

Abstract
Introduction:
Activation of the renin-angiotensin system (RAS) plays an important role in atrial electrical remodeling (AER). The purpose of the present study was to evaluate the effects of irbesartan on cardiac sodium current (INa) in a canine model of atrial fibrillation.
Materials and methods:
Eighteen dogs were randomized into sham, pacing or pacing+irbesartan groups (n = 6 in each group). The dogs in the pacing and irbesartan group were paced at 500 bpm for two weeks. Irbesartan (60 mg·kg−1·d−1) was administered orally in the pacing+irbesartan groups. INa was recorded using the whole-cell patch clamp technique from canine atrial myocytes. The expressions of cardiac Na+ channels (Nav1.5) mRNA were semi-quantified by reverse transcription-polymerase chain reaction.
Results:
Our results showed that INa density and Nav1.5 mRNA expression in the pacing group decreased significantly (p < 0.05 vs. sham). However, rapid atrial pacing had no effects on the half-activation voltage (V1/2act) and half-inactivation voltage (V1/2inact) of INa (p > 0.05 vs. sham). Irbesartan significantly increased INa densities and gene expression and hyperpolarized V1/2act without concomitant changes in V1/2inact.
Conclusions:
Irbesartan significantly increased INa densities, which contributed to improving intra-atrial conduction and prevented the induction and promotion of AF in atrial pacing dogs.
Keywords: Atrial electrical remodeling, irbesartan, renin-angiotensin system, sodium current, Nav1.5 α subunit
Introduction
Atrial fibrillation (AF) is a source of considerable morbidity and mortality. In 1995, Wijffels et al. revealed that AF modified atrial properties so that AF maintains itself more readily, a phenomenon called electrical remodeling and described as “AF begets AF.”1 The electric remodeling during AF involves decrease in the atrial effective refractory period (ERP), action potential (AP) duration,2-5 and/or ERP adaptation to heart rate,1,6 as well as decreased atrial conduction velocity (CV).2,3,5 Voltage-gated sodium channels are transiently activated on depolarization of the cardiac cell membrane and are responsible for the rapid upstroke of cardiac AP, and for rapid impulse conduction through cardiac tissue.7,8 Inhibition of cardiac sodium current (INa) slows down CV, which as a single factor reduces wavelength (defined as the product of CV and ERP) and should promote AF induction or atrial flutter.9
Activation of the renin-angiotensin system (RAS) has been suggested to play a role in the genesis and perpetuation of AF through atrial remodeling, and experimental studies have validated the utilization of RAS inhibition for AF prevention.10 Angiotensin II (Ang II), the major active component of RAS, can increase the intracellular free calcium, alters the potassium conductance, and induces the genesis of transient inward currents.11 Ang II can also decrease SCN5A transcription and current through production of H2O2 resulting in nuclear factor kappa B (NF-κB) binding to the sodium channel promoter.9 Therefore, Ang II has a high arrhythmogenic activity and may play an important role in the pathophysiology of atrial fibrillation, and inhibition of Ang II may prevent the development of atrial electrical remodeling (AER).10
Angiotensin II receptor blockers (ARBs) are medications frequently used in the treatment of congestive heart failure and hypertension and have several cardiac electrophysiological effects. Previous studies have shown that the ARB irbesartan was found to contribute to maintenance of sinus rhythm after successful electrical cardioversion and prevented the shortening of the atrial ERP during rapid atrial pacing.10,12 In addition, irbesartan suppresses atrial fibrosis and AF development in a canine atrial tachycardia remodeling model.13 Furthermore, irbesartan attenuated the hypotonic solution-induced increase in rectifier potassium currents induced by stretching atrial myocytes.14 Previous studies led to the conclusion that atrial electrical and structural remodeling could be prevented by irbesartan and Ang-(1–7); meanwhile, chronic rapid atrial pacing, a concomitant reduction in AF vulnerability in a canine model, could be observed.14-17 INa is essential for the heart conduction system, and slowing of intra-atrial conduction is one of the key factors for reentry and is necessary for AF induction or atrial flutter.6 Nevertheless, the effects of irbesartan on INa have not been fully explored, which naturally led us to the present study. Therefore, we tested the hypothesis that treatment with irbesartan increases INa densities, which contributes to improving intra-atrial conduction and prevents the induction and promotion of AF. Thus, the goal of this study was to examine the effects of irbesartan on INa and messenger RNA (mRNA) expression of sodium channel in a canine model of rapid atrial pacing.
