

Abstract
Introduction:
The aim of this study was to investigate the effects of plasma and tissue angiotensin-converting enzyme inhibitors (ACE-Is) against propofol-induced endothelial dysfunction and to elucidate the involved mechanisms in vitro.
Materials and methods:
We examined the effects of propofol (50 μM), quinaprilat and enalaprilat (10−5 M) on fibrinolysis (t-PA, PAI-1, TAFI antigen levels), oxidative stress parameters (H2O2 and MDA antigen levels and SOD and NADPH oxidase mRNA levels) and nitric oxide bioavailability (NO2/NO3 concentration and NOS expression at the level of mRNA) in human umbilical vein endothelial cells (HUVECs).
Results:
We found that both ACE-Is promoted similar endothelial fibrinolytic properties and decreased oxidative stress in vitro. Propofol alone increased the release of antifibrinolytic and pro-oxidative factors from the endothelium and increased mRNA iNOS expression. We also found that the incubation of HUVECs in the presence of propofol following ACE-Is pre-incubation caused weakness of the antifibrinolytic and pro-oxidative potential of propofol and this effect was similar after both ACE-Is.
Conclusions:
This observation suggests that the studied ACE-Is exerted protective effects against endothelial cell dysfunction caused by propofol, independently of hemodynamics.
Keywords: Human umbilical vein cells, quinaprilat, enalaprilat, propofol, fibrinolysis, nitric oxide, oxidative stress
Introduction
Angiotensin-converting enzyme inhibitors (ACE-Is) are called plasma or tissue according to their affinity for angiotensin-converting enzyme (ACE) circulating in plasma or localized in particular organs like vascular endothelium. It was established that tissue ACE-Is may influence hemostasis more significantly than plasma ACE-Is in normotensive rats.1 This assumption is not clearly obvious because an influence on thrombotic processes was also seen in captopril and enalapril-treated animals, which are considered to be plasma ACE-Is.2 This antithrombotic effect observed in normotensive rats was accompanied by Ang-(1–7), prostacyclin (PGI2) and nitric oxide (NO) concentration increase.2,3 Ang-(1–7), activating its own G-protein coupled receptors, Mass1–7, increases the production of NO and PGI2 in the endothelium.
Moreover, both groups of ACE-Is significantly inhibited platelet and erythrocyte aggregation, and decreased glycoprotein IIb/IIIa expression on the platelet surface.4–6 Tissue ACE-Is, both in normotensive and renovascular hypertensive (2K-1C) rats, activated significantly hemostasis, which was manifested as the extension of prothrombin time (PT), prolongation of activated partial thromboplastin time (APTT), and shortening of euglobulin clot lysis time (ECLT).1 Also, an increase in tissue plasminogen activator (t-PA) and decrease in tissue plasminogen activator inhibitor (PAI-1) concentration were observed.7
Analysis of the antithrombotic activity of ACE-Is in 2K-1C rats showed the involvement of different endothelial mechanisms. According to the literature data, endothelial dysfunction during hypertension was related to reduced NO bioavailability. Indeed, we have shown that quinapril decreased thrombosis and restored endothelium function in 2K-1C rats via reduction of platelet aggregation, decrease in iNOS expression, and fibrinolysis potentiation. Interestingly, eNOS expression in this model of hypertension was unchanged, thus it can be assumed that NO did not seem to be the leading vasorelaxant factor during chronic quinapril treatment in hypertension.7 It was shown in individuals with hypercholesterolemia that under the influence of bradykinin not only NO but also endothelium-derived hyperpolarizing factor (EDHF) was released, which, in case of decreased NO bioavailability, compensates vasodilating activity via activation of calcium-dependent potassium channels.8 Furthermore, literature data confirm the involvement of EDHF-dependent platelet inhibition during ACE-Is treatment.9–11 All the above-mentioned mechanisms occur in the presence of hemodynamic conditions such as vascular flow and vessel diameter. Therefore, the question arises if tissue or plasma ACE-Is differ in their influence on endothelium cells after elimination of the hemodynamic component.
In the case of propofol, there is a lack of studies evaluating its direct influence on the hemostatic potency of endothelium in vitro. However, its anti-inflammatory and antioxidative properties were described in human umbilical vein endothelial cells (HUVECs) as well as patients undergoing hepatectomy under propofol anesthesia.12–14 On the contrary, we demonstrated in 2K-1C rats the antifibrinolytic influence of propofol on the endothelium by lowering t-PA and increasing PAI-1 concentrations, which was accompanied by oxidative stress potentiation and hypotensive effect.7 The above discrepancies may be a result of different experimental protocols (species, conditions, propofol dosage). Therefore, some further experiments should be conducted to better learn the mechanism of propofol action.
Thus, the present study is an attempt to answer the question that appeared in our previous work whether the antifibrinolytic effect of propofol is the result of the direct impact of propofol on the endothelium or whether hemodynamic factors are of some significance. In order to eliminate the hemodynamic effect, we evaluated the influence of plasma (enalaprilat) and tissue (quinaprilat) ACE-Is and propofol on some hemostatic parameters in HUVECs. Furthermore, we evaluated the relationship between the fibrinolytic activity of the endothelium, NO bioavailability, and oxidative stress mechanism. At the same time, we answered the question whether an exclusion of the antihypertensive effect (vasodilatation), as an important factor reducing oxidative stress, is still associated with an influence of the tested drugs on the endothelium.
Materials and methods
Chemicals and drugs
Propofol (Plofed 1%, Polfa, Poland), quinaprilat (Pfizer, Germany), enalaprilat (Sigma, Poland), lipofundin (MCT/LCT 10%, Braun, Germany), aqua pro injection (Baxter, Poland), sodium hydroxide (Sigma, Poland), ethylenediaminetetraacetic acid (EDTA) (Sigma, Poland), low-serum growth supplement (LSGS, Cascade Biologics, UK), Medium 200 (M200, Cascade Biologics, UK), penicillin (Sigma, Poland), streptomycin (Sigma, Poland), trypsin (Sigma, Poland), trypan blue (Sigma, Poland), Oligotex Kit (Qiagen, USA), qPCRTM Mastermix, SYBR Green I (Eurogentec Seraing, Belgium), TaqMan Reverse Transcription Reagents Kit (Applied Biosystems, USA), Tris Buffer (Polish Chemical Reagents, Poland) and Trizol (Invitrogen Life Technologies, USA) were used in the study.
