T cells were transfected with appropriate plasmids using PEI reagent (Polysciences, Warrington, PA, USA) according to the manufacturer's instructions. The procedures for immunoprecipitation and Western blotting were as described in detail previously (Chen et al., 2013). The relative protein level was normalized against GAPDH by using ImageJ software.
Ubiquitin pull‐down assay
The HEK293T cells were lysed in urea buffer (10 mM Tris, pH 8.0, 8 M urea, 100 mM Na2HPO4, 0.2% Triton‐100 and 10 mM imidazole) for 30 min. The lysates were incubated with nickel–nitrilotriacetic acid (Ni‐NTA) acid beads (Qiagen, Germantown, MD, USA) for 3 h at room temperature. After incubation, the beads were washed twice in urea buffer (10 mM Tris, pH 6.3, 8 M urea, 100 mM Na2HPO4, 0.2% Triton X‐100 and 10 mM imidazole) and once in a wash buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 20% glycerol, 1 mM DTT and 10 mM imidazole). The ubiquitination levels were evaluated using Western blotting with specific antibodies as indicated.
Cytokine measurement
Cytokines in the colon tissue homogenates including IL‐6 (R&D, Minneapolis, MN, USA, DY406), TNF‐α (R&D, Minneapolis, MN, USA, DY410) and IFN‐γ (R&D, DY485) were measured using elisa kits according to the manufacturer's instructions. The detection limits of these elisa kits were as follows: IL‐6, from 15.60–1000 pg·mL−1; TNF‐α, from 31.20–2000 pg·mL−1; and IFN‐γ, from 31.20–2000 pg·mL−1.
Quantitative real‐time PCR
Tissues or cells were treated with TRIzol (Sigma‐Aldrich, St. Louis, MO, USA). Total RNA was isolated according to the manufacturer's instructions. The cDNA was synthesized using a reverse transcriptase kit (TaKaRa, Berkeley, CA, USA), followed by quantitative PCR analysis (SYBR Green; Applied Biosystems, Foster City, CA, USA). The level of mRNA was normalized to GAPDH expression, and the results were analysed using the 2−ΔΔCt method. The following primers were used in the present study: mouse IL‐6 forward, 5′‐CTGCAAGAGACTTCCATCCAGTT‐3′, IL‐6 reverse, 5′‐GAAGTAGGGAAGGCCGTGG‐3′; mouse TNF‐α forward, 5′‐CGAGTGACAAGCCTGTAGC‐3′, TNF‐α reverse, 5′‐GGTGTGGGTGAGGAGCACAT‐3′; mouse IFN‐γ forward, 5′‐CTACCTTCTTCAGCAACAGC‐3′, IFN‐γ reverse 5′‐GCTCATTGAATGCTTGGCGC‐3′; mouse GAPDH forward, 5′‐AAATCCCATCACCATCTTCC‐3′, GAPDH reverse, 5′‐TCACACCCTGACGAACA‐3′; human Foxp3 forward 5′‐TGCAAAAGGCTTCAGAGACA‐3′, human Foxp3 reverse 5′‐CTCTGTTGGGGTGAAAGGAG‐3′; human CD25 (also known as IL2RA) forward 5′‐GAGACGTCCATATTTACAACAG‐3′, human CD25 reverse 5′‐CCTTTGATTTCACTTGGGCTTC‐3′; human CTLA4 forward 5′‐TGGGGAATGAGTTGACCTTC‐3′, human CTLA4 reverse 5′‐GCACGGTTCTGGATCAATTA‐3′; human USP7 forward 5′‐GAGGAGGACATGGAGGATGA‐3′, human USP7 reverse 5′‐AAGCGTGGCATCACCATAAT‐3′; human IL‐10 forward 5′‐ACCTCTGATACCTCAACCCC‐3′, human IL‐10 reverse 5′‐TGGTCAGGCTTGGAATGGAA‐3′; human IL‐2 forward 5′‐GCAACTCCTGTCTTGCATTG‐3′, human IL‐2 reverse 5′‐CAGTTCTGTGGCCTTCTTGG‐3′; and human GAPDH forward 5′‐GAGTCAACGGATTTGGTCGT‐3′, human GAPDH reverse 5′‐GACAAGCTTCCCGTTCTCAG‐3′.
Polyubiquitin chain‐binding assay
The K48 polyUb or K63 polyUb chain‐binding assay was performed using a K48 polyUb or K63 polyUb chain‐binding protein identification kit (UBPBio, Aurora, CO, USA). According to the manufacturer's instructions, Treg cell lysates were incubated with the 6xHis‐Non‐cleavable K48 PolyUb Chain, 6xHis‐Non‐cleavable K63 PolyUb Chain or 6xHis‐Ubiquitin. The lysates were subsequently incubated with Ni resin followed by appropriate elution, and the supernatants, which contained non‐cleavable K48 polyUb chains or non‐cleavable K63 polyUb chains and binding proteins, were analysed using immunoblotting.
Histological analysis and immunohistochemistry
Once removed, the colons were immediately fixed in 10% buffered formalin and stained with haematoxylin and eosin. The histological scores were calculated as previously described (Lee et al., 2015). Briefly, assessment included reporting of oedema, extent of injury and crypt abscesses. In this grading system, inflammation severity was scored using a scale of 0–3 (0, no inflammation; 1, slight inflammation; 2, moderate inflammation; and 3, severe inflammation), similarly the extent of injury (0, no injury; 1, mucosal injury; 2, mucosal and submucosal injury; and 3, transmural injury). Crypt damage was scored using a scale of 0–4 (0, no damage; 1, basal third was damaged; 2, basal two‐thirds was damaged; 3, only the surface epithelium was intact; and 4, loss of entire crypt and epithelium). The total histopathological score was determined from the sum of the scores for each parameter to reflect the overall degree of inflammation within each specimen.
Immunohistochemistry (IHC) staining was performed as described previously (Leonhardt et al., 2003). Colon tissues were fixed in 10% formalin, embedded in paraffin and then cut into 4‐μm‐thick sections. The deparaffinized sections underwent antigen retrieval and endogenous peroxidase activity blocking and were then blocked in 5% BSA. Primary antibodies anti‐Foxp3, anti‐USP7, anti‐Pim‐1 proto‐oncogene serine/threonine‐protein kinase (PIM1) and anti‐s422 were all diluted (1:100) and incubated with the sections overnight at 4°C. After being washed with PBS (pH 7.4) three times for 10 min, the samples were incubated with secondary antibodies [anti‐mouse HRP (ab98467, Abcam) and anti‐rabbit HRP (ab97080, Abcam, Cambridge, UK), 1:200 dilution] at room temperature for 50 min. Then the slides were further washed with PBS three times for 10 min and developed with Pierce DAB substrate kit (3400; Thermo Scientific, Grand Island, NY, USA). Following which they were counterstained with haematoxylin solution (Harris modified, Sigma‐Aldrich) for 3 min and dehydrated, the images of all slides were visualized and captured using DP‐72 microscope (Olympus, Tokyo, Japan). The procedure also included negative controls with omission of the primary antibody; these controls did not show any immunoreaction (Alique et al., 2006).