Materials and methods
Animal model
Approval for use of the animals in this study was attained from the Experimental Animal Administration Committee of Tianjin Medical University and Tianjin Municipal Commission for Experimental Animal Control. Eighteen mongrel dogs of either sex, weighing between 11 and 15 kg, were randomly assigned to three groups. In the pacing+irbesartan group (n = 6), oral administration of irbesartan (60mg·kg−1·d−1) was continued to the end of the study. The dogs in the sham (n = 6) and pacing group (n = 6) did not receive irbesartan. Dogs within the pacing and pacing+irbesartan groups were paced at 500 beats per minute (bpm) (120 ms cycle length) with 0.2 ms square-wave pulses at twice-threshold current for 14 days. Sham animals were instrumented without pacing and were examined for 14 days following pacemaker insertion.
We used a previously described method to produce dogs with sustained AF.17 In brief, all dogs were anesthetized with sodium pentobarbital (30 mg·kg−1, intravenously). Under sterile technique, a unipolar screw-in St. Jude J pacing lead was inserted through the right jugular vein and the distal end of the lead was screwed in the right atrium. Initial atrial capture was verified using an external stimulator (TOP2001, Hongtong Co, Shanghai, China). The proximal end of the pacing lead was connected to a tachy-pacing generator (made in Shanghai Fudan University, China), which was inserted into a subcutaneous pocket in the neck. Twenty-four hours was allowed for the lead to stabilize. The pacemaker was programmed to stimulate the atrium at 500 bpm (120 ms cycle length). Surface electrocardiograms (ECGs) were verified after 24 hours, and then every other day, to ensure continuous 1:1 atrial capture. Direct systolic blood pressure was determined during anesthesia at baseline, and 14 days following atrial pacing.
Single-cell electrophysiology
The procedure for isolating cells was the same as used in previous research.17 On study days, the dogs were anesthetized with pentobarbital and ventilated as above. A median sternotomy was performed. At room temperature the hearts were removed and immersed in Ca2+-free Tyrode’s solution.
The left circumflex artery was cannulated, and the hearts were immersed in 4°C Ca2+-free Tyrode’s solution for 5 minutes until cardiac arrest and the effluent was clear. Any leaking arterial branches were ligated using silk thread to ensure adequate perfusion. The tissue was then perfused at 25 ml/min with Ca2+-free Tyrode’s solution for 20 minutes, followed by a 40-minute perfusion using the same solution containing collagenase (100 U/ml, CLSII, Worthington Biochemical, USA), and 0.5% bovine serum albumin (BSA) (Sigma Chemical Co, USA). The left atrium became opaque and mottled in appearance and tissue from a well-perfused region was removed using a forceps, gently triturated, and maintained at room temperature in a high-K+ storage solution.
High output of isolating atrial myocytes was obtained in our experiments. Only those quiescent rod-shaped cells lacking membrane deformities and showing clear cross-striations were studied. Five minutes after cell adhesion, cells were superfused with Tyrode’s solution (3 ml/min) for 5 minutes, followed by extracellular solution for the recording of INa at the same velocity and time of flow. Recordings of INa were obtained 5 minutes following membrane rupture.
Data acquisition
Whole-cell INa was recorded at room temperature using the patch-clamp technique as described previously.17 Borosilicate glass electrodes with tip resistances of 2.0 to 4.0 MΩ were connected to a patch clamp amplifier (Axopatch 200B, Axon Instruments, USA). Data were sampled by using an analog-to-digital converter (Digidata 1200, Axon Instruments, USA) and stored for subsequent analyses. Inactivation of current amplitude was measured on the basis of differences between the peak current and steady-state current at the end of a depolarizing pulse. After establishing the whole-cell configuration, series resistance (estimated from the decay of the capacitive transient) was typically two to three times the pipette resistance (<6 MΩ) and was electronically compensated.
Several minutes after seal formation, the membrane was ruptured by gentle suction to establish the whole-cell configuration for voltage clamping. Series resistance (Rs) was electrically compensated to minimize voltage errors. Voltage command pulses were generated from the Digidata 1200 and controlled using pClamp7.0 software. Current signals were low-pass filtered at 0.5 kHz and the sampling frequency was 10 Hz for recording of INa. To control for differences in cell size, current values were normalized to membrane capacitance (Cm), expressed as current densities (pA/pF).
Current-voltage relationships were determined in the extracellular solution over a voltage range of −80 to +60 mV with 10-mV increments. Voltage-dependent activation curves of INa were fitted with a Boltzmann equation: g/gmax (or I/Imax) = 1/{1+exp[(V–V1/2)/κ]}, where V1/2 is the potential at which half maximum activation or inactivation occurs and κ is the slope factor. Steady-state inactivation was measured utilizing a standard dual-pulse protocol. Cells were held at −120 mV prior to evoking a 1-second conditioning pulse immediately followed by a 40-ms pulse to −40 mV with 5-mV increments to measure sodium current. Normalized currents were plotted as a function of conditioning step voltage and were fitted to a standard Boltzmann equation. Cells with significant leak currents were rejected and leakage compensation was not used for further study.
