

Abstract
Hypertrophic cardiomyopathy (HCM) is a serious monogenic disease characterized by cardiac hypertrophy, fibrosis, sudden cardiac death, and heart failure. Previously, we identified that miR-139-5p was down-regulated in HCM patients. However, the regulatory effects of miR-139-5p remain unclear. Thus, we investigated the role of miR-139-5p in the regulation of cardiac hypertrophy. The expression of miR-139-5p in left ventricular tissues in HCM patients and mice subjected to transverse aortic constriction (TAC) was significantly down-regulated. Knockdown of miR-139-5p expression in neonatal rat cardiomyocytes (NRCMs) induced cardiomyocyte enlargement and increased atrial natriuretic polypeptide (ANP) expression. Overexpression of miR-139-5p antagonized isoproterenol (ISO)-induced cardiomyocyte enlargement and ANP/brain natriuretic peptide (BNP) up-regulation. More importantly, we found that c-Jun expression was inhibited by miR-139-5p in NRCMs. Knockdown of c-Jun expression significantly attenuated cardiac hypertrophy induced by miR-139-5p deprivation. Our data indicated that miR-139-5p was down-regulated in the hearts of HCM patients and that it inhibited cardiac hypertrophy by targetting c-Jun expression.
Keywords: c-Jun, hypertrophic cardiomyopathy, isoproterenol, miR-139-5p
Hypertrophic cardiomyopathy (HCM) is a monogenic disease with an estimated prevalence of 0.2% in the population [1]. This disease is characterized by cardiac hypertrophy, fibrosis, sudden cardiac death, and heart failure [2,3]. Although it has been revealed that HCM is mostly caused by mutations in sarcomere genes [4–6], the regulatory mechanisms of cardiac hypertrophy in HCM are not fully understood.
MicroRNAs (miRs) are intracellular small, non-coding RNAs that mainly down-regulate specific target genes by binding to the 3′-UTR of corresponding mRNAs [7]. Accumulating studies have revealed that miRs play important roles in regulating cardiac remodeling in HCM, thereby providing potential therapeutic targets for this disease [8–11]. By using microarray analyses, we previously screened for miRs in left ventricular tissues that were differentially expressed between HCM patients and healthy donors [9]. We identified that miR-139-5p was one of the most down-regulated miRs in the hearts of HCM patients [9]. However, whether miR-139-5p participates in the regulation of cardiac hypertrophy remains unclear.
The β-adrenergic receptor (β-AR) plays an important role in the regulation of cardiac excitation–contraction; however, hyperactivation of β-AR leads to cardiac remodeling [12,13]. The stimulation of β-AR activates the downstream activating protein-1 (AP-1), which then transmits signaling to induce cardiac hypertrophy [14,15]. AP-1 is a transcription factor consisting of a group of immediate early genes, such as c-Jun and c-Fos. They form homo- or heterodimers to regulate the expression of their target genes. By using online bioinformative assays (www.targetscan.org), we predicted that c-Jun might be a target of miR-139-5p in cardiomyocytes, which might participate in the regulation of cardiac hypertrophy. Therefore, we investigated this aspect in the present study. We found that isoproterenol (ISO), a potent β-AR agonist, strongly induced the expression of c-Jun, whereas exogenous addition of miR-139-5p mimic antagonized its expression and attenuated ISO-induced cardiac hypertrophy.
Materials and methods
Ethics statement
All protocols pertaining to human subjects were approved by the Ethics Committee of Fuwai Hospital (Beijing, China) in accordance with the Declaration of Helsinki. Written informed consent was obtained from all the participants. Left ventricular tissues were obtained as previously described [9]. All the protocols pertaining to animals were approved by the Ethics Committee for Animal Study of Fuwai Hospital (Beijing, China) (approval number: 0088-R/M-300/200-GZ(X)).
Transverse aortic constriction operation
Transverse aortic constriction (TAC) operation in mice was performed as previously described [16]. In brief, 6–8 weeks old male C57BL/6 mice were weighed and anesthetized with 2.5% avertin (0.018 ml/g) through intraperitoneal injection. The transverse aorta was ligated for TAC operation. Sham-operated mice served as controls. All mice were killed at 4 weeks post operation, and the hearts were harvested for miR-139-5p expression analysis.