RNA‐seq analysis
Procedures for RNA preparation, library construction and sequencing on the BGISEQ‐500 platform have been described in detail previously (Xin et al., 2017). The fold changes were also estimated according to the fragments per kilobase of exon per million fragments mapped (FPKM) in each sample. The differentially expressed genes were selected using the following filter criteria: false discovery rate ≤0.05 and fold change ≥2. We have uploaded the raw data of this RNA‐seq to the Sequence Read Archive (SRA) database. The BioProject accession is PRJNA418063, the BioSample accession is SAMN08014122 and the SRA submission number is SUB3216027.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All the experimental data are presented as mean ± SEM. The statistical significance of the differences between two groups was determined using Student's unpaired t‐test, and multiple comparisons were analysed using one‐way ANOVA followed by Dunnett's post hoc test using GraphPad Prism 5.0 software. P < 0.05 was taken to indicate statistical significance.
Materials
Cambogin (Figure 1A) was isolated from the G. esculenta Y. H. Li. The twigs of G. esculenta Y. H. Li were collected in Nujiang, Yunnan Province, China, in August 2010. The plant material was identified by Prof. Yuanchuan Zhou, Yunnan University of Traditional Chinese Medicine. A voucher specimen (herbarium no. 20100801) was deposited at the Engineering Research Centre of Shanghai Colleges for TCM New Drug Discovery, Shanghai University of Traditional Chinese Medicine. Cambogin's structure was determined using 1H‐NMR and 13C‐NMR spectral analysis, and the purity of this compound was more than 98% based on HPLC analysis (Figure 1C, D). 1H‐NMR and 13C‐NMR spectra were measured on a Bruker AV‐400 spectrometer and calibrated by the solvent peak used (pyridine‐d 6). Ultra performance liquid chromatography (UPLC) was performed using a Waters Acquity UPLC I class system (Waters, Milford, MA, USA), equipped with a binary solvent delivery system, an autosampler and a photodiode array detection system. Chromatography was performed on a Waters ACQUITY BEH C18 column (2.1 mm × 100 mm I.D., 1.7 μm, Waters). The mobile phase consisted of (i) 0.1% formic acid in water and (ii) acetonitrile. The UPLC eluting conditions were as follows: 60–70% B (0–10 min), 70–90% B (10–15 min) and 90–100% B (15–18 min). The flow rate was maintained at 0.4 mL·min−1. The column and autosampler were maintained at 40 and 10°C respectively. The detection wavelengths were set at 231, 276 and 310 nm (Zhang et al., 2014).
Figure 1.
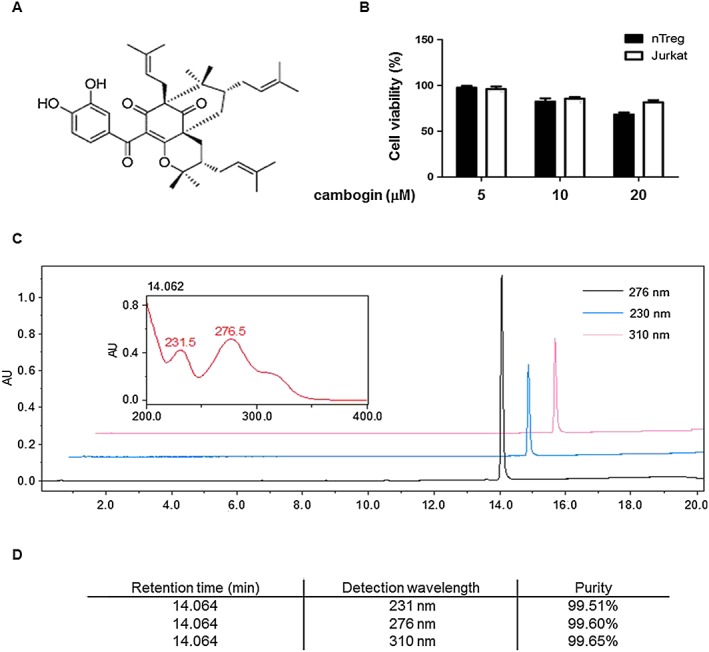
Chemical study of cambogin. (A) Chemical structure of cambogin. (B) Effect of cambogin on cell viability. Human Treg cells and Jurkat cells were incubated in the presence or absence of different concentrations of cambogin for 24 h. Cell viability was assessed using an MTT assay. Data are presented as the means ± SEM from five independent experiments. (C) UPLC chromatogram of cambogin. Column: Waters ACQUITY UPLC® BEH C18 (1.7 μm, 2.1 * 100 mm); mobile phase: (C) acetonitrile and (D) 0.1% formic acid in water, in gradient mode as follows: 0–10 min, 60–70% (C); 10–15 min, 70–90% (C); 15–18 min, 90–100% (C); and 18–20 min, 100–60% (C); detection wavelength: 231, 276 and 310 nm; and flow rate: 0.4 mL·min−1. (D) Purity detection of cambogin.
DSS (MW: 36 000–50 000 Da) was purchased from MP Biochemicals, Solon, OH, USA. PMA (P1585), ionomycin (I0634), LPS (L2880, Escherichia coli O55 : B5) and Flag (F3165) antibody were purchased from Sigma‐Aldrich. MG132 (474790) was purchased from Calbiochem (San Diego, CA, USA). The USP7 inhibitor P5091 was purchased from Selleckchem (Houston, TX, USA). The Myd88 (4283, 1:1000), phospho‐IKKα/β (2681, 1:1000), phospho‐IκBα (2859, 1:1000), IκBα (4812, 1:1000), phospho‐ERK1/2 (4377, 1:1000), ERK (4695, 1:1000), phospho‐JNK (9255, 1:1000), JNK (9252, 1:1000), phospho‐p38 (9211, 1:1000) and p38 (9212, 1:1000) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The toll‐like receptor 4 (TLR4) (sc293072, 1:1000), IKKα/β (sc7607, 1:1000), USP7 (sc30164, 1:1000), Stub1 (sc133066, 1:1000), c‐Myc (sc40, 1:500), PIM1 (sc13513, 1:500) and GAPDH (sc32233, 1:1000) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phospho‐Ser422‐Foxp3 site‐specific polyclonal antibodies were generated by Abmart (Arlington, MA, USA) with the phosphorylated peptide 418SQRP(pS)RCSN426 (Li et al., 2014). The Foxp3 (700914, 1:1000) antibody was purchased from Thermo Scientific. For in vitro experiments, cambogin was dissolved in DMSO at 10 mM concentration. We used 0.5% DMSO in cell experiments and always set up a DMSO buffer as a control.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Results
Cambogin administration ameliorated DSS‐induced colitis
We first evaluated the purity and toxicity of cambogin. The cytotoxic effect of cambogin on human Treg cells and Jurkat cells was measured using an MTT assay, revealing that cambogin did not affect cell viability at 20 μM (Figure 1B). To investigate the potential pharmacological effect of cambogin, particularly anti‐inflammatory and immune regulatory effects, we used an ovalbumin‐induced asthma model and IgE‐induced PSA model. Cambogin did not show any obvious protective effect in these experimental models (Supporting Information Figure S1). In contrast, cambogin suppressed DSS‐induced chronic colitis (Figure 2). During the study, the clinical signs of colitis, including body weight, stool consistency and rectal bleeding, were monitored daily, and the length and histology of the colons were examined on day 28. Although the body weight did not show a significant change throughout the experiment following DSS exposure, colon shortening and increased production of inflammatory cytokines, such as IFN‐γ, IL‐6 and TNF‐α, were observed. Consistent with the exacerbated clinical signs, we also observed increased histological damage, such as disrupted tissue architecture, the disappearance of intestinal crypts, infiltrated lymphocytes and oedema following DSS exposure (Figure 2A). Although the administration of cambogin did not affect the body weight of the mice (Figure 2B), it ameliorated the colon shortening (Figure 2C), histological damage (Figure 2D) and increased DAI (Figure 2E) and suppressed the expression and production of inflammatory cytokines (Figure 2F, G). These data suggest that cambogin treatment ameliorates the clinical parameters, histological damage and inflammatory cytokine production in DSS‐induced colitis.