Cell culture
The cryopreserved human umbilical vein endothelial cells (HUVECs) were purchased from Cascade Biologics Inc (Mansfield, Nottinghamshire, UK). The cells were grown in Medium 200 supplemented with penicillin/streptomycin (100 units/ml penicillin and 100 mg/ml streptomycin) and low-serum growth supplement at 37°C in a 95% humidified atmosphere of 5% CO2. The medium was replaced every two to three days. At confluence, the cells were subcultured by trypsinization (0.025% trypsin − 0.01% EDTA), after which the cells were seeded with a split ratio of 1:3. Cellular viability was determined with the application of the trypan blue staining method.15 Cultures at four to five passages were used in the experiments.
The HUVECs were divided into experimental groups, cultured at 37°C for 24 hours in the presence of a drug or its vehiculum in the medium. The study protocol is presented in Table 1. Enalaprilat (Ena) and quinaprilat (Quin) were added to the medium at similar molar concentrations (10−5 M) according to their molar mass, pharmacokinetics, and in vitro data.16,17 Propofol (Pro) was added to the medium at a concentration of 50 μM and the time of incubation was 30 minutes, which corresponds with concentrations of propofol observed in the blood of patients during short anesthesia and time of premedication.18
Table 1.
The study protocol.
| Group | ACE-Is |
Propofol |
ACE-Is + propofol |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | VIII | IX | X | |
|
Drug/vehiculum
added to medium |
Enalaprilat 10−5 M |
Aqua pro inj. | Quinaprilat 10−5M |
NaOH 0.1 M |
Propofol 50 µM |
Lipofundin | Enalaprilat 10−5 M, Propofol 50 µM |
Quinaprilat 10−5 M, Propofol 50 µM |
Aqua pro inj., Lipofundin |
NaOH 0.1 M, Lipofundin |
| Abbreviation | Ena | Veh Ena | Quin | Veh Quin | Pro | Lipo | Ena + Pro | Quin + Pro | Veh Ena + Lipo | Veh Quin + Lipo |
| Time of incubation | 24 h | 24 h | 24 h | 24 h | 30 min | 30 min | 24 h/30 min | 24h/30min | 24 h/30 min | 24 h/30 min |
ACE-Is: angiotensin-converting enzyme inhibitors; h: hours; min: minutes.
The human umbilical vein endothelial cells (HUVECs) were divided into experimental groups, cultured at 37°C for 24 h in the presence of a drug or its vehiculum in the medium. Active metabolites of ACE-IS: enalaprilat (Ena) or quinaprilat (Quin) were added to the medium at similar molar concentration (10-5M) and volume. In the control group the vehiculum of Ena (Aqua pro inj.) or vehiculum of Quin (0.1 M NaOH) were added to the medium in the same manner as ACE-Is. Propofol (Pro) was added to the medium at a concentration of 50 μM and the time of incubation was 30 min. In the control group the vehiculum of Pro, lipofundin (Lipo) was added to the medium in the same manner as Pro. In the HUVECs treated both with ACE-I and Pro, Pro was added to the medium 30 minutes before the end of a 24-hour incubation with ACE-I. The conditions of HUVECs incubation (temperature, humidity, medium molarity, pH) were the same in all experimental groups.
Real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
To evaluate the effect of the tested drugs on nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, superoxide dismutase (SOD), endothelial and inducible nitric oxide synthase (eNOS and iNOS) expression at the level of messenger RNA (mRNA), HUVECs were grown on 24-well microplates. mRNA levels were determined by real-time qPCR as previously described.19,20 Briefly, total RNA was extracted from the cells using Trizol reagent and was processed directly to complementary DNA (cDNA) synthesis using the TaqMan Reverse Transcription Reagents kit according to the manufacturer’s protocol. cDNA was subsequently amplified by MasterAmp Pfl DNA polymerase using the ABI Prism 7000 detection system (Applied Biosystems, USA). Specific primer sequences were designed based on primary mRNA sequences from GenBank. β-actin was used as an active and endogenous reference to correct for differences in the amount of total RNA added to the reaction and to compensate for different levels of inhibition during RT of RNA and during PCR. To compensate for variations in input RNA amounts and efficiency of RT, β-actin mRNA was quantified and the results were normalized to these values. Relative mRNA expression was calculated using the formula: 2−∆∆Ct (2−(Cttarget gene – Ctreference gene)), where Ct is a copy threshold.
Determination of fibrinolysis
To determine the influence of ACE-Is and propofol on fibrinolytic parameters in HUVECs, tissue plasminogen activator (t-PA), plasminogen activator inhibitor (PAI-I), and thrombin activatable fibrinolysis inhibitor (TAFI) antigen levels were measured in culture supernatants using commercial enzyme-linked immunosorbent assay (ELISA) test kits (Innovative Research, USA; ImmunoKontact AMS Biotechnology, Germany).
Determination of plasma NO level
Since NO is an extremely labile molecule and decomposes rapidly in biological solutions into nitrite (NO2−) and nitrate (NO3−), these stable metabolites of NO were analyzed in culture supernatants as indirect markers for NO plasma bioavailability.21
In our study, the NO level was measured colorimetrically as NO2−/NO3− concentration with a commercially available kit (Correlate Assay Nitric Oxide NO-2/NO-3 Kit, Assay Designs, USA).
Determination of NOS mRNA level
To determine the influence of ACE-Is and Pro administration on NOS activity, eNOS and iNOS mRNA levels were measured in the supernatant using the real-time PCR technique.19,20 The following PCR primers were designed: 5’-CATCGGCGTGCTGCGGGATCAG-3’ and 5’-GGGCTGTTGGTGTCTGAGCCGG-3’, 5’-CCAACAATGGCAACATCAGG-3’ and 5’-TCGTGCTTGCCATCACTCC-3’, specific for the mRNA of eNOS and iNOS, respectively. The reaction for each probe was conducted in the same conditions as a multiplex PCR with two pairs of primers (eNOS and iNOS) at the same time. The amount of eNOS and iNOS mRNA was quantified.