Cell culture
Primary neonatal rat cardiomyocytes (NRCMs) were cultured as previously described [17]. In brief, ventricles from neonatal Wistar rats were isolated and cut into small pieces. The tissues were washed with PBS and digested with 0.06% collagenase II (Worthington Biochemical Corporation, Lakewood, NJ, U.S.A.) at 37°C. The cell suspension was centrifuged and resuspended with Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS. For differential adhesion, the cells were maintained in an atmosphere with 5% CO2 at 37°C for 90 min. The supernatants were resuspended with complete DMEM containing 0.1 mM bromodeoxyuridine, seeded in corresponding plates and routinely cultured for 24 h. Then, the medium was replaced with DMEM containing 1% FBS for experiments.
Cell transfection and treatment
NRCMs were transiently transfected with miR-139-5p mimics or inhibitors (GenePharma, Suzhou, China) at a final concentration of 100 nM. For the knockdown of c-Jun expression, a predesigned specific siRNA was used (Silencer® Select, s127979, Thermo Fisher, Carlsbad, CA, U.S.A.). All the above small RNAs were transfected by using Lipofectamine® RNAiMAX Reagent (Thermo Fisher) according to the manufacturer’s protocol. For inducing cardiac hypertrophy in vitro, NRCMs were treated with 100 nmol/l ISO for 24 h [18].
Determination of cell surface area
NRCMs (5 × 105 cells/ml) were seeded in 24-well plates and routinely cultured. The cells were fixed with 4% paraformaldehyde and permeabilized with 1% Triton X-100 in PBS for 5 min. The cells were then washed with PBS and stained with Texas Red-Phalloidin (Thermo Fisher) for 30 min at room temperature. The nuclei were stained with 0.1 μg/ml DAPI for 5 min. The cells were visualized with a fluorescence microscope, and the cell surface area was measured by analyzing 30 NRCMs from at least five random fields with ImagePro Plus 6.0 Software (Media Cybernetics, Bethesda, MD, U.S.A.).
Quantitative real-time PCR analysis
Total RNA was extracted by using TRIzol reagent (Thermo Fisher), quantitated, and normalized. To analyze the expression of miR-139-5p, a specific primer containing a stem loop structure was used for cDNA synthesis with a cDNA synthesis kit (Takara, Dalian, China): 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACTGGAGA-3′. The expression of miR-139-5p was determined using the following primers: forward, 5′-ACACTCCAGCTGGGTCTACAGTGCACGTGTC-3′ and reverse, 5′-TGGTGTCGTGGAGTCG-3′, and normalized to U6 expression determined with the following primers: forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse, 5′-AACGCTTCACGAATTTGCGT-3′.
For the routine quantitation of mRNAs, cDNA was synthesized with a cDNA Synthesis Kit (Takara). The following specific primers for real-time PCR were used: atrial natriuretic polypeptide (ANP) forward, 5′-GGGCTCCTTCTCCATCAC-3′ and reverse, 5′-CGGCATCTTCTCCTCCAG-3′; brain natriuretic peptide (BNP) forward, 5′-AGAACAATCCACGATGCAGAAG-3′ and reverse, 5′-AAACAACCTCAGCCCGTCACA-3′; and 18S rRNA forward, 5′-CTTAGAGGGACAAGTGGCG-3′ and reverse, 5′-GGACATCTAAGGGCATCACA-3′. The relative expression of target genes was determined by using the ΔΔCt (cycle threshold) method.
Western blotting
Total protein was extracted with RIPA lysis buffer, quantitated, and normalized. Approximately 20 μg protein from each sample was separated using SDS/PAGE and blotted on to a nitrocellulose membrane. The membrane was blocked with 5% BSA at room temperature for 1 h and then incubated with the following primary antibodies at 4°C overnight: rabbit anti-Akt, anti-p-Akt, anti-β-catenin, anti-GAPDH (Cell Signaling Technology, Beverly, MA, U.S.A.), anti-c-Jun, anti-IGF-1 receptor (anti-IGF-1R) (Santa Cruz Technology, Dallas, TX, U.S.A.), anti-myocardin (anti-MyoCD) (Sigma, St. Louis, MO, U.S.A.), and anti-Wnt1 (Abcam, Cambridge, MA, U.S.A.). Then, the membrane was washed three times with TBS Tween-20 (TBS-T) and incubated with goat anti-rabbit secondary antibody (Cell Signaling Technology) at room temperature for 1 h. The bands were visualized and analyzed using Quantity One software V 4.6.2 (Bio–Rad, Hercules, CA, U.S.A.).