Figure 2.

Cambogin ameliorated colitis in DSS‐treated mice. (A) Representative photomicrographs of colon sections stained with haemotoxylin and eosin (H&E) were examined. (B) Body weight was measured every day from day 1. (C) Colon length was measured after the mice had been killed on day 29. (D) Histological score and (E) DAI were calculated. (F) Cytokine production and (G) expression in the colon were measured. All data are presented as the means ± SEM of n = 7 in each group. Compared with normal control group, #P < 0.05; compared with DSS control group, *P < 0.05.
Effect of cambogin on immune cells following colitis induction
MLN and spleen cells were collected from mice with DSS‐induced colitis, subsequently stained with anti‐CD4, anti‐Foxp3, anti‐IL‐17A, anti‐IFN‐γ, anti‐T‐bet, anti‐RORγt and anti‐CTLA4 antibodies and analysed using flow cytometry. In both the spleen and MLN, DSS treatment decreased the proportion of Foxp3+ cells among the total CD4+ cell population, and cambogin administration increased the proportion of these cells, suggesting an increased number of Foxp3+ cells, as the total CD4+ numbers did not change significantly. Cambogin administration also inhibited Th1‐type cytokine IFN‐γ production and the expression of the Th1 transcription factor T‐bet. However, it did not affect IL‐17 production and RORγt expression. We also examined CTLA4 expression in Foxp3+ cells and observed decreased CTLA4 levels after DSS treatment; cambogin administration reversed this trend (Figure 3).
Figure 3.
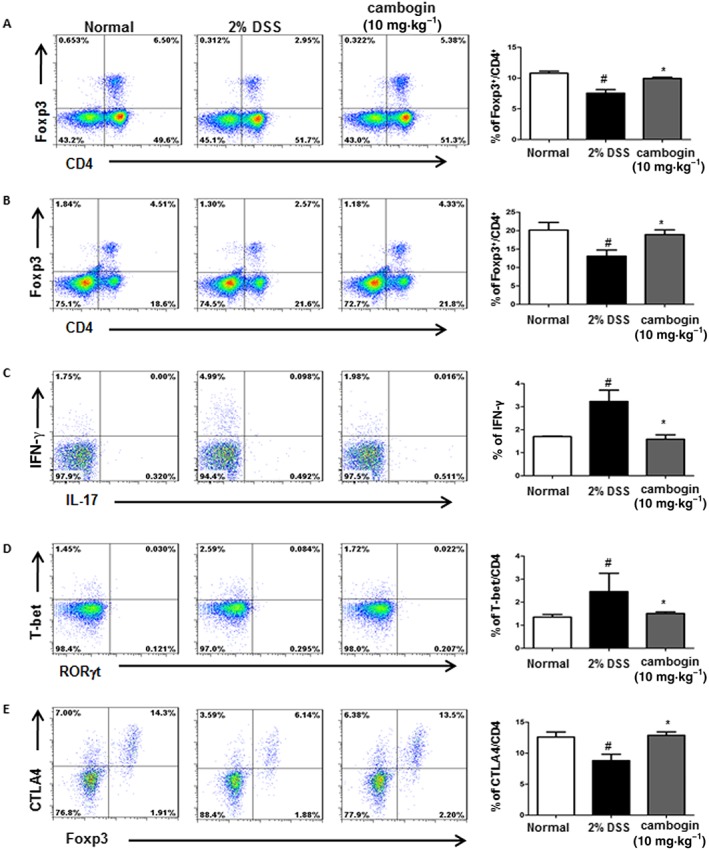
Effect of cambogin on immune cells following DSS exposure. (A and D) MLN cells and (B–E) spleen cells isolated from the colitis model were stimulated with PMA and ionomycin for 4 h, subsequently stained with antibodies against CD4, IL‐17, IFN‐γ, Foxp3, T‐bet, RORγt and CTLA4 and analysed using flow cytometry. The numbers in the corners of the FACS dot plots represent the percentage of each cell population within that quadrant as a fraction of the total number of cells. All data are presented as the means ± SEM and n = 7 mice per group. Compared with normal control group, #P < 0.05; compared with DSS control group, *P < 0.05.
Cambogin administration prevented Foxp3 loss both in vitro and in vivo
Since we observed a decreased percentage of Foxp3+ cells in both MLN and spleen cells, we considered that Treg cells might participate in the pathological process of DSS‐induced colitis. A previous study showed that Treg cells lose Foxp3 expression during inflammation (Chen et al., 2013); therefore, we examined the effect of cambogin on Treg cells during LPS stimulation. We used a Jurkat T‐cell line stably expressing Flag‐tagged Foxp3 and exposed these cells to LPS. Foxp3 protein expression was noticeably decreased upon exposure to LPS (Figure 4A), but cambogin pretreatment prevented Foxp3 loss. The proteasome inhibitor MG132 was used as a control drug, as the Foxp3 loss was proteasome‐dependent (Chen et al., 2013; van Loosdregt et al., 2013; Li et al., 2016). Similar results were observed in CD4+CD25hiCD127lo human primary Treg cells (Figure 4B). To determine how cambogin prevented Foxp3 loss, we determined the effect of cambogin on both transcription and post‐translational modification in human primary Treg cells. Interestingly, Foxp3 mRNA expression in Treg cells was unchanged by cambogin (Supporting Information Figure S1), but the mRNA expression of other Foxp3 downstream genes, such as CD25, CTLA4 and IL‐10 were up‐regulated, in accord with the down‐regulated IL‐2 level (Supporting Information Figure S2). These data suggest that the effect of cambogin on Foxp3 loss is not mediated through transcription. Next, we focused on post‐translational modification. Protein deubiquitination is an equally well‐regulated process modulated by a large family of deubiquitinating enzymes (DUBs). DUBs catalyse the removal of ubiquitin from specific protein substrates, thereby preventing protein degradation, resulting in increased target protein expression (Nijman et al., 2005). In previous studies, we showed that Foxp3 could be ubiquitinated and degraded by the E3 ubiquitin ligase Stub1 (STIP1 homology and U‐Box containing protein 1) or deubiquitinated and stabilized by the deubiquitinase USP21 (ubiquitin‐specific peptidase 21) (Chen et al., 2013; Yang et al., 2015; Li et al., 2016). In this study, we evaluated which of these factors were affected by cambogin. As shown in (Figure 4B), the treatment of Flag‐Foxp3 Jurkat cells and human Treg cells with LPS resulted in the loss of Foxp3 protein concurrently with a down‐regulation of USP7. We also determined the Foxp3 and USP7 protein levels in the DSS‐induced colitis model. In colon tissues, the Foxp3 and USP7 protein levels were down‐regulated after DSS exposure and recovered after cambogin administration (Figure 5). In a previous study, we demonstrated that PIM1 interacts with and phosphorylates Foxp3 at Ser422, thereby negatively regulating Foxp3 DNA‐binding activity (Li et al., 2014); thus, in the present study, we assessed the PIM1 level and Ser422 phosphorylation of Foxp3 in colon tissues. We first confirmed that cambogin treatment suppressed the phosphorylation of Foxp3 at Ser422 in HEK293T cells (Supporting Information Figure S3). The IHC analysis showed increased levels of PIM1 and Ser422 phosphorylation of Foxp3 after DSS exposure, and this effect was ameliorated by cambogin administration (Figure 5). Taken together, we observed a loss of Foxp3 in Treg cells treated with LPS and in the DSS‐induced colitis model, and cambogin administration ameliorated these effects. Thus, we hypothesized that cambogin prevents this loss of Foxp3 through post‐translational modifications, particularly through USP7‐mediated deubiquitination.