Solutions
Ca2+-free Tyrode’s solution contained (mM) KCl 5.4, MgCl2 0.8, NaH2PO4 0.33, NaCl 136, HEPES 10, glucose 10, and pH 7.4 adjusted with NaOH. High-K+ storage solution contained (mM) KCl 20, KH2PO4 10, taurine 10, L-glutamic acid 70, EGTA 10, glucose 10, BSA 0.5% (5 mg/ml, Roche Group), and pH 7.4 adjusted with KOH. INa extracellular solution contained (mM) choline-Cl 110, CsCl 20, MgCl2 1.0, HEPES 10, glucose 10, NaCl 10, CaCl2 1.0, and pH 7.4 adjusted using NaOH. The pipette solution for INa recording was (mM) CsCl 20, tetraethyl ammonium chloride 20, aspartic acid 80, CsOH 80, MgCl2 1, HEPES 10, EGTA 10, Mg2ATP 5, Na3GTP 0.1, Na2-phosphocreatine 5, and pH 7.25 adjusted with CsOH. All solutions used for dissection and perfusion were equilibrated using 100% O2.
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR) analyses
Following cardiectomy, the left atria were excised for subsequent molecular biological studies. Specimens were snap-frozen in liquid nitrogen and stored separately at −80°C for further analyses. One aliquot of each tissue was used to investigate the mRNA expression of the Nav1.5 α subunit. Total RNA was extracted from 100 mg atrial tissues using a total RNA extraction kit (New Industry Co, Canada). Integrity of RNA was confirmed by agarose gel electrophoresis, the quantity of RNA was assessed spectrophotometrically (UV-240, Shimadzu Co, Japan) at a wavelength of 260 nm, and RNA was stored at −80°C for later analysis.
Specific oligonucleotide primer pairs for the amplification of Nav1.5 were designed according to GeneBank sequences. β-actin was utilized as the loading control. GeneBank accession numbers were NM-001002994 and AF021873 (β-actin). Gene-specific primers were designed using the Gene Runner software package. The specific primers were as follows: 5’-TGAATGTCCTCCTCGTCTG-3’/5’-TGTTGGTTGAAGTTGTCG-3’ (Nav1.5 α subunit, 424 bp), 5’-GGACCTGACTGACTACCTC-3’/5’-ACTCCTGCTTGCTGATCC-3’ (β-actin, 536 bp). Primers were purchased from Shanghai Yingjun Gen-Tech Ltd (Shanghai, China). A total of 200 ng of total RNA underwent RT-PCR using a commercially available kit (TaKaRa Co, Japan). PCR involved the following steps: initial denaturation at 94°C for 5 minutes followed by 37 cycles of denaturation for 40 seconds at 94°C, annealing for 40 seconds at 51°C, and elongation for 30 seconds at 72°C with a final extension at 72°C for 5 minutes. Product (5 μl) was analyzed using 1% agarose gel electrophoresis. The resultant target gene sequence was identified using Shanghai Yingjun Gen-Tech Ltd (Shanghai, China).
Statistical analyses
In order to ensure data validity from voltage-clamp studies, similar numbers of cells from each heart were studied utilizing the same protocol (i.e. cells were distributed evenly across dogs). Grouped data are presented as mean±SEM. Statistical comparisons among groups were performed by analysis of variance. Differences with a two-tailed p < 0.05 were considered statistically significant. All authors had full access to the data and take responsibility for their integrity.
Results
Hemodynamic parameters
No significant differences in ventricular rate or systolic blood pressure among the three groups were observed at baseline (p > 0.05), with no differences found during pacing (p > 0.05, Table 1). Heart rate and blood pressure were also unchanged by rapid pacing within each group (p > 0.05).
Table 1.
Hemodynamic parameters before and after pacing in each group.
| Grouping (n = 6 in each group) | Heart rate (bpm) |
Systolic blood pressure (mmHg) |
||||
|---|---|---|---|---|---|---|
| Before pacing | Paced for 14 days | p | Before pacing | Paced for 14 days | p | |
| Sham | 165.8±9.6 | 187.0±13.0 | >0.05 | 134.0±7.0 | 138.0±6.0 | >0.05 |
| Pacing | 171.6±11.9 | 188.0±20.0 | >0.05 | 140.0±10.0 | 138.0±8.0 | >0.05 |
| Irbesartan | 168.0±10.5 | 186.0±11.0 | >0.05 | 135.0±10.0 | 136.0±9.0 | >0.05 |
| P | >0.05 | >0.05 | >0.05 | >0.05 | ||
Sham: sham group; Pacing: pacing group; Irbesartan: pacing+irbesartan group; bpm: beats per minute.
INa densities
To avoid contaminating the effects of time-dependent changes in INa I-V, all protocols were performed by utilizing the same sequence: beginning with the INa density-voltage relationship, immediately followed by analyses of voltage-dependent inactivation.