Oxidative stress parameters
To determine the influence of ACE-Is and Pro on oxidative stress parameters, the concentrations of hydrogen peroxide (H2O2) and malondialdehyde (MDA) were measured in culture supernatant with commercially available kits (Hydrogen Peroxide Colorimetric Detection Kit, Assay Designs, USA; MDA Adducts ELISA Kit, Cell Biolabs, USA). Moreover, the amounts of mRNA of NADPH oxidase and SOD were measured in supernatant using the real-time PCR technique.19,20 The following PCR primers were designed: NADPH oxidase (5’-CATCCAGCTGTACCTCAGTC-3’ and 5’-GAAAGACTCTTTATTGTATTG-3’) and SOD (5’-GGAAACGCTGGAAGTCGTTTG-3’ and 5’-CTCACTA CAGGTACTTTAAAG-3’).
Statistical analysis
The data were shown as mean ± SEM. All multiple comparison data were analyzed using analysis of variance (ANOVA) with post-hoc Dunn’s multiple comparisons test, and direct group-group comparisons were carried out using the Mann-Whitney test. qRT-PCR results were analyzed using ANOVA, followed by multiple comparisons with the Newman-Keuls post hoc test. A p value of <0.05 was considered statistically significant. Statistical analyses were performed with SPSS 16.0 (SPSS, Chicago, IL, USA).
Results
Assessment of endothelial activity exposed to different vehiculum
Different substances (vehiculum (Veh)) dedicated to the examined drugs were used as the control in the present study (Table 1). There were no differences in the concentration of hemostatic and oxidative stress parameters in supernatant of HUVECs after incubation with different control substances (vehiculum) (Tables 2 and 3).
Table 2.
Hemostatic parameters and nitric oxide bioavailability in supernatant of HUVECs after incubation with different control substances.
|
t-PA (ng/ml) |
PAI-1 (ng/ml) |
TAFI (µg/ml) |
eNOS (2−ΔΔCT) |
iNOS (2−ΔΔCT) |
NO2/NO3 (μM/l) |
|
|---|---|---|---|---|---|---|
| Veh Ena | 2.54±0.11 | 5.11±0.9 | 2.20±0.09 | 0.25±0.03 | 0.48±0.02 | 9.02±0.9 |
| Veh Quin | 2.63±0.19 | 5.05±0.7 | 2.12±0.11 | 0.24±0.06 | 0.45±0.01 | 8.89±0.8 |
| Lipo | 2.58±0.05 | 4.99±1.03 | 2.14±0.07 | 0.27±0.04 | 0.43±0.01 | 8.79±0.3 |
| Veh Ena + Lipo | 2.48±0.11 | 5.17±0.9 | 2.15±0.11 | 0.24±0.03 | 0.46±0.01 | 8.97±0.4 |
| Veh Quin + Lipo | 2.53±0.25 | 5.09±0.4 | 2.13±0.03 | 0.25±0.05 | 0.46±0.04 | 9.01±0.6 |
p = NS for all control groups.
HUVECs: human umbilical vein endothelial cells; t-PA: tissue plasminogen activator; PAI-1: tissue plasminogen activator inhibitor; TAFI: thrombin activatable fibrinolysis inhibitor; eNOS: endothelial nitric oxide synthase; iNOS: inducible nitric oxide synthase; NO2/NO3: nitrite/nitrate; Veh Ena: Aqua pro injection; Veh Quin: NaOH 0.1 M; Lipo: Lipofundin; Veh Ena + Lipo: Aqua pro injection, Lipofundin; Veh Quin + Lipo: NaOH 0.1 M, Lipofundin.
Table 3.
Oxidative stress parameters in supernatant of HUVECs after incubation with different control substances.
|
H2O2 (ng/ml) |
MDA (pM/mg protein) |
SOD (2−ΔΔCT) |
NADPH oxidase (2−ΔΔCT) |
|
|---|---|---|---|---|
| Veh Ena | 117±3 | 0.187±0.07 | 0.931±0.04 | 1.078±0.07 |
| Veh Quin | 128±0.1 | 0.179±0.05 | 1.09±0.04 | 1.112±0.04 |
| Lipo | 109±0.9 | 0.178±0.08 | 0.996±0.04 | 1.101±0.06 |
| Veh Ena + Lipo | 119±1.2 | 0.189±0.08 | 0.914±0.04 | 1.101±0.03 |
| Veh Quin + Lipo | 125±0.7 | 0.199±0.02 | 0.923±0.05 | 1.091±0.02 |
p = NS for all of the control groups.
HUVECs: human umbilical vein endothelial cells; H2O2: hydrogen peroxide; MDA: malondialdehyde; SOD: superoxide dismutase; NADPH: nicotinamide adenine dinucleotide phosphate; Veh Ena: Aqua pro injection; Veh Quin: NaOH 0.1 M; Lipo: Lipofundin; Veh Ena + Lipo: Aqua pro injection, Lipofundin; Veh Quin + Lipo: NaOH 0.1 M, Lipofundin.
Fibrinolytic activity of endothelial cells after ACE-Is or/and Pro incubation
Pro alone decreased the t-PA antigen level in supernatant (1.08±0.09 ng/ml vs 2.58±0.05 ng/ml, 42%, p < 0.001) when compared with Veh (Lipo), while ACE-Is increased it significantly (Ena 3.08±0.09 ng/ml vs 2.54±0.11 ng/ml, 121%, p < 0.01; Quin 3.14±0.15 ng/ml vs 2.63±0.19 ng/ml, 120%, p < 0.01) in comparison with their Veh, respectively.
Apparently, the co-incubation with Pro and Ena or Quin significantly reduced t-PA concentration in supernatant when compared with Ena or Quin alone (p < 0.001). Although, the t-PA level after Pro in supernatant of HUVECs pretreated with ACE-Is was still higher than after Pro alone (p < 0.05) (Figure 1).
Figure 1.