Figure 4.
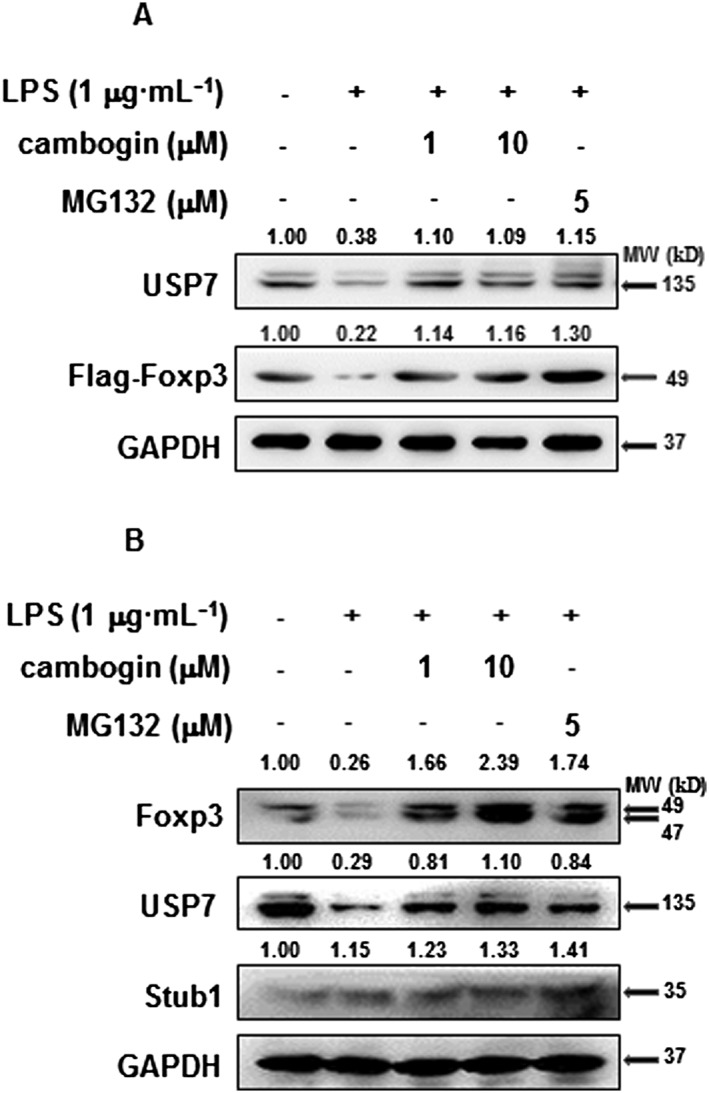
Cambogin prevents Foxp3 loss in LPS treated cells. (A) Flag‐Foxp3 or (B) human Treg cells were treated with LPS (1 μg·mL−1) for 24 h in the absence or after cambogin pretreatment for 1 h. The cells were harvested for Western blotting as indicated. Data were quantified from five independent experiments. The relative protein levels were normalized to GAPDH by using ImageJ software.
Figure 5.
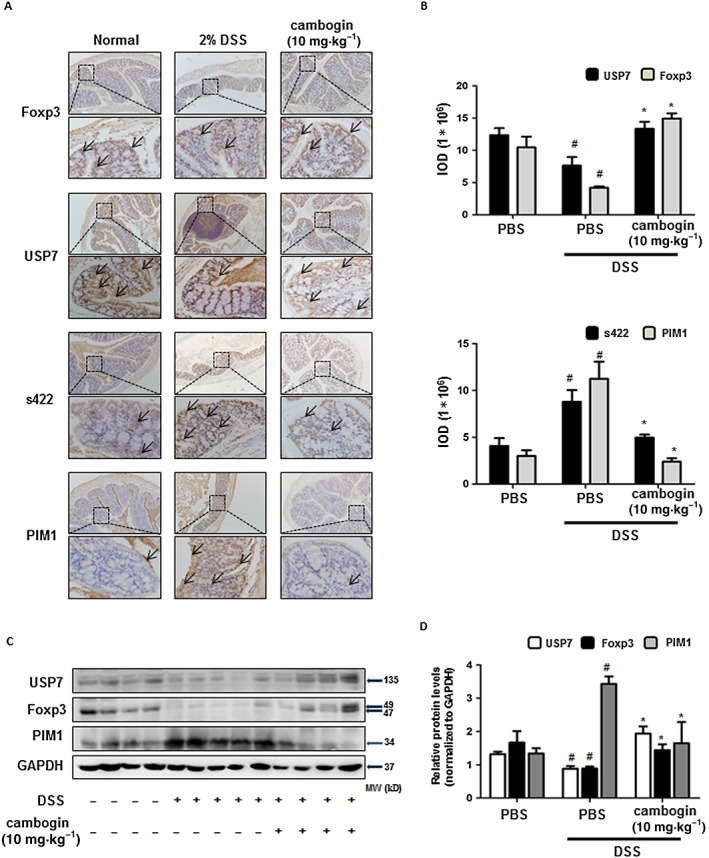
Cambogin prevents Foxp3 loss in Treg cells following DSS treatment in vivo. (A) The IHC of Foxp3, USP7, phospho‐Ser422 and PIM1 were measured. The threshold values show the quantification of the integrated OD (IOD) in the different groups using Image Pro‐Plus 6.0. (C) Cells from colon tissues were used for Western blotting. The protein levels of Foxp3, USP7 and PIM1 were normalized to GAPDH by using ImageJ software. All data are presented as the means ± SEM and n = 7 in each group. Compared with normal control group, #P < 0.05; compared with DSS control group, *P < 0.05.