Figure 1 illustrates the overall results for INa current densities. Peak INa density is shown as a function of test potential. The peak INa densities (as is shown in Table 1) were −61.56±14.17 pA/pF (14 cells) in the sham, –32.65±10.92 pA/pF (28 cells) in the pacing, and −65.24±14.79 pA/pF (13 cells) in the pacing+irbesartan groups. INa was significantly decreased at all voltages (–60 mV to +35 mV) in the pacing group but the form of the I-V curve did not change compared with the sham group (p < 0.05). The reduction of mean INa density was significantly prevented by irbesartan, and peak current densities increased by 99.82% compared with the pacing group (p < 0.05). Cell capacitance averaged 69.15±19.16 pF (n = 14) in the sham, 73.48±13.01 pF (n = 28) in the pacing and 76.46±22.79 pF (n = 13) in the pacing+irbesartan groups.
Figure 1.
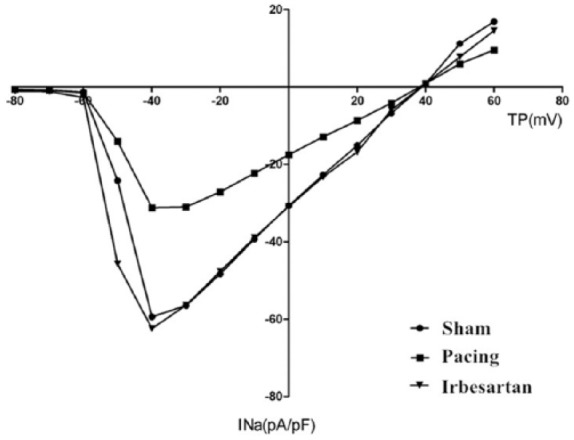
Mean current-voltage relationship of INa in the sham (n = 14), pacing (n = 28), and pacing+irbesartan (n = 13) groups. Currents were elicited from a high potential of −90 mV with 40 ms depolarizing steps to TP between −80 and +60 mV. INa was significantly decreased at all voltages (–60 mV to +35 mV) in the pacing group but the form of the I-V curve did not change compared with the sham group (p < 0.05). The reduction of mean INa density was significantly prevented by irbesartan and peak current densities increased by 99.82% compared with the pacing group (p < 0.05). INa: cardiac sodium current; Sham: sham group; Pacing: pacing group; Irbesartan: pacing+irbesartan group; TP: test potential; pA/pF: current densities.
Activation and inactivation characteristics of INa in isolated atrial myocytes
Figure 2 depicts the steady-state activation curve of INa. Table 2 summarizes the shifts in activation and inactivation variables. Voltage-dependent activation data were well-fitted using the Boltzmann equation. In the present experiment, rapid pacing did not alter half-activation voltage (V1/2act) (membrane voltage (Vm) at which half-activation occurs) compared with the sham group (p > 0.05). V1/2act in the pacing+irbesartan group was more negative compared with the sham or pacing groups (p < 0.05). The activation slope factor κact, V1/2inact (Vm at which half-inactivation occurs) and inactivation slope factor κinact were not significantly different among the three groups, indicating that rapid pacing and irbesartan have no significant effects on κact, V1/2inact or κinact of INa (Figure 3).
Figure 2.
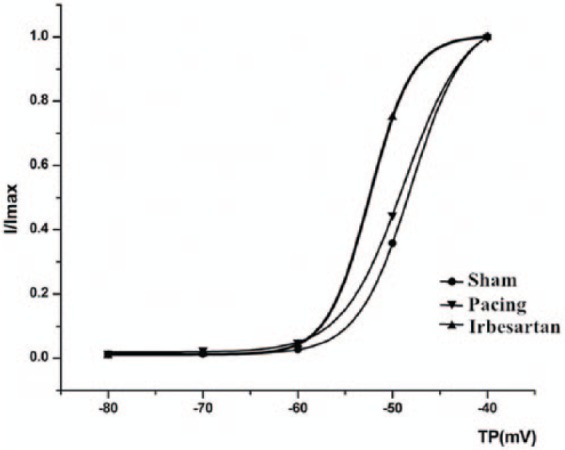
Mean values of voltage dependence of INa activation characteristics (I/Imax). Rapid pacing did not alter the half activation voltage (V1/2act) compared with the sham group (p > 0.05). V1/2act in the pacing+irbesartan group was more negative compared with the sham or pacing groups (p < 0.05). The activation slope factor κact was not significantly different among the three groups. Sham: sham group, Pacing: pacing group, Irbesartan: pacing+irbesartan group; TP: test potential.
Table 2.