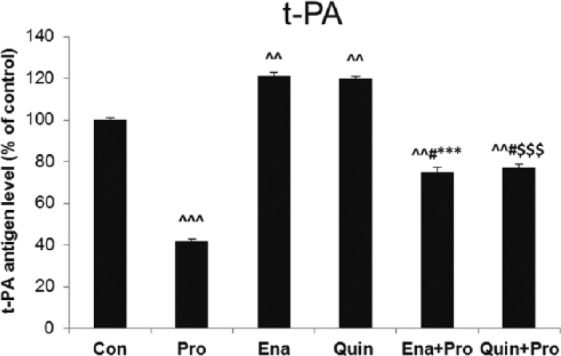
The effect of propofol and angiotensin-converting enzyme inhibitors (ACE-Is) on tissue plasminogen activator (t-PA) antigen level in supernatant of human umbilical vein endothelial cells (HUVECs). The results are presented as percentage of control (Con: adequate vehiculum). Pro: propofol; Ena: enalaprilat; Quin: quinaprilat; Ena+Pro: enalaprilat and propofol; Quin+Pro: quinaprilat and propofol. ^^p < 0.01, ^^^p < 0.001 vs adequate vehiculum for each substance; #p < 0.05 vs Pro; ***p < 0.001 vs Ena; $$$p < 0.001 vs Quin.
Antifibrinolytic activity of endothelial cells after ACE-Is and/or Pro incubation
Incubation of HUVECs with Pro resulted in increased TAFI and PAI-1 concentration in supernatant when compared with Veh: 3.89±0.12 ng/ml vs 2.14±0.07 ng/ml, (182%, p < 0.001) and 8.11±0.9 ng/ml vs 4.99±1.03 ng/ml (162%, p < 0.001), respectively. Exposure of HUVECs to 24-hour incubation with Ena increased TAFI antigen level in comparison with its Veh (3.42±0.05 ng/ml vs 2.34±0.09 ng/ml, 146%, p < 0.01). Quin did not remarkably influence TAFI level. Both ACE-Is similarly decreased PAI-1 concentration; Ena: 4.77±1.1 ng/ml vs 5.11±0.9 ng/ml, (93%, p < 0.05); Quin: 4.64±1.0 ng/ml vs 5.05±0.7 ng/ml (92%, p<0.05), when compared with their Veh, respectively. Pro addition to Ena or Quin did not change TAFI and PAI-1 levels when compared with Ena and Quin alone. However, TAFI and PAI-1 levels remainded lower than in the group treated with Pro alone (Figure 2(a), (b)).
Figure 2.
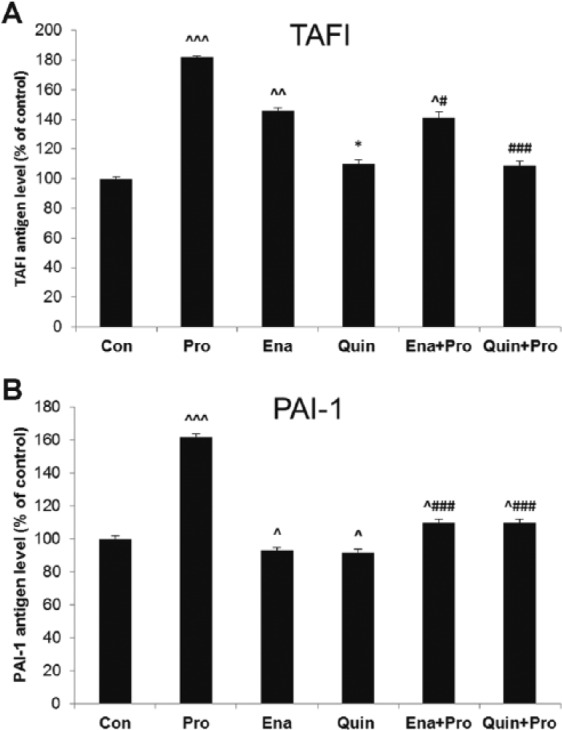
The effect of propofol and angiotensin-converting enzyme inhibitors (ACE-Is) on (a) tissue activatable fibrinolysis inhibitor (TAFI) and (b) plasminogen activator inhibitor (PAI-1) antigen levels in supernatant of human umbilical vein endothelial cells (HUVECs). The results are presented as percentage of control (Con: adequate vehiculum). Pro: propofol; Ena: enalaprilat; Quin: quinaprilat; Ena+Pro: enalaprilat and propofol; Quin+Pro: quinaprilat and propofol. ^p < 0.05, ^^p < 0.01, ^^^p < 0.001 vs adequate vehiculum for each substance; #p < 0.05, ###p < 0.001 vs Pro; *p < 0.05 vs Ena.
NO bioavailability (expression of eNOS and iNOS and NO2/NO3 concentration)
Incubation of HUVECs with Pro increased iNOS mRNA expression remarkably when compared with Veh (0.61±0.08 2−ΔΔct vs 0.43±0.01 2−ΔΔct, 142%, p < 0.01), but we did not observe a significant rise in eNOS mRNA level and NO2/NO3 concentration in supernatant. ACE-Is reduced iNOS mRNA expression (Ena 0.31±0.01 2−ΔΔct vs 0.48±0.005 2−ΔΔct, 64%; Quin 0.31±0.02 2−ΔΔct vs 0.45±0.01 2−ΔΔct, 69%, p < 0.01 both), when compared with their Veh. ACE-Is did not influence the mRNA of eNOS significantly. Among ACE-Is, only Ena decreased NO2/NO3 levels (7.21±0.5 μM/L vs 9.02±0.9 μM/l, 80%, p < 0.05 vs Veh). Only Quin prevented an increase in iNOS expression after Pro (p < 0.05 vs Pro). There were no changes in eNOS mRNA expression and NO2/NO3 level after Pro and ACE-Is co-incubation (Figure 3(a), (b)).
Figure 3.