Cambogin promotes USP7‐mediated Foxp3 deubiquitination, resulting in increased Foxp3 protein expression
In a previous study, van Loosdregt et al. (2013) showed that USP7 interacted with and deubiquitinated Foxp3, thereby increasing Foxp3 protein levels. We first determined whether cambogin affects the interaction between USP7 and Foxp3. Myc‐tagged USP7 and Flag‐tagged Foxp3 were cotransfected into HEK293T cells for analysis using a coimmunoprecipitation assay. The results revealed that cambogin treatment promoted the interaction between USP7 and Foxp3, and the USP7 inhibitor completely inhibited this interaction (Supporting Information Figure S4A). Moreover, we confirmed an endogenous protein interaction between USP7 and Foxp3 in human primary Treg cells after T‐cell receptor treatment (Supporting Information Figure S4B). Next, we investigated whether cambogin affects the USP‐mediated deubiquitination of Foxp3. We cotransfected Stub1, USP7, Foxp3 and His‐ubiquitin into HEK293T cells followed by coimmunoprecipitation. The results revealed that the polyubiquitination of Foxp3 was reduced through wild‐type USP7 but not the USP7CS mutant, and cambogin further reduced the polyubiquitination of Foxp3 (Figure 6A, B). Further examination of the polyubiquitination of Foxp3 in human primary Treg cells treated with cambogin confirmed this phenomenon (Figure 7A). However, cambogin did not affect Stub1‐mediated ubiquitination of Foxp3. Previous studies have shown that Lys‐48‐linked polyubiquitin chains result in the proteasomal degradation of modified proteins, whereas Lys‐63‐linked chains represent non‐proteolytic signals in several intracellular pathways (Pickart and Fushman, 2004; Han et al., 2014). To determine which lysine residue was required for Foxp3 deubiquitination by USP7, we mutated all of the lysines of ubiquitin to arginines with the exception of Lys‐48 (48K) or Lys‐63 (63K), and we observed that the USP7‐mediated deubiquitination of Foxp3 occurred via the K48 and K63 linkages (Supporting Information Figure S5). We also performed an in vitro polyubiquitin chain‐binding assay to confirm the effect of cambogin on the direct interactions between K48 or K63 polyUb chains and Foxp3 (Figure 7B), and the results indicate that cambogin treatment directly inhibited the interactions between K48 or K63 polyUb chains and Foxp3. Taken together, these results suggest that cambogin promoted USP7‐mediated Foxp3 deubiquitination through Lys‐48‐linked and Lys‐63‐linked polyubiquitination.
Figure 6.

Effect of cambogin on USP7‐mediated Foxp3 deubiquitination. HEK293 T‐cells were transfected with (A) Flag‐USP7, Myc‐Foxp3 and His‐ubiquitin or (B) Flag‐Foxp3, Myc‐USP7 (WT and C223S) and His‐ubiquitin and treated with 20 μM MG132 for 4 h prior to harvest. Cambogin or the USP7 inhibitor, P5091, was administered for 48 h after transfection. Pull down using Ni‐NTA beads; ubiquitinated Foxp3 was visualized through IB using anti‐Myc Ab or anti‐Flag Ab. The relative protein levels were normalized to GAPDH by using ImageJ software. Data were quantified from five independent experiments.
Figure 7.

Cambogin promotes USP7‐mediated Foxp3 deubiquitination through Lys‐48‐ and Lys‐63‐linked deubiquitination. Primary human Treg cells were pretreated with cambogin or USP7 inhibitor, P5091, for 1 h and stimulated with LPS (1 μg·mL−1) for 24 h. MG132 was administered for 8 h prior to harvest. Immunoprecipitation was performed as indicated. The relative protein levels were normalized to GAPDH by using ImageJ software. Data were quantified from five independent experiments.
RNA‐seq analyses
To investigate the global effects of cambogin on Treg cells, transcriptomic analyses were used. After sequencing and a series of analyses, the results showed cambogin treatment dramatically up‐regulated CCL3L1, USP18, IFIT1, SLC30A3, KITLG, IRF7, ATF3, PHLDA3, TRIM22, IFI6 and IFI44 and down‐regulated SGK3, FADS2, SCD, PLA2G4B, C4a, U2AF1L5, Kua‐UEV, ABCA1, ABCG1 and PFKFB2 on Treg cells compared with the DMSO sample (Figure 8A). We also determined whether cambogin affects the TLR4 signalling pathway stimulated by LPS treatment (Figure 8B). The results revealed that cambogin treatment did not change the expression of TLR4, MYD88 directly. However, cambogin treatment inhibited the phosphorylation of IKKα/β, IκBα, JNK and p38, indicating that it suppressed the NF‐κB and MAPK pathways.
Figure 8.
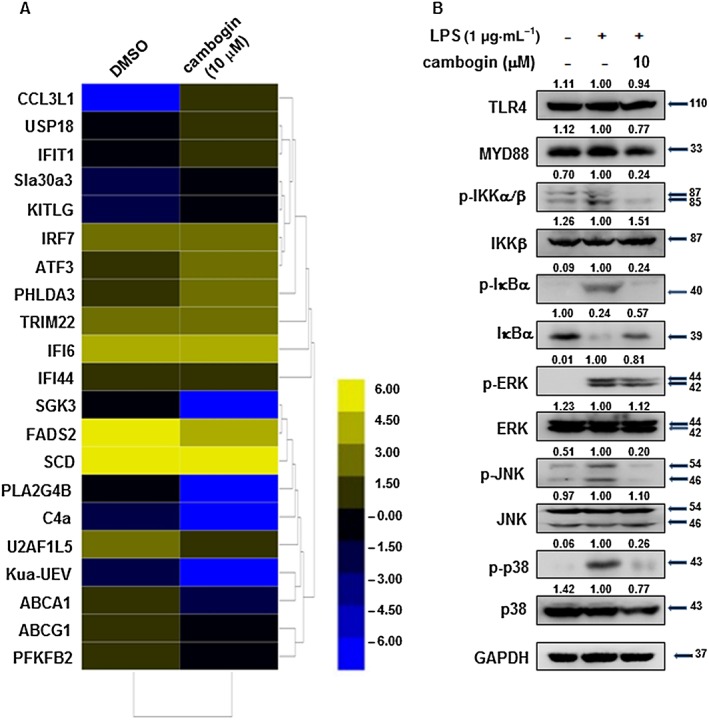
Effect of cambogin on differentially expressed genes and the TLR4 signalling pathway. (A) Heat map of the transcriptional changes identified in cambogin‐treated Treg cells compared with normal Treg cells. Yellow and blue represent high‐ and low‐levels of expression of the indicated genes respectively. The colours indicate the value of log2 fold change. (B) Effect of cambogin on TLR4 signalling pathway was assessed by determining the levels of key‐related proteins. Human Treg cells were treated with LPS (1 μg·mL−1) for 15 min after being pretreated with cambogin for 1 h. The relative protein levels were normalized to GAPDH by using ImageJ software. The Western blot data were quantified from five independent experiments.
Discussion
Treg cells are major components of the immune system, responsible for suppressing immune responses and implicated in preventing allergic, autoimmune and inflammatory disorders (Mottet et al., 2003; DiPaolo et al., 2005; Scalapino et al., 2006; Wang et al., 2006). Therefore, it is important to understand the mechanism of Treg cells in vivo under different pathophysiological processes. In the well‐characterized model of IBD in Rag1−/− mice, the infusion of CD4+CD25−CD45RBhigh effector T‐cells leads to the development of colitis, whereas Treg cells have been reported to be protective against the development of colitis (Bartczak et al., 2017). Foxp3 is the master regulator of Treg cell development, and the deletion of the gene encoding Foxp3 results in multi‐organ tissue inflammation together with a loss of Treg cell function and an excessive generation of effector T‐cells, which leads to premature death in mice (Wu et al., 2017). Du et al. (2013) found that p300 leads to dramatic effects on phenotypic changes in Treg cells, and they found the natural compound garcinol induces physical degradation of p300 through the lysosomal degradation processes, which could be used as a cancer therapy. Treg cells can also lose Foxp3 expression during inflammation through the ubiquitin–proteasome system (Chen et al., 2013; van Loosdregt et al., 2013). Van Loosdregt et al. (2013) and Li et al. (2016) showed that Foxp3 protein expression is regulated through the deubiquitinases USP7 and USP21. By interacting with and deubiquitinating Foxp3, these deubiquitinases increased Foxp3 protein levels and thus promote Treg‐cell‐mediated suppression of inflammation both in vitro and in vivo. In the present study, the natural compound cambogin stabilized Foxp3 expression in LPS treated cells and promoted the K48‐ and K63‐linked polyubiquitination of Foxp3 in a USP7‐dependent manner. Furthermore, the administration of cambogin promoted the stability of Foxp3, thereby enhancing Treg cell functionality and consequently alleviating the symptoms of colitis.