Statistical analysis
Statistical analyses were performed with IBM SPSS 19.0 Software (SPSS, Inc., Chicago, IL, U.S.A.). Two-tailed Student’s t test or ANOVA was used to determine the differences. The data are expressed as the mean ± S.E.M., and P<0.05 was considered statistically significant.
Results
miR-139-5p is down-regulated in hypertrophic hearts
To validate whether miR-139-5p is down-regulated in hypertrophic hearts, we detected its expression in left ventricular tissues from 16 HCM patients and 8 healthy donors. We found that the expression of miR-139-5p in left ventricular tissues of HCM patients was down-regulated to 57.6% of that in the healthy donors (P<0.05, power: 0.505) (Figure 1A). We then performed TAC operation to induce cardiac hypertrophy in mice. Consistently, the expression of miR-139-5p in the hearts of TAC-operated mice was significantly down-regulated compared with that in sham-operated mice, indicating that miR-139-5p is down-regulated in hypertrophic hearts (P<0.01, power: 0.765) (Figure 1B).
Figure 1. miR-139-5p is down-regulated in hypertrophic hearts.
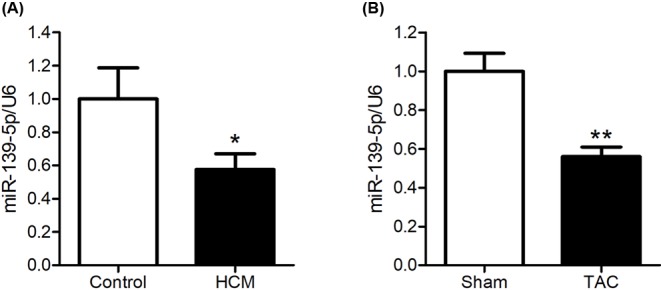
(A) Expression of miR-139-5p in 16 HCM patients and 8 healthy donors (Control). (B) Expression of miR-139-5p in mice subjected to TAC (n=8); sham-operated mice were used as controls (n=8). Real-time PCR assay was performed; and U6 was employed as an internal control. Data represent the mean ± S.E.M. *P<0.05 and **P<0.01 compared with the control group.
miR-139-5p is required for maintaining normal cell size in cardiomyocytes
To investigate the role of miR-139-5p in cardiac hypertrophy, we knocked down its expression in NRCMs by transfecting a specific miR inhibitor. The levels of miR-139-5p were down-regulated to 25.4% of that in control NRCMs at 48 h post transfection (Figure 2A). We then measured changes in cell surface area by staining NRCMs with Texas Red-conjugated Phalloidin. We found that the cell surface area was significantly increased in miR-139-5p-deprived NRCMs (Figure 2B). ANP and BNP are two known biomarkers of cardiac hypertrophy. The knockdown of miR-139-5p induced a 2.06-fold increase in ANP expression (P<0.05); however, the expression of BNP was unchanged (Figure 2C). These results indicated that miR-139-5p is required to maintain normal cell size in cardiomyocytes.
Figure 2. Knockdown of miR-139-5p expression induces cardiac hypertrophy.

(A–C) NRCMs were transfected with miR-139-5p inhibitor or the inhibitor control RNA for 48 h. Intracellular F-actin was stained with Texas Red-Phalloidin, and nuclei were stained with DAPI; scale bars: 100 μm (A). Cell surface area was quantitated with ImagePro Plus 6.0 from at least 30 cells in three independent experiments. Data are expressed as the mean ± S.E.M. *P<0.05 compared with the controls (B). Expression of ANP and BNP were quantitated by using real-time PCR. 18S rRNA was used as the loading control. Relative expression was indicated as the mean ± S.E.M. (n=4), and P-values are shown as indicated (C).