INa densities and steady-state activation and inactivation of INa.
| Grouping (n = 6 in each group) | INa densities |
Activation |
Inactivation |
|||||
|---|---|---|---|---|---|---|---|---|
| (pA/pF) n | κact | V1/2act (mV) | n | κinact | V1/2inact (mV) | n | ||
| Sham | −61.56±14.17 | 14 | 0.75±0.99 | −50.68±4.45 | 14 | 4.16±0.81 | −84.89±4.05 | 12 |
| Pacing | −32.65±10.92a | 28 | 1.77±1.31 | −49.43±4.44 | 28 | 4.57±0.48 | −89.16±5.40 | 21 |
| Irbesartan | −65.24±14.79b | 13 | 0.85±1.09 | −55±5.35c | 13 | 4.61±0.76 | −88.31±9.12 | 9 |
All data shown represent meant±SEM. ap < 0.01 vs. corresponding value in the sham group. bp < 0.05 vs. corresponding value in the pacing group. cp < 0.05 vs. corresponding value in the sham and pacing groups. Sham: sham group; Pacing: pacing group; Irbesartan: pacing+irbesartan group. INa: cardiac sodium current; pA/pF: current densities; κact: activation slope factor; V1/2act: half activation voltage.
Figure 3.
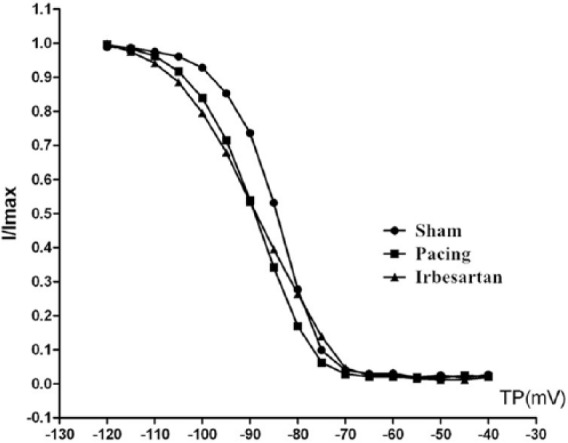
Mean values of voltage dependence of INa inactivation (I/Imax). Sham: sham group; Pacing: pacing group; Irbesartan: pacing+irbesartan group; TP: test potential. The V1/2inact and κinact were not significantly different among the three groups. V1/2inact: half-inactivation voltage; κinact: inactivation slope factor.
Nav1.5 α subunit mRNA expression
INa in the heart is primarily generated by Nav1.5.18 The α subunit is the principal component of Nav1.5 forming the pore and all essential gating elements, and is sufficient by itself for generating INa in heterologous expression systems. Figure 4(a) shows a gel obtained by semiquantitative RT-PCR for the Nav1.5 α subunit. Lanes S, P and IB are PCR products from the sham, pacing and pacing+irbesartan groups obtained by the addition of 5 µl of the internal standard along with 5 µl of Nav1.5 α subunit PCR product, respectively. DL2000 is the DNA marker. The upper band in each lane represents the Nav1.5 α subunit mRNA product (424 bp), and the lower band is the internal standard signal (β-actin, 536 bp). Figure 4(b) shows the mean±SEM of Nav1.5 α subunit mRNA levels in six hearts (one independent determination per heart) from each group of dogs. Nav1.5 α subunit mRNA expression was decreased by 54.83% after 14 days of rapid atrial pacing (p < 0.05 vs. sham). The observed changes in mRNA levels are parallel to changes in INa densities. Irbesartan prevented the decrease of Nav1.5 α subunit mRNA expression compared with the paced group (p < 0.05).
Figure 4.
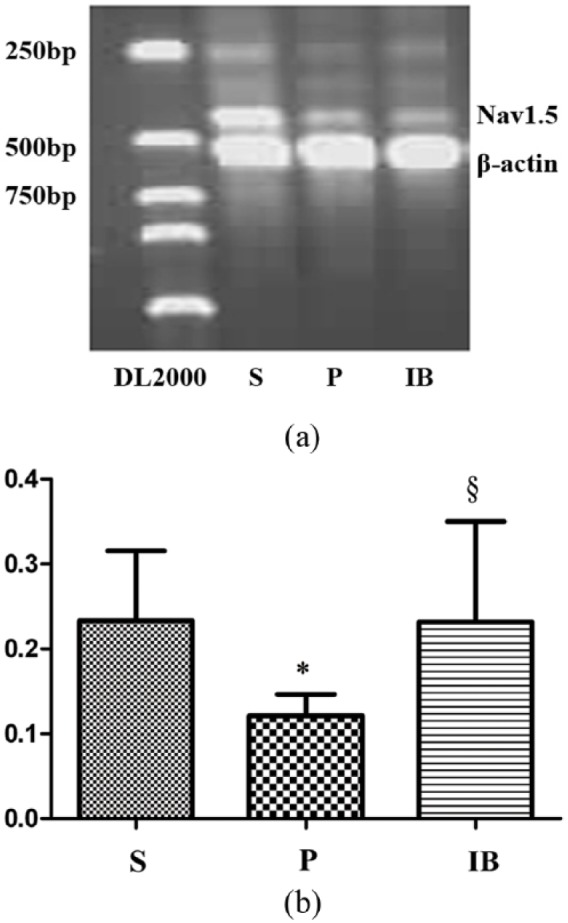
(a) Atrial pacing decreased Nav1.5 α subunit mRNA expression in canine atria. Irbesartan increased Nav1.5 α subunit mRNA in paced atria. Columns and error bars indicate mean±SEM, respectively. β-actin was utilized as the loading control. (b) Atrial pacing decreased Nav1.5 α subunit mRNA expression in canine atria. Irbesartan increased Nav1.5 α subunit mRNA in paced atria. Columns and error bars indicate mean±SEM, respectively. *p < 0.05 vs. corresponding value in the sham group, §p < 0.05 vs. corresponding value in the pacing group. DL2000: DNA marker; S: sham group; P: pacing group; IB: pacing+irbesartan group (n = 6 in each group). Nav1.5: cardiac Na+ channels; mRNA: messenger RNA.