In a previous study, we demonstrated that the PIM1‐mediated phosphorylation of Foxp3 at Ser422 decreased its DNA binding activity, thereby negatively regulating Foxp3‐mediated transcriptional regulation and the suppressive activity of Treg cells (Li et al., 2014). We also observed that the inflammatory cytokine IL‐6 could induce PIM1 expression and Foxp3 phosphorylation at Ser422 in Treg cells. In colon tissue, IL‐6 production and expression were dramatically high in the DSS treated group. We speculated that this up‐regulated IL‐6 level induced PIM1 expression, which was confirmed using IHC and Western blotting (Figure 5). Although the phosphorylation of Foxp3 at Ser422 was difficult to detect in colon tissue, we observed decreased CTLA4 expression and increased IFN‐γ production followed by decreased Foxp3 and USP7 protein levels, suggesting impaired Treg cell function. Cambogin administration reversed these phenomena in vivo. Furthermore, the in vitro experiments demonstrated that cambogin treatment inhibited the interaction between PIM1 and Foxp3 and phosphorylation of Foxp3 at Ser422. Cambogin treatment also enhanced the Foxp3‐mediated transcriptional activation or repression of Treg‐associated genes, including CD25, CTLA4, IL‐10 and IL‐2, in in vitro‐expanding Treg cells, whereas the Foxp3 expression level remained unchanged. In addition to observing that LPS could disrupt the association between USP7 and Foxp3, Yang et al. (2012) also found that in colon tumours, IL‐6 inhibits USP7 expression in a STAT3‐dependent manner, and the USP7‐Foxp3 association was prolonged by this reduction in USP7 expression. These findings indicate that in inflammatory micro‐environments in vivo, both LPS and IL‐6 can affect the expression of USP7 and Foxp3 and disrupt the association between USP7‐Foxp3. Taken together, we propose that cambogin regulates Treg cell functionality through the PIM1‐mediated phosphorylation of Foxp3 at the Ser422 site.
The natural compound garcinol isolated from Garcinia indica fruit rind has shown anti‐inflammatory and anticancer effects. For example, Liao et al. (2004) demonstrated that garcinol suppresses inducible NOS and COX‐2 by down‐regulating NF‐κB pathway. Also Hong et al. (2007) showed that garcinol and its derivatives have potent growth‐inhibitory effects on all intestinal cells. But the immunoregulation effect of natural compounds from Garcinia species have not been studied yet. In this study, we found that cambogin, isolated from G. esculenta Y. H. Li prevented the decrease in Foxp3 in Treg cells during inflammation. Since natural compounds consistently display numerous multi‐target effects on pathophysiological processes, in addition to USP7 and PIM1, we supposed that cambogin might affect other signalling pathways or the expression of other genes in Treg cells. We performed RNA sequencing and compared the gene expression profiles of Treg cells with those of normal and cambogin‐treated Treg cells. The results showed that cambogin treatment dramatically up‐regulated CCL3L1, USP18, IFIT1, SLC30A3, KITLG, IRF7, ATF3, PHLDA3, TRIM22, IFI6 and IFI44 and down‐regulated SGK3, FADS2, SCD, PLA2G4B, C4a, U2AF1L5, Kua‐UEV, ABCA1, ABCG1 and PFKFB2 on Treg cells compared with the DMSO sample. Most of these genes have been implicated in immunoregulatory, infection and inflammatory processes or lipid metabolism (Struyf et al., 2001; Fang et al., 2002; Miyazaki et al., 2003; Ghosh et al., 2006; Terenzi et al., 2006; Kent et al., 2008; Ciancanelli et al., 2015; Ketscher et al., 2015), suggesting the potential effect of cambogin on infectious, immunological and metabolic‐related diseases. We also examined several major signalling molecules in Treg cell transduction, which respond to LPS stimulation. Cambogin did not alter the expression of TLR4 and Myd88, but cambogin suppressed the MAPK and IKK/IκB pathways, which play leading roles in the inflammatory process. Indeed, it is complicated and difficult to determine the specific mechanism of cambogin on Foxp3 expression, as the role of transcription factors, such as AP‐1, NF‐κB, NF‐AT and stat5, in Foxp3 expression during Treg cell development and functional regulation is still debatable (Bettelli et al., 2005; Lee et al., 2008; Tone et al., 2014; Harusato et al., 2017). Nevertheless, the results of the present study demonstrated that cambogin promotes the expression of Foxp3 expression in an inflammatory environment.
In summary, cambogin was shown to promote the expression of Foxp3 and Treg cell function both in vitro and in vivo, which could provide a potential mechanism for developing a novel therapeutic agent for Treg cell‐associated autoimmune diseases, inflammation, infections and cancer.
Author contributions
Y.L. designed the study and wrote the manuscript. Y.L., N.‐M.K., Y.‐W.J., F.‐X.Z. and R.L. performed the screening and mechanism study of cambogin in vitro and in vivo. H.Z. and D.Z. performed the compound synthesis. H.‐X.X. and B.L. supervised the study and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 (A) Seven‐week‐old female Balb/c mice were sensitized on days 0 and 14 by intraperitoneal (i.p.) injection with 20 mg of OVA (Sigma‐Aldrich, St. Louis, MO) in PBS mixed with equal volumes of alum as an adjuvant in a total volume of 200 mL. On days 22, 23 and 24, the mice were exposed to aerosolized OVA (1% OVA in PBS) or PBS for 30 min. Cambogin was administered 14 times orally every 12 h from one day before the first challenge, meanwhile the control group mice were administered with PBS. Concentration of OVA‐specific IgE in the serum was measured by ELISA. (B) In PSA test, mice were sensitized by i.v. injection of 2 mg IgE in 100 ml saline or treated with saline alone. After 24 h, the mice were challenged i.v with 2 mg DNP‐HSA in 200 ml saline after oral administration of 10 mg•kg‐1 cambogin for 1 h. Blood was collected 5 min after Ag challenge, and serum histamine concentration was determined by ELISA. All data are the means ± s.e.m. # P < 0.05 compared to non‐treated mice. The experiments were performed twice with similar results and used a minimum of seven mice in each group.
Figure S2 Effect of cambogin on gene expression in primary human Treg cells. Human Treg cells were isolated from the PBMCs of healthy donors. The mRNA was were prepared from these samples and used for the detection of Foxp3, USP7, CD25, CTLA4, IL‐10 and IL‐2 through qPCR. Data represent five independent experiments, and the error bars represent the means ± SEM. Compared with untreated cells, *P < 0.05.
Figure S3 HEK293 T cells were transfected with Myc‐Foxp3 and Flag‐PIM1, Cambogin was administered for 48 h after transfection. The indicated proteins were measured. The relative protein level were normalized to GAPDH by using Image J software. Data are representative of five independent experiments.
Figure S4 Cambogin promoted the interaction between USP7 and Foxp3. (A) HEK293T cells were transfected with Flag‐Foxp3 and Myc‐USP7. Co‐IP was performed using either anti‐Flag antibody or anti‐Myc antibody. (B) Primary human Treg cells were stimulated using anti‐CD3 and anti‐CD28 antibodies for 1 day after cambogin or USP7 inhibitor pretreatment. The cells were harvested and lysed using IP assay buffer. The cells lysate was immunoprecipitated with an anti‐USP7 antibody. Immune blotting was performed with the indicated antibodies. The relative protein level were normalized to GAPDH by using Image J software. Data are representative of five independent experiments.