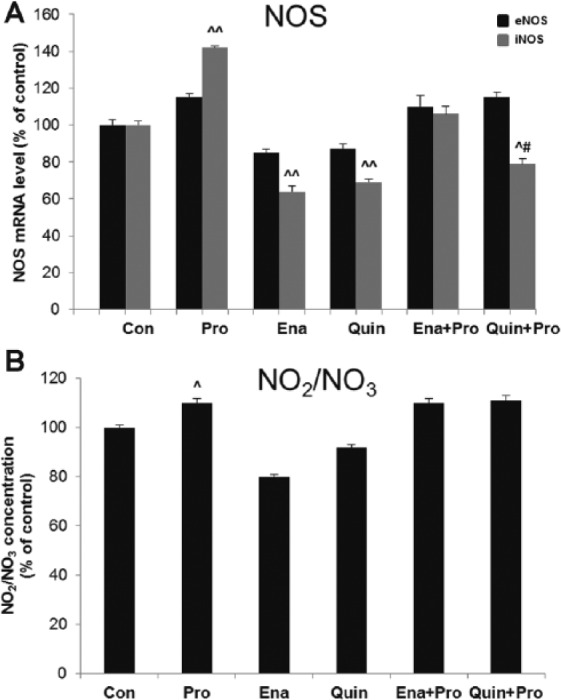
The effect of propofol and angiotensin-converting enzyme inhibitors (ACE-Is) on (a) the messenger RNA (mRNA) level of nitric oxide synthase (eNOS and iNOS) and (b) nitric oxide metabolites concentration (NO2/NO3) in supernatant of human umbilical vein endothelial cells (HUVECs). The results are presented as percentage of control (Con: adequate vehiculum). Pro: propofol; Ena: enalaprilat; Quin: quinaprilat; Ena+Pro: enalaprilat and propofol; Quin+Pro: quinaprilat and propofol. ^p < 0.05, ^^p < 0.01 vs adequate vehiculum for each substance; #p < 0.05 vs Pro.
Oxidative stress in endothelial cells after ACE-Is and/or Pro incubation
H2O2 concentration was increased in supernatant of HUVECs stimulated with Pro when compared with Veh (213±1.0 ng/ml vs 109±0.9 ng/ml, 195%, p < 0.001). Moreover, H2O2 concentration was the highest in Pro among all the studied groups. Among ACE-Is, only Quin significantly reduced the concentration of H2O2 (98±2 ng/ml vs 128±0.1 ng/ml, 76%, p < 0.01). The effect of Pro on H2O2 concentration was reduced in supernatant of HUVECs pretreated with Ena or Quin when compared with Pro alone (p < 0.001), although it was still higher when compared with ACE-Is (p < 0.01) (Figure 4(a)).
Figure 4.
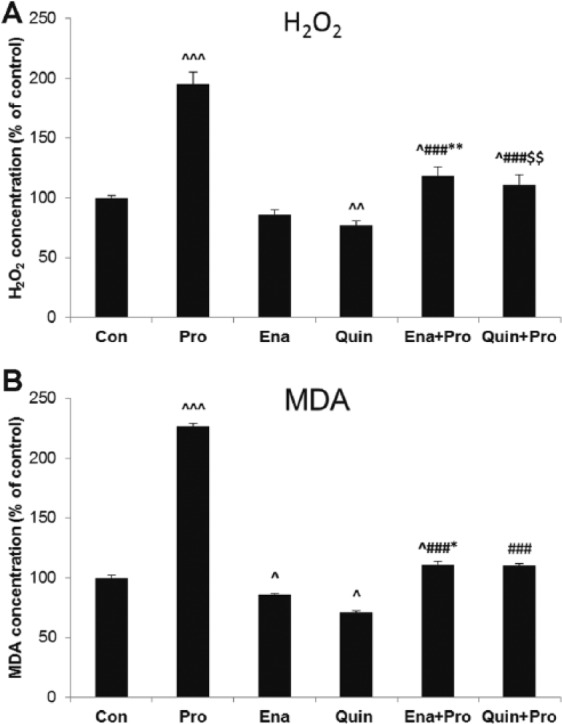
The effect of propofol and angiotensin-converting enzyme inhibitors (ACE-Is) on (a) hydrogen peroxide (H2O2) and (b) malondialdehyde concentrations (MDA) in supernatant of human umbilical vein endothelial cells (HUVECs). The results are presented as percentage of control (Con: adequate vehiculum). Pro: propofol; Ena: enalaprilat; Quin: quinaprilat; Ena+Pro: enalaprilat and propofol; Quin+Pro: quinaprilat and propofol. ^p < 0.05, ^^p < 0.01, ^^^p < 0.001 vs adequate vehiculum for each substance; ###p < 0.001 vs Pro; *p < 0.05, **p < 0.01 vs Ena; $$p < 0.01 vs Quin.
MDA concentration increased significantly in the Pro-treated group (0.41±0.11 pmol/mg vs 0.18±0.08 pmol/mg, 227%, p < 0.001 vs Veh). Conversely, ACE-Is decreased MDA concentration (Ena 0.16±0.07 pmol/mg vs 0.19±0.07 pmol/mg, 84% and Quin 0.14±0.04 pmol/mg vs 0.18±0.05 pmol/mg, 77%, p < 0.05) when compared with their Veh. The effect of Pro on MDA concentration was reduced in supernatant of HUVECs pretreated with Ena or Quin when compared with Pro alone (p < 0.001), although it was still higher when compared with ACE-Is (p < 0.05) (Figure 4(b)).
An increase in SOD mRNA expression was observed in supernatant of HUVECs treated with Pro (1.22±0.04 2−ΔΔct vs 0.99±0.04 2−ΔΔct, 123%, p < 0.01 vs Veh). Twenty-four-hour treatment of HUVECs with ACE-Is decreased SOD mRNA expression (Ena 0.55±0.05 2−ΔΔct vs 0.93±0.04 2−ΔΔct, 58%, p < 0.001 and Quin 0.53±0.03 2−ΔΔct vs 1.09±0.04 2−ΔΔct, 47%, p < 0.001) when compared with their Veh. Co-incubation with Pro and Quin evoked the highest increase in SOD expression when compared with Pro alone (p < 0.001) (Figure 5(a)).
Figure 5.