Discussion
Key findings
A brief summary can be drawn from the key findings of the present study, that is, following pacing, INa density and mRNA expression of the Nav1.5 α subunit were decreased; irbesartan increased INa densities and hyperpolarized V1/2act. Meanwhile, it did not significantly affect κact or the inactivation characteristics of INa when compared with the pacing group. Irbesartan prevented downregulation of mRNA expression of the Nav1.5 α subunit in the pacing group.
Effects of rapid atrial pacing on INa and Nav1.5 α subunit gene expression
As the key structure of excitability in conduction of electrical impulses in cardiac tissue, the voltage-gated sodium channel is targeted by a series of anti-arrhythmics. The Nav1.5 α subunit is the principal component of cardiac Na+ channels, forming the pore and all essential gating elements and is sufficient to generate INa by itself in heterologous expression systems. Our previous research has shown that reductions in INa density and Nav1.5 α subunit gene expression potentially accounted for slowing of atrial conduction in a canine model of chronic AF.17 Decreased conduction and ERP results in shorter wavelength (defined as the product of CV and ERP) and increases the number of wavelengths that can coexist in the given atrial dimension. Thus, it increases the likelihood of sustained AF.
Effects of irbesartan on INa and Nav1.5 α subunit mRNA expression
Activation of the RAS contributes to development and maintenance of AF.19,20 Ang II has been shown to stimulate various growth factors inducing development and maintenance of AER by acting on type 1 receptors10 and may have cellular electrophysiologic effects on the modulation of ion channels in cardiac myocytes.21,22 Ang II may increase intracellular calcium concentration23,24 and intracellular calcium overload can downregulate Na+ channels, which may lead to decrease of INa and conduction delay deficits.25 It is also reported that Ang II antagonists may exert favorable anti-arrhythmic effects in AF.26 Li et al. found that imidapril markedly increases INa density of ventricular myocytes from healed myocardial infarction in rabbits.27 Thus, they suggested that increased INa density may underlie the anti-arrhythmic effects of imidapril. We previously reported that Ang-(1–7) treatment significantly increased INa densities but had no effect on the expression of the Nav1.5 gene.17 However, the effects of irbesartan on INa or Nav1.5 mRNA expression are not clear. Our present data suggest that irbesartan can increase INa and Nav1.5 α subunit mRNA expression in a canine model of atrial tachycardia. We speculate that Ang II-induced intracellular calcium overload, which leads to decrease of INa and conduction delay deficits, may be partially responsible for this effect.25
AF is generally considered to be a re-entrant arrhythmia, and the stability of AF is classically related to wavelength. As previously discussed, wavelength is defined as the product of CV and ERP. Therefore, modification of ERP or CV can affect AF vulnerability and stability.28 Irbesartan increased INa and contributed to the increase of CV, the lengthening of reentry wavelength as well as the decreasing in the number of wavelengths. Thus, treatment with irbesartan could have the potential to reduce AF incidence. We also found that irbesartan upregulated Nav1.5 α subunit mRNA expression. These findings suggest that increased Nav1.5 gene levels by irbesartan, at least in part, are potentially responsible for changes in INa densities.
Effects of irbesartan on activation and inactivation characteristics of INa
Our previous research has shown that rapid atrial pacing has no effects on voltage-dependent activation of INa, and Ang-(1–7) resulted in more negative test potentials.17 The present study revealed that irbesartan had a similar effect as Ang-(1–7), which suggested an increased conductance at a particular membrane potential.29 Such shifts induced faster activation kinetics and current augmentation at potentials closer to the threshold for activation and could actually indicate an enhancement of the current amplitude.29 And this could be speculated as a key reason why the agent increased INa densities.