Figure S5 HEK293 T cells were transfected with Myc‐USP7, His‐ubiquitin, Flag‐Foxp3 or His‐ubiquitin (WT, 48 K, and 63 K) and treated with 20 M MG132 for 4 h prior to harvest. Pull‐down using Ni‐NTA beads; ubiquitinated Foxp3 was visualized through IB using anti‐Flag Ab. The relative protein level were normalized to GAPDH by using Image J software. Data are representative of five independent experiments.
Acknowledgements
This work was financially sponsored by a grant from the Shanghai Sailing Programme (Y.L., 17YF1419500) and Shanghai Academic Research Leader Programme (B.L., 16XD1403800).
Lu, Y. , Kim, N.‐M. , Jiang, Y.‐W. , Zhang, H. , Zheng, D. , Zhu, F.‐X. , Liang, R. , Li, B. , and Xu, H.‐X. (2018) Cambogin suppresses dextran sulphate sodium‐induced colitis by enhancing Treg cell stability and function. British Journal of Pharmacology, 175: 1085–1099. doi: 10.1111/bph.14150.
Contributor Information
Bin Li, Email: binli@shsmu.edu.cn.
Hong‐Xi Xu, Email: xuhongxi88@gmail.com.
References
- Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to Pharmacology 2017/18: overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alique M, Lucio FJ, Herrero JF (2006). Vitamin A active metabolite, all‐trans retinoic acid, induces spinal cord sensitization. II. Effects after intrathecal administration. Br J Pharmacol 149: 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartczak A, Zhang J, Adeyi O, Amir A, Grant D, Gorczynski R et al (2017). Overexpression of fibrinogen‐like protein 2 protects against T cell‐induced colitis. World J Gastroenterol 23: 2673–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Dastrange M, Oukka M (2005). Foxp3 interacts with nuclear factor of activated T cells and NF‐κB to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A 102: 5138–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma G, Strober W (2003). The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol 3: 521–533. [DOI] [PubMed] [Google Scholar]
- Carpenter HA, Talley NJ (2000). The importance of clinicopathological correlation in the diagnosis of inflammatory conditions of the colon: histological patterns with clinical implications. Am J Gastroenterol 95: 878–896. [DOI] [PubMed] [Google Scholar]
- Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y et al (2013). The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 39: 272–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S et al (2015). Infectious disease. Life‐threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348: 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPaolo RJ, Glass DD, Bijwaard KE, Shevach EM (2005). CD4+CD25+ T cells prevent the development of organ‐specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J Immunol 175: 7135–7142. [DOI] [PubMed] [Google Scholar]
- Du T, Nagai Y, Xiao Y, Greene MI, Zhang H (2013). Lysosome‐dependent p300/FOXP3 degradation and limits Treg cell functions and enhances targeted therapy against cancers. Exp Mol Pathol 95: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB (2002). Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor‐mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C‐dependent intracellular pathway. Mol Cell Biol 22: 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubion WA, Jr., Loftus EV, Jr., Harmsen WS, Zinsmeister AR, & Sandborn WJ (2001). The natural history of corticosteroid therapy for inflammatory bowel disease: a population‐based study. Gastroenterology 121: 255–260. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Loper R, Gelb MH, Leslie CC (2006). Identification of the expressed form of human cytosolic phospholipase A2beta (cPLA2beta): cPLA2beta3 is a novel variant localized to mitochondria and early endosomes. J Biol Chem 281: 16615–16624. [DOI] [PubMed] [Google Scholar]
- Guilbert TW, Morgan WJ, Zeiger RS, Mauger DT, Boehmer SJ, Szefler SJ et al (2006). Long‐term inhaled corticosteroids in preschool children at high risk for asthma. N Engl J Med 354: 1985–1997. [DOI] [PubMed] [Google Scholar]
- Han L, Yang J, Wang X, Wu Q, Yin S, Li Z et al (2014). The E3 deubiquitinase USP17 is a positive regulator of retinoic acid‐related orphan nuclear receptor gammat (RORgammat) in Th17 cells. J Biol Chem 289: 25546–25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haribhai D, Chatila TA, Williams CB (2016). Immunotherapy with iTreg and nTreg cells in a murine model of inflammatory bowel disease. Methods Mol Biol 1422: 197–211. [DOI] [PubMed] [Google Scholar]
- Harrison OJ, Powrie FM (2013). Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harusato A, Abo H, Ngo VL, Yi SW, Mitsutake K, Osuka S et al (2017). IL‐36gamma signaling controls the induced regulatory T cell‐Th9 cell balance via NFkappaB activation and STAT transcription factors. Mucosal Immunol 10: 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Kwon SJ, Sang S, Ju J, Zhou JN, Ho CT et al (2007). Effects of garcinol and its derivatives on intestinal cell growth: inhibitory effects and autoxidation‐dependent growth‐stimulatory effects. Free Radic Biol Med 42: 1211–1221. [DOI] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS (2010). Inflammatory bowel disease. Annu Rev Immunol 28: 573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent D, Copley M, Benz C, Dykstra B, Bowie M, Eaves C (2008). Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin Cancer Res 14: 1926–1930. [DOI] [PubMed] [Google Scholar]
- Ketscher L, Hannss R, Morales DJ, Basters A, Guerra S, Goldmann T et al (2015). Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci U S A 112: 1577–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Gao B, Fang D (2008). FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA‐binding activity of AP‐1. Blood 111: 3599–3606. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY et al (2015). Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS One 10: e0135858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt A, Glaser A, Wegmann M, Schranz D, Seyberth H, Nusing R (2003). Expression of prostanoid receptors in human ductus arteriosus. Br J Pharmacol 138: 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu Y, Wang S, Han Z, Zhu F, Ni Y et al (2016). USP21 prevents the generation of T‐helper‐1‐like Treg cells. Nat Commun 7: 13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S et al (2014). PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem 289: 26872–26881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CH, Sang S, Liang YC, Ho CT, Lin JK (2004). Suppression of inducible nitric oxide synthase and cyclooxygenase‐2 in downregulating nuclear factor‐kappa B pathway by Garcinol. Mol Carcinog 41: 140–149. [DOI] [PubMed] [Google Scholar]
- Liu W, Guo W, Wu J, Luo Q, Tao F, Gu Y et al (2013). A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium‐induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem Pharmacol 85: 1504–1512. [DOI] [PubMed] [Google Scholar]
- Lohr J, Knoechel B, Wang JJ, Villarino AV, Abbas AK (2006). Role of IL‐17 and regulatory T lymphocytes in a systemic autoimmune disease. J Exp Med 203: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cai S, Nie J, Li Y, Shi G, Hao J et al (2016). The natural compound nujiangexanthone A suppresses mast cell activation and allergic asthma. Biochem Pharmacol 100: 61–72. [DOI] [PubMed] [Google Scholar]
- Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A et al (2005). Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 128: 1868–1878. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Jacobson MJ, Man WC, Cohen P, Asilmaz E, Friedman JM et al (2003). Identification and characterization of murine SCD4, a novel heart‐specific stearoyl‐CoA desaturase isoform regulated by leptin and dietary factors. J Biol Chem 278: 33904–33911. [DOI] [PubMed] [Google Scholar]