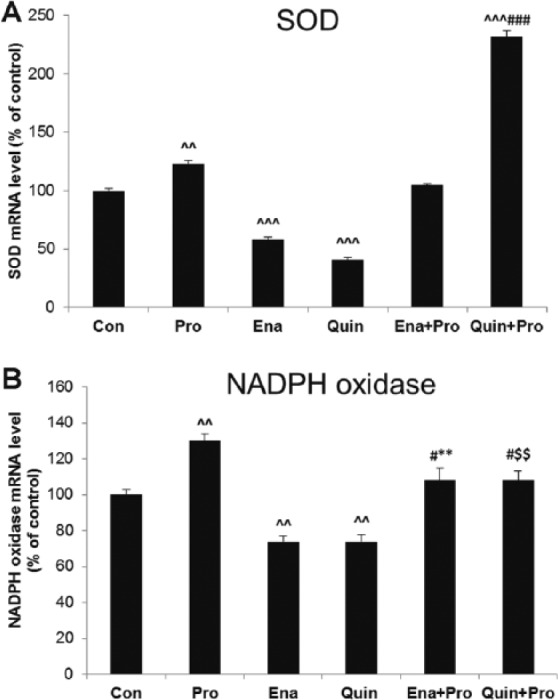
The effect of propofol and angiotensin-converting enzyme inhibitors (ACE-Is) on the messenger RNA (mRNA) levels of (a) superoxide dismutase (SOD) and (b) nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in supernatant of human umbilical vein endothelial cells (HUVECs). The results are presented as percentage of control (Con: adequate vehiculum). Pro: propofol; Ena: enalaprilat; Quin: quinaprilat; Ena+Pro: enalaprilat and propofol; Quin+Pro: quinaprilat and propofol. ^^p < 0.01, ^^^p < 0.001 vs adequate vehiculum for each substance; #p < 0.05, ###p < 0.001 vs Pro; **p < 0.01 vs Ena; $$p < 0.01 vs Quin.
NADPH oxidase expression was increased in the Pro group (1.43±0.3 2−ΔΔct vs 1.1±0.06 2−ΔΔct, 130%, p < 0.01 vs Veh). In contrast, ACE-Is reduced this pro-oxidative enzyme expression (Ena 0.80±0.03 2−ΔΔct vs 1.08±0.07 2−ΔΔct, and Quin 0.82±0.03 2−ΔΔct vs 1.12±0.04 2−ΔΔct, 74%, p < 0.01 for both). In the supernatant of HUVECs pretreated with both Pro and ACE-I, decreased NADPH oxidase expression was observed when compared with Pro alone (p < 0.05), although it was still higher when compared with ACE-Is (p < 0.01) (Figure 5(b)).
Discussion
The present in vitro study is an attempt to find out the role of endothelium in the antifibrinolytic and pro-oxidative effect of propofol and the influence of ACE-Is on its action. We evaluated the same parameters of fibrinolysis (t-PA, PAI-1, TAFI), NO bioavailability (NO2/NO3, NOS) and oxidative stress (H2O2, MDA, NADPH oxidase, SOD) as previously in our in vivo study.7 There are crucial correlations between those parameters of oxidative stress and fibrinolysis. It was shown that H2O2-induced oxidative stress in HUVECs caused an increase in PAI-1, urokinase plasminogen activator receptor (u-PAR), t-PA and urokinase-type plasminogen activator (u-PA) expression at the levels of mRNA, protein and promoter activity.22 It was also observed in HUVECs that NADPH oxidase is a major source of reactive oxygen species (ROS) in endothelial cells responsible for PAI-1 upregulation.23 Moreover, in vivo studies confirm the role of oxidative stress in hemostatic disorders and endothelial dysfunction leading to thrombosis. SOD administration inhibited platelet aggregation and improved endothelial function in the coronary arteries of dogs.24 Furthermore, SOD diminished venous thrombosis in rats, while reduction in NO bioavailability augmented this process.25 In our study we decided also to evaluate NO, since its bioavailability strictly depends on oxidative balance, thus NO could be considered as another oxidative stress marker.26
We found that propofol alone increased the release of antifibrinolytic and pro-oxidative factors from endothelium. We also found that 30-minute incubation of HUVECs in the presence of propofol following ACE-Is pre-incubation caused weakness in the antifibrinolytic and pro-oxidative potential of the propofol, and this effect was similar after both ACE-Is. We also confirmed that both ACE-Is increased endothelial fibrinolytic activity and decreased oxidative stress in HUVECs.
ACE-Is
The profibrinolytic activity of ACE-Is, including increase of t-PA, and decrease of PAI-1 and TAFI concentrations, was proved in our previous studies in normotensive and 2K-1C rats.1,2,7 Now we have proved that ACE-Is stimulated an increase of t-PA release from the endothelium, which suggests their effect on tissue ACE.
We observed that ACE-Is enhanced the fibrinolytic activity of the endothelium but did not influence eNOS and NO levels. Correspondingly, we showed that the hypotensive and profibrinolytic effect of ACE-Is was not connected with increased eNOS expression and NO level in hypertensive rats.7 Our in vitro results indicate the NO-independent activity of ACE-Is. Similar conclusions came from an in vivo study with ACE-Is-induced bradykinin-dependent thrombolysis in rats, which was mediated mainly by PGI2, and NO played a minor role.27
Therefore, it may be assumed that the release of t-PA from endothelial cells is not correlated with an increase of NO but is mediated by other factors. According to others, bradykinin-dependent t-PA release from the endothelium was solely mediated by EDHF signaling pathways, through a calcium and G protein-dependent way.28 The authors emphasized the lack of contribution of NO to bradykinin-stimulated t-PA release. The in vitro results confirmed data in vivo that the dominant mechanism of profibrinolitic activity of ACE-Is is increasing t-PA concentration by bradykinin-EDHF stimulation and decreasing its degradation through PAI-1.28–31 We also observed that ACE-Is reduced TAFI concentration in HUVECs. There are very few and contradictory studies reporting the expression of TAFI in HUVECs.32,33 Although we found quinaprilat reduced TAFI concentration, the mechanism is still unclear and seems to be independent of PAI-1.34
In our study, a significant decrease of iNOS mRNA was observed, which may suggest the anti-inflammatory effect of ACE-Is. It was suggested that the mechanism of ACE-Is-attenuated iNOS expression may be associated with a reduction of superoxide production.35 Similarly, quinapril downregulated iNOS with a possible tumor necrosis factor alpha (TNF-α)-mediated mechanism in normotensive rats.36 The present study allowed us to highlight the significant participation of endothelium in decreasing SOD, NADPH oxidase, H2O2, and MDA concentrations after ACE-Is. It seems that the antioxidant function of quinaprilat and enalaprilat is modified through lowering Ang II formation or promoting Ang-(1–7) production, which reduces the increase in ROS and phosphorylation of c-Src kinase by Ang II in intact human endothelial cells.37
Propofol
The effect of propofol on hemostasis in HUVECs has not been evaluated so far. We observed that propofol increased PAI-1 and TAFI and decreased t-PA concentration, with simultaneous growth of iNOS expression and increased levels of oxidative stress parameters. These results are similar to those obtained in our previous study in 2K-1C rats infused with propofol at a dose of 15 mg/kg.7 Propofol given in doses of 10–15 mg/kg achieved a concentration in rat plasma of 3 μg/ml, lasting up to 30 minutes, which enables achieving effective anesthesia.38 The concentration of propofol (50 μM) used in this study and time of incubation (30 minutes) corresponded also with the concentration of propofol in the blood of patients (3–6 μg/ml) during short anesthesia.39