Overexpression of miR-139-5p attenuates ISO-induced cardiac hypertrophy
Since miR-139-5p participates in the regulation of cell size in cardiomyocytes, we wanted to explore whether the exogenous administration of miR-139-5p is able to antagonize cardiac hypertrophy. ISO is a β-AR agonist that is widely used to induce cardiac hypertrophy. Treating NRCMs with 100 nmol/l ISO for 24 h induced a significant increase in cell size (Figure 3A,B). Pre-transfection of NRCMs with the miR-139-5p mimic for 48 h attenuated ISO-induced cellular enlargement (Figure 3A,B). Consistently, the expression of ANP and BNP was up-regulated by ISO treatment by 2.11- and 7.68-fold, respectively (Figure 3C), whereas the transfection of NRCMs with the miR-139-5p mimic attenuated the ISO-induced increase in ANP and BNP expression (Figure 3C). In addition, miR-139-5p did not affect cell surface area or ANP/BNP levels under basal conditions (Figure 3A–C). These data indicated that miR-139-5p attenuates ISO-induced cardiac hypertrophy in vitro.
Figure 3. Overexpression of miR-139-5p attenuates ISO-induced cardiac hypertrophy.

(A–C) NRCMs were transfected with the miR-139-5p mimic or scrambled control small RNAs for 48 h, followed by ISO treatment for 24 h. Intracellular F-actin and nuclei were stained with Texas Red-Phalloidin and DAPI, respectively. Scale bars: 100 μm (A). Cell surface area was measured using ImagePro Plus 6.0 software from at least 30 cells in three independent experiments (B). Expression of ANP and BNP were quantitated by using real-time PCR and normalized to the level of 18S rRNA (n=3 per group) (C). Data are indicated as the mean ± S.E.M., and P-values are shown as indicated.
miR-139-5p down-regulates c-Jun expression in ISO-induced cardiac hypertrophy
miRs specifically bind to the 3′-UTR of its target mRNAs to regulate their expression. By using TargetScan (http://www.targetscan.org/vert_71/), an online tool for predicting miR target genes, we identified c-Jun, IGF-1R, MyoCD, Wnt1, and β-catenin as candidate target genes of miR-139-5p. We transfected NRCMs with the miR-139-5p mimic for 48 h and found that the levels of c-Jun, IGF-1R, and β-catenin were all down-regulated, whereas the levels of MyoCD and Wnt1 were unaffected (Figure 4A,B). Interestingly, miR-139-5p overexpression inhibited the ISO-induced up-regulation of c-Jun expression (Figure 4A,B). In contrast, the knockdown of endogenous miR-139-5p increased the level of c-Jun (Figure 4E,F). These results indicated that miR-139-5p inhibits the expression of c-Jun in cardiomyocytes and thus might play an important role in the regulation of cardiac hypertrophy.
Figure 4. miR-139-5p targets c-Jun expression in cardiomyocytes.

(A–D) NRCMs were treated as indicated. Levels of c-Jun, IGF-1R, MyoCD, Wnt1, and β-catenin (A) and the phosphorylation status of Akt (B) were detected using Western blotting. The relative expression intensity (B,D) was obtained from three independent experiments were performed. (E–I) siRNAs for knocking down (KD) c-Jun expression were co-transfected into to NRCMs with the miR-139-5p inhibitor or the inhibitor control RNA for 48 h. Expression of c-Jun was determined using Western blotting (E) and the relative expression of c-Jun was quantitated (F). F-actin and nuclei were stained with Texas Red-Phalloidin and DAPI, respectively. Scale bars: 100 μm (G). Cell surface area was quantitated from at least 30 cells in three independent experiments (H). Expression of ANP was quantitated by using real-time PCR and normalized to the level of 18S rRNA (n=3 per group) (I). Data are indicated as the mean ± S.E.M., *P<0.05 and **P<0.01 compared between the indicated groups.
The expression of IGF-1R was down-regulated by miR-139-5p in NRCMs, although ISO alone slightly down-regulated IGF-1R expression (Figure 4A,B). The IGF-1R signaling pathway has been reported to regulate cardiac hypertrophy via PI3K/Akt [19]. As expected, miR-139-5p suppressed ISO-induced hyperphosphorylation of Akt in NRCMs, indicating that the IGF-1R/Akt pathway might also contribute to the antihypertrophic effects of miR-139-5p (Figure 4C,D). Wnt1/β-catenin has been reported to be regulated by miR-139-5p [20]. We observed that miR-139-5p indeed down-regulated β-catenin expression under basal conditions, but it failed to inhibit Wnt1 or β-catenin expression in response to ISO stimulation (Figure 4A,B).