- Mottet C, Uhlig HH, Powrie F (2003). Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol 170: 3939–3943. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna‐Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK et al (2005). A genomic and functional inventory of deubiquitinating enzymes. Cell 123: 773–786. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Fushman D (2004). Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol 8: 610–616. [DOI] [PubMed] [Google Scholar]
- Rosen D, Kathy Hoffstadter T, Bao R, Tomaino J, Ceballos C, Russell GJ et al (2012). Analysis of current treatments used in clinical practice in a pediatric summer camp population for children with inflammatory bowel disease. Inflamm Bowel Dis 18: 1818–1824. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S (2005). Naturally arising Foxp3‐expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non‐self. Nat Immunol 6: 345–352. [DOI] [PubMed] [Google Scholar]
- Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI (2006). Suppression of disease in New Zealand Black/New Zealand White lupus‐prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol 177: 1451–1459. [DOI] [PubMed] [Google Scholar]
- Shen K, Lu F, Xie J, Wu M, Cai B, Liu Y et al (2016). Cambogin exerts anti‐proliferative and pro‐apoptotic effects on breast adenocarcinoma through the induction of NADPH oxidase 1 and the alteration of mitochondrial morphology and dynamics. Oncotarget 7: 50596–50611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Xie J, Wang H, Zhang H, Yu M, Lu F et al (2015). Cambogin induces caspase‐independent apoptosis through the ROS/JNK pathway and epigenetic regulation in breast cancer cells. Mol Cancer Ther 14: 1738–1749. [DOI] [PubMed] [Google Scholar]
- Struyf S, Menten P, Lenaerts JP, Put W, D'Haese A, De Clercq E et al (2001). Diverging binding capacities of natural LD78beta isoforms of macrophage inflammatory protein‐1alpha to the CC chemokine receptors 1, 3 and 5 affect their anti‐HIV‐1 activity and chemotactic potencies for neutrophils and eosinophils. Eur J Immunol 31: 2170–2178. [DOI] [PubMed] [Google Scholar]
- Terenzi F, Hui DJ, Merrick WC, Sen GC (2006). Distinct induction patterns and functions of two closely related interferon‐inducible human genes, ISG54 and ISG56. J Biol Chem 281: 34064–34071. [DOI] [PubMed] [Google Scholar]
- Tone Y, Kidani Y, Ogawa C, Yamamoto K, Tsuda M, Peter C et al (2014). Gene expression in the Gitr locus is regulated by NF‐kappaB and Foxp3 through an enhancer. J Immunol 192: 3915–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE et al (2013). Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg‐cell‐suppressive capacity. Immunity 39: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xu H, Wang Y, Ma J, Mao C, Shao Q et al (2006). Regulatory T cells induced by rAAV carrying the forkhead box P3 gene prevent autoimmune thyroiditis in mice. Int J Mol Med 18: 1193–1199. [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF (2007). Chemically induced mouse models of intestinal inflammation. Nat Protoc 2: 541–546. [DOI] [PubMed] [Google Scholar]
- Wu C, Chen Z, Dardalhon V, Xiao S, Thalhamer T, Liao M et al (2017). The transcription factor musculin promotes the unidirectional development of peripheral Treg cells by suppressing the TH2 transcriptional program. Nat Immunol 18: 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin B, Tao F, Wang Y, Liu H, Ma C, Xu P (2017). Coordination of metabolic pathways: enhanced carbon conservation in 1,3‐propanediol production by coupling with optically pure lactate biosynthesis. Metab Eng 41: 102–114. [DOI] [PubMed] [Google Scholar]
- Yamada A, Arakaki R, Saito M, Tsunematsu T, Kudo Y, Ishimaru N (2016). Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J Gastroenterol 22: 2195–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Xu P, Han L, Guo Z, Wang X, Chen Z et al (2015). Cutting edge: ubiquitin‐specific protease 4 promotes Th17 cell function under inflammation by deubiquitinating and stabilizing RORgammat. J Immunol 194: 4094–4097. [DOI] [PubMed] [Google Scholar]
- Yang Z, Huo S, Shan Y, Liu H, Xu Y, Yao K et al (2012). STAT3 repressed USP7 expression is crucial for colon cancer development. FEBS Lett 586: 3013–3017. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang DD, Lao YZ, Fu WW, Liang S, Yuan QH et al (2014). Cytotoxic and anti‐inflammatory prenylated benzoylphloroglucinols and xanthones from the twigs of Garcinia esculenta . J Nat Prod 77: 1700–1707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Seven‐week‐old female Balb/c mice were sensitized on days 0 and 14 by intraperitoneal (i.p.) injection with 20 mg of OVA (Sigma‐Aldrich, St. Louis, MO) in PBS mixed with equal volumes of alum as an adjuvant in a total volume of 200 mL. On days 22, 23 and 24, the mice were exposed to aerosolized OVA (1% OVA in PBS) or PBS for 30 min. Cambogin was administered 14 times orally every 12 h from one day before the first challenge, meanwhile the control group mice were administered with PBS. Concentration of OVA‐specific IgE in the serum was measured by ELISA. (B) In PSA test, mice were sensitized by i.v. injection of 2 mg IgE in 100 ml saline or treated with saline alone. After 24 h, the mice were challenged i.v with 2 mg DNP‐HSA in 200 ml saline after oral administration of 10 mg•kg‐1 cambogin for 1 h. Blood was collected 5 min after Ag challenge, and serum histamine concentration was determined by ELISA. All data are the means ± s.e.m. # P < 0.05 compared to non‐treated mice. The experiments were performed twice with similar results and used a minimum of seven mice in each group.
Figure S2 Effect of cambogin on gene expression in primary human Treg cells. Human Treg cells were isolated from the PBMCs of healthy donors. The mRNA was were prepared from these samples and used for the detection of Foxp3, USP7, CD25, CTLA4, IL‐10 and IL‐2 through qPCR. Data represent five independent experiments, and the error bars represent the means ± SEM. Compared with untreated cells, *P < 0.05.
Figure S3 HEK293 T cells were transfected with Myc‐Foxp3 and Flag‐PIM1, Cambogin was administered for 48 h after transfection. The indicated proteins were measured. The relative protein level were normalized to GAPDH by using Image J software. Data are representative of five independent experiments.
Figure S4 Cambogin promoted the interaction between USP7 and Foxp3. (A) HEK293T cells were transfected with Flag‐Foxp3 and Myc‐USP7. Co‐IP was performed using either anti‐Flag antibody or anti‐Myc antibody. (B) Primary human Treg cells were stimulated using anti‐CD3 and anti‐CD28 antibodies for 1 day after cambogin or USP7 inhibitor pretreatment. The cells were harvested and lysed using IP assay buffer. The cells lysate was immunoprecipitated with an anti‐USP7 antibody. Immune blotting was performed with the indicated antibodies. The relative protein level were normalized to GAPDH by using Image J software. Data are representative of five independent experiments.
Figure S5 HEK293 T cells were transfected with Myc‐USP7, His‐ubiquitin, Flag‐Foxp3 or His‐ubiquitin (WT, 48 K, and 63 K) and treated with 20 M MG132 for 4 h prior to harvest. Pull‐down using Ni‐NTA beads; ubiquitinated Foxp3 was visualized through IB using anti‐Flag Ab. The relative protein level were normalized to GAPDH by using Image J software. Data are representative of five independent experiments.