We suggest a multiple mechanism of propofol antifibrinolytic action. It is known that propofol, as a lipophilic agent, may disturb the continuity of cell membranes mechanically and reduce the glycocalyx barrier on endothelial cells.40 Thus the direct effect of propofol on endothelial cells, depending strictly on its physicochemical properties, may further disturb the release of the endothelial mechanism of t-PA, PAI-1 and other factors. Moreover, there are some studies indicating a direct effect of propofol on shape and viscoelasticity of erythrocytes and platelets related to intercalation of propofol in the cell membrane.41 We also cannot exclude a simple direct effect of propofol on t-PA synthesis, since there are some data indicating that short incubation with propofol (30, 60 minutes) may change the expression of different endothelial factors at a protein level.42,43 Finally, the antifibrinolytic effect of propofol may be a result of t-PA and PAI-1 interaction, leading to inactive complex44 formulation. A similar conclusion was reached by others in a study with cultured rat astrocytes incubated with propofol.45
In our study propofol increased iNOS expression without affecting NO metabolites level and eNOS expression. It was shown in HUVECs that propofol (50 µM) up-regulated eNOS expression by increasing phosphorylation of eNOS at Ser1177, which is responsible for endothelial-derived NO production.46 It was proved that propofol stimulated NO production in a concentration-dependent manner (0.03–1 mM) in cultured porcine aortic endothelial cells.47 However, in our study the changes in NO concentration were not observed, probably because of a longer time of incubation with propofol (30 minutes vs 10 minutes) and possible increased degradation of NO. Furthermore, in the face of an increase in NADPH oxidase activity observed in the propofol group, NO may be converted into superoxide anions, e.g. peroxynitrite (ONOO−). The upregulation of iNOS expression after incubation with propofol may be a source of ROS, which is in line with the H2O2 and MDA concentration increase observed here. Similarly, in 2K-1C rats, we observed an increase of iNOS expression and oxidative stress parameters, but also increased NO level, after propofol administration.7 Thus, we suggest that NO release from the endothelium was shear stress dependent.
Interestingly, propofol also increased SOD activity in HUVECs, which remains in line with our previous results. Higher SOD activity catalyzed dismutation of the strongly toxic superoxide anion to less toxic hydrogen peroxidase. Moreover, the antioxidative effects of propofol in the ischemic liver tissue of rats, as a decreased MDA level and increased SOD level, was detected.48 The authors assumed that a low concentration of NO may serve to promote cell survival and protect the liver against ischemia/reperfusion injury. Also in HUVECs, the protective effect of propofol against heat stress-induced cell injury is associated with the induction of manganese (Mn)SOD expression.49 This more pronounced antioxidant effect of propofol seems to be dose and time dependent, because in both studies the dose (two- to three-fold higher) and time of incubation (six hours vs 30 minutes) were higher than in our study. A possible explanation of the partial antioxidant properties of propofol is its structural similarity to alfa-tocoferol, which increases SOD activity.49
ACE-Is and propofol
We found that ACE-Is caused weakness of the antifibrinolytic potential of propofol, since we observed an increase of t-PA and decrease of TAFI and PAI-1 levels. Moreover, co-incubation with quinaprilat and propofol reduced propofol-induced iNOS expression. This may be a result of the antioxidative effect of ACE-Is, since in the propofol group pre-incubated with ACE-Is we observed a significant reduction in H2O2 and MDA levels and NADPH oxidase expression. Interestingly, quinaprilat potentiated propofol-induced SOD expression, which together with a reduced iNOS expression may suggest a catalyzed dismutation of ROS. Similarly, in our previous study in 2K-1C rats, quinapril decreased the unfavorable effects of propofol in fibrinolysis and its pro-oxidative potential.7
Our observation suggests that the studied ACE-Is exerted at least partial protective effects against endothelial cell dysfunction caused by propofol, independently of hemodynamics. The above effect of ACE-Is on propofol activity did not depend on NO bioavailability.
On the other hand, bearing in mind our in vivo and in vitro results, the replacement of propofol by another intravenous anesthetic in patients treated with ACE-Is may be essential to maintain the pleiotropic (profibrinolytic and antioxidative) effects of ACE-Is in hemostasis. However, there are very few studies evaluating the effect of other intravenous anesthetics (ketamine, etomidate) on oxidative balance and hemostasis. What is more, the results of these studies are inconsistent (pro-oxidative and antioxidative effects as well), which may be a result of different experimental protocols used (species, doses, time of treatment, concomitant pathological state, tissue where oxidative stress parameters are measured). Nevertheless, further studies regarding the effects of anesthetics and ACE-Is co-treatment may have important clinical implications.
Conclusion
In the present study in HUVECs, we showed for the first time that propofol diminished profibrinolytic and antioxidative activity of endothelial cells. ACE-Is exerts partial protective effects against endothelial dysfunction caused by propofol, in the NO-independent mechanism. The complexity of the endothelial activity of both studied groups of drugs, which are commonly used in patients undergoing surgery, requires further exploration.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the projects No N/ST/MN/16/001/2226 and No N/ST/ZB/16/002/2226 of the Medical University of Bialystok.