Inhibition of c-Jun activation suppresses miR-139-5p knockdown-induced cardiac hypertrophy
As the expression of c-Jun was found to be regulated by miR-139-5p in NRCMs, we proposed that the up-regulation of c-Jun might be required in miR-139-5p deprivation-induced cardiac hypertrophy. To investigate this aspect, we knocked down c-Jun expression by transfecting c-Jun-specific siRNAs into miR-139-5p-deprived NRCMs. The knockdown of miR-139-5p expression in NRCMs significantly increase c-Jun levels, whereas the knockdown of c-Jun expression prevented this increase (Figure 4E,F). As expected, the increase in cell surface area and the up-regulation of ANP expression induced by miR-139-5p deprivation were restored by c-Jun knockdown (Figure 4G–I). These results indicated that c-Jun is indispensable in miR-139-5p deprivation-mediated cardiac hypertrophy in vitro.
Discussion
Pathological cardiac remodeling in HCM is associated with the deregulated expression of many intracellular miRs [8–11]. In the present study, we reported that miR-139-5p was down-regulated in heart tissues of HCM patients. The knockdown of miR-139-5p expression in NRCMs induced cardiac hypertrophy, whereas the exogenous overexpression of miR-139-5p antagonized ISO-induced cardiac hypertrophy. These findings indicated that miR-139-5p acts an anti-hypertrophic miR in the heart.
HCM is a monogenic myocardial disease, which is characterized by cardiac hypertrophy, sarcomere disarrangement, cardiac fibrosis, heart failure, and cardiac sudden death [2]. By using microarray analysis, we found that miR-139-5p was significantly down-regulated in the left ventricular tissues of HCM patients. Moreover, the expression of miR-139-5p was down-regulated in the hearts of TAC-operated mice, and knockdown of miR-139-5p in NRCMs induced cardiomyocyte enlargement. These data indicated that miR-139-5p is indispensable in normal cardiomyocytes.
The β-AR signaling pathway, which is classically activated by catecholamines, is one of the well-known pathways in the induction of cardiac hypertrophy [12,13]. The activation of β-AR regulates cardiac excitation–contraction and leads to the increase in heart rate and cardiomyocyte contractility [21–23]. The hyperactivation of β-AR results in cardiac hypertrophy, fibrosis, senescence, inflammation, and cardiomyocyte apoptosis and necrosis [24–29]. The treatment of NRCMs with ISO caused cardiomyocyte enlargement and increased the expression of ANP and BNP. We observed that miR-139-5p overexpression attenuated ISO-induced cardiac hypertrophy, which indicated that miR-139-5p plays an antihypertrophic role in the heart. It has been reported that the stimulation of β-AR can induce the activation of AP-1, which is associated with the activation of the CaMKII, ERK, and JNK pathways [14,15]. AP-1 acts as a transcription factor for a group of immediate early genes, including c-Jun, c-Fos, JunB, and JunD. These proteins form homo- or heterodimers and bind to the promoters of various genes to regulate their expression, which subsequently induces cardiac hypertrophy [14,30–32]. In the present study, we observed that c-Jun expression was strongly induced by ISO-treatment, whereas miR-139-5p inhibited this increase. Consistently, the knockdown of c-Jun expression significantly attenuated miR-139-5p deprivation-induced cardiac hypertrophy. The miR-139-5p/c-Jun axis has been reported to form a feedback loop in human gastric cancer cells [33]. Our data indicated that the down-regulation of c-Jun is indispensable for the anti-hypertrophic effects of miR-139-5p in cardiomyocytes.