There is no significant difference in comparison of voltage-dependent inactivation of INa among the three groups, which is consistent with the previous study.6 However, in human AF, myocardium in the inactivation curve was shifted to more positive voltages, which could increase sodium channel availability at the resting potential and increase CV.30 There are many reasons contributing to these contrasting results of human and animal models of AF: differences of AF types (experimental vs. clinical AF), underlying cardiac disease, anti-arrhythmic drugs, or interspecies differences.
Li and colleagues demonstrated that imidapril hyperpolarized V1/2inact of INa in rabbit ventricular myocytes.27 Kim et al. reported that telmisartan significantly delayed the inactivation of the voltage-gated Na+ channel, causing cytosolic Na+ overload, prolonged AP duration, and subsequent Ca2+ overload in ventricular cardiomyocytes of male Sprague Dawley rats.31 However, we did not find any difference in our data. We thus concluded that irbesartan did not have obvious effects on the inactivation characteristics of INa. Therefore, our conclusion varied from the above-mentioned one. The difference in conclusions might be caused by several issues, such as interspecies differences, differences in the animal models, time of drug administration, different drugs, or differences between atrial and ventricular myocardium. In addition, there was no alteration of slope factor κact and κinact shown in the three groups, which suggested that the intrinsic voltage of the channel was unaffected by pacing or drug treatment.29
Study limitations
One of the limitations of the present study is that we did not assess ventricular function using echocardiography. Since ventricular rate and blood pressure were not significantly changed by rapid atrial pacing among the three groups, ventricular tachycardia-induced left ventricular dysfunction likely did not have a significant effect on the results of these experiments. Furthermore, in our study only the left atria were analyzed. Therefore, responses of the right atrium to irbesartan remain unknown. Research on the sodium channel protein expression is still under way.
Conclusions
Irbesartan is a powerful activator of atrial sodium channels by selectively shifting the voltage-activation relationship of INa to hyperpolarized potential or upregulating Nav1.5 mRNA expression. Therefore, irbesartan may contribute to the improvement of intra-atrial conduction and the prevention of induction and promotion of AF in atrial pacing dogs.
Footnotes
Declaration of conflicting interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Program of the Natural Science Foundation of China (grant number 30770863).
References
- 1. Wijffels MC, Kirchhof CJ, Dorland R, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995; 92: 1954–1968. [DOI] [PubMed] [Google Scholar]
- 2. Gaspo R, Bosch RF, Talajic M, et al. Functional mechanisms underlying tachycardia-induced sustained atrial fibrillation in a chronic dog model. Circulation 1997; 96: 4027–4035. [DOI] [PubMed] [Google Scholar]
- 3. Daoud EG, Bogun F, Goyal R, et al. Effect of atrial fibrillation on atrial refractoriness in humans. Circulation 1996; 94: 1600–1606. [DOI] [PubMed] [Google Scholar]
- 4. Goette A, Honeycutt C, Langberg JJ. Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation 1996; 94: 2968–2974. [DOI] [PubMed] [Google Scholar]
- 5. Yue L, Feng J, Gaspo R, et al. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res 1997; 81: 512–525. [DOI] [PubMed] [Google Scholar]
- 6. Gaspo R, Bosch RF, Bou-Abboud E, et al. Tachycardia-induced changes in sodium current in a chronic dog model of atrial fibrillation. Cir Res 1997; 81: 1045–1052. [DOI] [PubMed] [Google Scholar]
- 7. Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 2004; 84: 431–488. [DOI] [PubMed] [Google Scholar]
- 8. Ding GL, Hu D, Liu X, et al. Suppression of sodium current by arachidonic acid in atrial myocytes from patients with coronary heart disease. Pacing Clin Electrophysiol 2000; 23: 1820–1822. [DOI] [PubMed] [Google Scholar]
- 9. Shang LL, Sanyal S, Pfahnl AE, et al. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol 2008; 294: C372–C379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakashima H, Kumagai K, Urata H, et al. Angiotensin Ⅱ antagonist prevents electrical remodeling in atrial fibrillation. Circulation 2000; 101: 2612–2617. [DOI] [PubMed] [Google Scholar]