The activation of β-AR induced the phosphorylation of Akt/GSK3β in NRCMs, which is associated with the up-regulation of ANP expression [34]. In colitis-associated tumorigenesis, miR-139-5p inhibits the cross-talk between the PI3K/Akt and Wnt pathways by targetting IGF-1R [35]. Our results indicated that overexpression of miR-139-5p in NRCMs antagonizes ISO-induced activation of Akt. miR-139-5p also inhibits the expression of IGF-1R in cells both under basal and ISO-stimulated conditions, indicating that the down-regulation of IGF-1R and inhibition of Akt might be involved in preventing cardiac hypertrophy. Mi et al. [20] reported that Wnt1 and β-catenin were directly targetted by miR-139-5p in C2C12 cells. In our study, we observed that miR-139-5p indeed down-regulated β-catenin expression under basal conditions; however, it failed to antagonize the ISO-induced up-regulation of β-catenin in NRCMs. The expression of Wnt1 was not altered by miR-139-5p overexpression in NRCMs. These data indicated that the Wnt1/β-catenin pathway might not be associated with the antihypertrophic effects of miR-139-5p. In addition, we found that the predicted MyoCD was not a target of miR-139-5p in NRCMs, as its expression was not altered under basal or ISO-stimulated conditions.
Taken together, our study highlights that miR-139-5p is a novel antihypertrophic miR in cardiomyocytes. miR-139-5p attenuates cardiac hypertrophy possibly through the down-regulation of c-Jun expression in vitro. Our data indicated that the exogenous delivery of the miR-139-5p mimic to the heart might be a potential therapeutic strategy for pathological cardiac hypertrophy. These findings provide evidences that the miR-139-5p/c-Jun axis is a novel target for the prevention and treatment for cardiac remodeling. The future study will focus on the mechanism of miR-139-5p expression and its association with c-Jun.
Abbreviations
- ANP
atrial natriuretic polypeptide
- AP-1
activating protein-1
- BNP
brain natriuretic peptide
- DMEM
Dulbecco’s modified Eagle’s medium
- HCM
hypertrophic cardiomyopathy
- IGF-1R
IGF-1 receptor
- ISO
isoproterenol
- miR
microRNA
- MyoCD
myocardin
- NRCM
neonatal rat cardiomyocyte
- TAC
transverse aortic constriction
- β-AR
β-adrenergic receptor
Author contribution
W.J.-z. and W.H. designed the research. S.M., Q.W., and L.J.-h. performed the experiments. H.R.-t., S.L., and J.M. contributed to the analysis of the data. S.M. and W.S.-y. wrote this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 81500236].
Competing interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Ommen S.R. (2011) Hypertrophic cardiomyopathy. Curr. Probl. Cardiol. 36, 409–453 10.1016/j.cpcardiol.2011.06.001 [DOI] [PubMed] [Google Scholar]
- 2.Marston S., Copeland O., Gehmlich K., Schlossarek S. and Carrier L. (2012) How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J. Muscle Res. Cell Motil. 33, 75–80 10.1007/s10974-011-9268-3 [DOI] [PubMed] [Google Scholar]
- 3.Maron B.J. and Maron M.S. (2013) Hypertrophic cardiomyopathy. Lancet 381, 242–255 10.1016/S0140-6736(12)60397-3 [DOI] [PubMed] [Google Scholar]
- 4.Schramm C., Fine D.M., Edwards M.A., Reeb A.N. and Krenz M. (2012) The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am. J. Physiol. Heart Circ. Physiol. 302, H231–H243 10.1152/ajpheart.00665.2011 [DOI] [PubMed] [Google Scholar]
- 5.Niimura H., Bachinski L.L., Sangwatanaroj S., Watkins H., Chudley A.E., McKenna W. et al. (1998) Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N. Engl. J. Med. 338, 1248–1257 10.1056/NEJM199804303381802 [DOI] [PubMed] [Google Scholar]
- 6.Liu X., Ye B., Miller S., Yuan H., Zhang H., Tian L. et al. (2012) Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol. 32, 4493–4504 10.1128/MCB.01092-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robins H., Li Y. and Padgett R.W. (2005) Incorporating structure to predict microRNA targets. Proc. Natl. Acad. Sci. U.S.A. 102, 4006–4009 10.1073/pnas.0500775102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ntelios D., Meditskou S., Efthimiadis G., Pitsis A., Nikolakaki E., Girtovitis F. et al. (2017) Elevated plasma levels of miR-29a are associated with hemolysis in patients with hypertrophic cardiomyopathy. Clin. Chim. Acta. 471, 321–326 10.1016/j.cca.2017.07.004 [DOI] [PubMed] [Google Scholar]
- 9.Song L., Su M., Wang S., Zou Y., Wang X., Wang Y. et al. (2014) MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 18, 2266–2274 10.1111/jcmm.12380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernardo B.C., Gao X.M., Tham Y.K., Kiriazis H., Winbanks C.E., Ooi J.Y. et al. (2014) Silencing of miR-34a attenuates cardiac dysfunction in a setting of moderate, but not severe, hypertrophic cardiomyopathy. PLoS ONE 9, e90337 10.1371/journal.pone.0090337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danielson L.S., Park D.S., Rotllan N., Chamorro-Jorganes A., Guijarro M.V., Fernandez-Hernando C. et al. (2013) Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 27, 1460–1467 10.1096/fj.12-221994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi D.J., Koch W.J., Hunter J.J. and Rockman H.A. (1997) Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increased beta-adrenergic receptor kinase. J. Biol. Chem. 272, 17223–17229 10.1074/jbc.272.27.17223 [DOI] [PubMed] [Google Scholar]
- 13.Hakamata N., Hamada H., Ohsuzu F. and Nakamura H. (1997) Cardiac beta-adrenergic signaling pathway alteration in isoproterenol-induced cardiac hypertrophy in male Sprague–Dawley rats. Jpn. Heart J. 38, 849–857 10.1536/ihj.38.849 [DOI] [PubMed] [Google Scholar]
- 14.Mani S.K., Egan E.A., Addy B.K., Grimm M., Kasiganesan H., Thiyagarajan T. et al. (2010) beta-Adrenergic receptor stimulated Ncx1 upregulation is mediated via a CaMKII/AP-1 signaling pathway in adult cardiomyocytes. J. Mol. Cell Cardiol. 48, 342–351 10.1016/j.yjmcc.2009.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang N., Guan P., Zhang J.P., Li Y.Q., Chang Y.Z., Shi Z.H. et al. (2011) Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways. J. Cell. Biochem. 112, 1920–1929 10.1002/jcb.23112 [DOI] [PubMed] [Google Scholar]
- 16.Rockman H.A., Ross R.S., Harris A.N., Knowlton K.U., Steinhelper M.E., Field L.J. et al. (1991) Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 88, 8277–8281 10.1073/pnas.88.18.8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su M., Wang J., Wang C., Wang X., Dong W., Qiu W. et al. (2015) MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 22, 986–999 10.1038/cdd.2014.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo D, Kassiri Z, Basu R, Chow FL, Kandalam V, Damilano F et al. (2010) Loss of PI3Kγ enhances cAMP-dependent MMP remodeling of the myocardial N-cadherin adhesion complexes and extracellular matrix in response to early biomechanical stress. Circ. Res. 107, 1275–1289 10.1161/CIRCRESAHA.110.229054 [DOI] [PubMed] [Google Scholar]
- 19.Huynh K., McMullen J.R., Julius T.L., Tan J.W., Love J.E., Cemerlang N. et al. (2010) Cardiac-specific IGF-1 receptor transgenic expression protects against cardiac fibrosis and diastolic dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes 59, 1512–1520 10.2337/db09-1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi L., Li Y., Zhang Q., Zhao C., Peng Y., Yang G. et al. (2015) MicroRNA-139-5p regulates C2C12 cell myogenesis through blocking Wnt/beta-catenin signaling pathway. Biochem. Cell. Biol. 93, 8–15 10.1139/bcb-2014-0079 [DOI] [PubMed] [Google Scholar]