- 11. Touyz RM, Sventek P, Larivière R, et al. Cytosolic calcium changes induced by angiotensin II in neonatal rat atrial and ventricular cardiomyocytes are mediated via angiotensin II subtype I receptors. Hypertension 1996; 27: 1090–1096. [DOI] [PubMed] [Google Scholar]
- 12. Madrid AH, Bueno MG, Rebollo JM, et al. Use of irbesartan to maintain sinus rhythm in patients with long-lasting persistent atrial fibrillation: A prospective and randomized study. Circulation 2002; 106: 331–336. [DOI] [PubMed] [Google Scholar]
- 13. Kataoka N, Nishida K, Kinoshita K, et al. Effect of irbesartan on development of atrial fibrosis and atrial fibrillation in a canine atrial tachycardia model with left ventricular dysfunction, association with p53. Heart Vessels 2016; 31: 2053–2060. [DOI] [PubMed] [Google Scholar]
- 14. Wu J, Ding WG, Zhao J, et al. Irbesartan-mediated AT1 receptor blockade attenuates hyposmotic-induced enhancement of IKs current and prevents shortening of action potential duration in atrial myocytes. J Renin Angiotensin Aldosterone Syst 2014; 15: 341–347. [DOI] [PubMed] [Google Scholar]
- 15. Liu E, Xu Z, Li J, et al. Enalapril, irbesartan, and angiotensin-(1–7) prevent atrial tachycardia-induced ionic remodeling. Int J Cardiol 2011; 146: 364–370. [DOI] [PubMed] [Google Scholar]
- 16. Liu E, Yang S, Xu Z, et al. Angiotensin-(1–7) prevents atrial fibrosis and atrial fibrillation in long-term atrial tachycardia dogs. Regul Pept 2010; 162: 73–78. [DOI] [PubMed] [Google Scholar]
- 17. Wang X, Li G. Angiotensin-(1–7) prevent atrial tachycardia induced sodium channel remodeling. Pacing Clin Electrophysiol 2014; 37: 1349–1356. [DOI] [PubMed] [Google Scholar]
- 18. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005; 85: 1205–1253. [DOI] [PubMed] [Google Scholar]
- 19. Willems R, Sipido KR, Holemans P, et al. Different patterns of angiotensin II and atrial natriuretic peptide secretion in a sheep model of atrial fibrillation. J Cardiovasc Electrophysiol 2001; 12: 1387–1392. [DOI] [PubMed] [Google Scholar]
- 20. Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1–7): An evolving story in cardiovascular regulation. Hypertension 2006; 47: 515–521. [DOI] [PubMed] [Google Scholar]
- 21. De Mello WC. Intracellular angiotensin II regulates the inward calcium current in cardiac myocytes. Hypertension 1998; 32: 9769–9782. [DOI] [PubMed] [Google Scholar]
- 22. Daleau P, Turgeon J. Angiotensin II modulates the delayed rectifier potassium current of guinea pig ventricular myocytes. Pflugers Arch 1994; 427: 553–555. [DOI] [PubMed] [Google Scholar]
- 23. Allessie MA, Boyden PA, Camm AJ, et al. Pathophysiology and prevention of atrial fibrillation. Circulation 2001; 103: 769–777. [DOI] [PubMed] [Google Scholar]
- 24. Lea JP, Jin SG, Roberts BR, et al. Angiotensin II stimulates calcineurin activity in proximal tubule epithelia through AT-1 receptor-mediated tyrosine phosphorylation of the PLC-gammal isoform. J Am Soc Nephrol 2002; 13: 1750–1756. [DOI] [PubMed] [Google Scholar]
- 25. Duff HJ, Offord J, West J, et al. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel alpha subunit in rat cardiac muscle. Mol Pharmacol 1992; 42: 570–574. [PubMed] [Google Scholar]
- 26. Hsieh YC, Hung CY, Li CH, et al. Angiotensin-receptor blocker, angiotensin-converting enzyme inhibitor, and risks of atrial fibrillation: A nationwide cohort study. Medicine (Baltimore) 2016; 95: e3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Y, Niu HY, Liu N, et al. Effect of imidapril on the effective refractory period and sodium current of ventricular noninfarction zone in healed myocardial infarction. Yao Xue Xue Bao 2005; 40: 654–658. [PubMed] [Google Scholar]
- 28. Nattel S, Shiroshita-Takeshita A, Brundel BJ, et al. Mechanisms of atrial fibrillation: Lessons from animal models. Prog Cardiovasc Dis 2005; 48: 9–28. [DOI] [PubMed] [Google Scholar]
- 29. Weigt HU, Kwok WM, Rehmert GC, et al. Voltage-dependent effects of volatile anesthetics on cardiac sodium current. Anesth Analg 1997; 84: 285–293. [DOI] [PubMed] [Google Scholar]
- 30. Chen L, Li QY, Yang Y, et al. Inhibitory effects of tetrandrine on the Na(+) channel of human atrial fibrillation myocardium. Acta Pharmacol Sin 2009; 30: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim HK, Youm JB, Lee SR, et al. The angiotensin receptor blocker and PPAR-γ agonist, telmisartan, delays inactivation of voltage-gated sodium channel in rat heart: Novel mechanism of drug action. Pflugers Arch 2012; 464: 631–643. [DOI] [PubMed] [Google Scholar]