- 21.Pereira L., Bare D.J., Galice S., Shannon T.R. and Bers D.M. (2017) beta-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J. Mol. Cell Cardiol. 108, 8–16 10.1016/j.yjmcc.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bobin P., Varin A., Lefebvre F., Fischmeister R., Vandecasteele G. and Leroy J. (2016) Calmodulin kinase II inhibition limits the pro-arrhythmic Ca2+ waves induced by cAMP-phosphodiesterase inhibitors. Cardiovasc. Res. 110, 151–161 10.1093/cvr/cvw027 [DOI] [PubMed] [Google Scholar]
- 23.Deng J., Liu W., Wang Y., Dong M., Zheng M. and Liu J. (2012) Polydatin modulates Ca(2+) handling, excitation-contraction coupling and beta-adrenergic signaling in rat ventricular myocytes. J. Mol. Cell Cardiol. 53, 646–656 10.1016/j.yjmcc.2012.08.009 [DOI] [PubMed] [Google Scholar]
- 24.Hong H.Q., Lu J., Fang X.L., Zhang Y.H., Cai Y., Yuan J. et al. (2017) G3BP2 is involved in isoproterenol-induced cardiac hypertrophy through activating the NF-κB signaling pathway. Acta. Pharmacol. Sin. 39, 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun R., Zhu B., Xiong K., Sun Y., Shi D., Chen L. et al. (2017) Senescence as a novel mechanism involved in beta-adrenergic receptor mediated cardiac hypertrophy. PLoS ONE 12, e0182668 10.1371/journal.pone.0182668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanriverdi L.H., Parlakpinar H., Ozhan O., Ermis N., Polat A., Vardi N. et al. (2017) Inhibition of NADPH oxidase by apocynin promotes myocardial antioxidant response and prevents isoproterenol-induced myocardial oxidative stress in rats. Free Radic. Res., 51, 772–786 [DOI] [PubMed] [Google Scholar]
- 27.Xiao H., Li H., Wang J.J., Zhang J.S., Shen J., An X.B. et al. (2017) IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart J., 39, 60–69, [DOI] [PubMed] [Google Scholar]
- 28.Jiang S., Huo D., Wang X., Zhao H., Tan J., Zeng Q. et al. (2017) Beta-adrenergic receptor-stimulated cardiac myocyte apoptosis: role of cytochrome P450 omega-hydroxylase. J. Cardiovasc. Pharmacol. 70, 94–101 10.1097/FJC.0000000000000499 [DOI] [PubMed] [Google Scholar]
- 29.Lee G.J., Yan L., Vatner D.E. and Vatner S.F. (2015) Mst1 inhibition rescues beta1-adrenergic cardiomyopathy by reducing myocyte necrosis and non-myocyte apoptosis rather than myocyte apoptosis. Basic Res. Cardiol. 110, 7 10.1007/s00395-015-0461-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gang C., Qiang C., Xiangli C., Shifen P., Chong S. and Lihong L. (2015) Puerarin suppresses angiotensin II-induced cardiac hypertrophy by inhibiting NADPH oxidase activation and oxidative stress-triggered AP-1 signaling pathways. J. Pharm. Pharm. Sci. 18, 235–248 10.18433/J3N318 [DOI] [PubMed] [Google Scholar]
- 31.Hill C., Wurfel A., Heger J., Meyering B., Schluter K.D., Weber M. et al. (2013) Inhibition of AP-1 signaling by JDP2 overexpression protects cardiomyocytes against hypertrophy and apoptosis induction. Cardiovasc. Res. 99, 121–128 10.1093/cvr/cvt094 [DOI] [PubMed] [Google Scholar]
- 32.Tu V.C., Sun H., Bowden G.T. and Chen Q.M. (2007) Involvement of oxidants and AP-1 in angiotensin II-activated NFAT3 transcription factor. Am. J. Physiol. Cell Physiol. 292, C1248–C1255 10.1152/ajpcell.00624.2005 [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y., Shen W.L., Shi M.L., Zhang L.Z., Zhang Z., Li P. et al. (2015) Involvement of aberrant miR-139/Jun feedback loop in human gastric cancer. Biochim. Biophys. Acta. 1853, 481–488 10.1016/j.bbamcr.2014.12.002 [DOI] [PubMed] [Google Scholar]
- 34.Morisco C., Zebrowski D., Condorelli G., Tsichlis P., Vatner S.F. and Sadoshima J. (2000) The Akt-glycogen synthase kinase 3beta pathway regulates transcription of atrial natriuretic factor induced by beta-adrenergic receptor stimulation in cardiac myocytes. J. Biol. Chem. 275, 14466–14475 10.1074/jbc.275.19.14466 [DOI] [PubMed] [Google Scholar]
- 35.Maoa R., Zou F., Yang L., Lin S., Li Y., Ma M. et al. (2015) The loss of miR-139-5p promotes colitis-associated tumorigenesis by mediating PI3K/AKT/Wnt signaling. Int. J. Biochem. Cell Biol. 69, 153–161 10.1016/j.biocel.2015.10.008 [DOI] [PubMed] [Google Scholar]