References
- 1. Wojewodzka-Zelezniakowicz M, Chabielska E, Mogielnicki A, et al. Antithrombotic effect of tissue and plasma type angiotensin converting enzyme inhibitors in experimental thrombosis in rats. J Physiol Pharmacol 2006; 57: 231–245. [PubMed] [Google Scholar]
- 2. Chabielska E, Mogielnicki A, Kramkowski K, et al. Antithrombotic effect of captopril and enalapril in old rats. Pharmacol Rep 2005; 57: 135–137. [PubMed] [Google Scholar]
- 3. Kucharewicz I, Pawlak R, Matys T, et al. Antithrombotic effect of captopril and losartan is mediated by Angiotensin-(1–7). Hypertension 2002; 40: 774–779. [DOI] [PubMed] [Google Scholar]
- 4. Islim IF, Bareford D, Beevers DG. A single (investigator)-blind randomised control trial comparing the effects of quinapril and nifedipine on platelet function in patients with mild to moderate hypertension. Platelets 2001; 12: 274–288. [DOI] [PubMed] [Google Scholar]
- 5. Zurbano MJ, Anguera I, Heras M, et al. Captopril administration reduces thrombus formation and surface expression of platelet glycoprotein IIb/IIa in early postmyocardial infarction stage. Arterioscler Thromb Vasc Biol 1999; 19: 1791–1795. [DOI] [PubMed] [Google Scholar]
- 6. Korbut RA, Madej J, Adamek-Guzik T, et al. Secretory dysfunction of vascular endothelium limits the effect of angiotensin converting enzyme inhibitor quinapril on aggregation of erythrocytes in experimental hypertension. J Physiol Pharmacol 2003; 54: 397–408. [PubMed] [Google Scholar]
- 7. Wojewodzka-Zelezniakowicz M, Kisiel W, Kramkowski K, et al. Quinapril decreases antifibrinolytic and prooxidative potential of propofol in arterial thrombosis in hypertensive rats. J Renin Angiotensin Aldosterone Syst 2016; 17: 1470320316647239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ozkor MA, Murrow JR, Rahman AM, et al. Endothelium-derived hyperpolarizing factor determines resting and stimulated forearm vasodilator tone in health and in disease. Circulation 2011; 123: 2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kähönen M, Mäkynen H, Wu X, et al. Endothelial function in spontaneously hypertensive rats: Influence of quinapril treatment. Br J Pharmacol 1995; 115: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi N, Honda T, Yoshida K, et al. Critical role of bradykinin-eNOS and oxidative stress-LOX-1 pathway in cardiovascular remodeling under chronic angiotensin-converting enzyme inhibition. Atherosclerosis 2006; 187: 92–100. [DOI] [PubMed] [Google Scholar]
- 11. Moinuddin G, Inamdar MN, Kulkarni KS, et al. Modulation of hemodynamics, endogenous antioxidant enzymes, and pathophysiological changes by Angiotensin-converting enzyme inhibitors in pressure-overload rats. Hellenic J Cardiol 2011; 52: 216–226. [PubMed] [Google Scholar]
- 12. Corcoran TB, O’Shea A, Engel A, et al. The influence of propofol on P-selectin expression and nitric oxide production in re-oxygenated human umbilical vein endothelial cells. Acta Anaesthesiol Scand 2006; 50: 348–354. [DOI] [PubMed] [Google Scholar]
- 13. Ozgul U, Ucar M, Erdogan MA, et al. Effects of isoflurane and propofol on hepatic and renal functions and coagulation profile after right hepatectomy in living donors. Transplant Proc 2013; 45: 966–970. [DOI] [PubMed] [Google Scholar]
- 14. Wang B, Luo T, Chen D, et al. Propofol reduces apoptosis and up-regulates endothelial nitric oxide synthase protein expression in hydrogen peroxide-stimulated human umbilical vein endothelial cells. Anesth Analg 2007; 105: 1027–1033. [DOI] [PubMed] [Google Scholar]
- 15. Freshney RI. Culture of animal cells: A manual of basic technique. New York: Alan R. Liss Inc, 1987. [Google Scholar]
- 16. Mailloux A, Deslandes B, Vaubourdolle M, et al. Captopril and enalaprilat decrease antioxidant defences in human endothelial cells and are unable to protect against apoptosis. Cell Biol Int 2003; 27: 825–830. [DOI] [PubMed] [Google Scholar]
- 17. Hayase N, Satomi M, Hara A, et al. Protective effects of quinaprilat and trandolaprilat, active metabolites of quinapril and trandolapril, on hemolysis induced by lysophosphatidylcholine in human erythrocytes. Biol Pharm Bull 2003; 26: 712–716. [DOI] [PubMed] [Google Scholar]
- 18. Vasileiou I, Xanthos T, Koudouna E, et al. Propofol: A review of its non-anaesthetic effects. Eur J Pharmacol 2009; 1: 1–8. [DOI] [PubMed] [Google Scholar]
- 19. Winer J, Jung CK, Shackel I, et al. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem 1999; 15: 41–49. [DOI] [PubMed] [Google Scholar]
- 20. Gromotowicz A, Szemraj J, Stankiewicz A, et al. Study of the mechanisms of aldosterone prothrombotic effect in rats. J Renin Angiotensin Aldosterone Syst 2011; 12: 430–439. [DOI] [PubMed] [Google Scholar]
- 21. Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med 2007; 43: 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oszajca K, Bieniasz M, Brown G, et al. Effect of oxidative stress on the expression of t-PA, u-PA, u-PAR, and PAI-1 in endothelial cells. Biochem Cell Biol 2008; 86: 477–486. [DOI] [PubMed] [Google Scholar]
- 23. Jaulmes A, Sansilvestri-Morel P, Rolland-Valognes G, et al. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells. Thromb Res 2009; 124: 439–446. [DOI] [PubMed] [Google Scholar]
- 24. Yao SK, Ober JC, Gonenne A, et al. Active oxygen species play a role in mediating platelet aggregation and cyclic flow variations in severely stenosed and endothelium-injured coronary arteries. Circ Res 1993; 73: 952–967. [DOI] [PubMed] [Google Scholar]
- 25. Peire MA, Puig-Parellada P. Oxygen-free radicals and nitric oxide are involved in the thrombus growth produced by iontophoresis of ADP. Pharmacol Res 1998; 38: 353–356. [DOI] [PubMed] [Google Scholar]
- 26. Pierini D, Bryan NS. Nitric oxide availability as a marker of oxidative stress. Methods Mol Biol 2015; 1208: 63–71. [DOI] [PubMed] [Google Scholar]
- 27. Gryglewski RJ, Swies J, Uracz W, et al. Mechanisms of angiotensin-converting enzyme inhibitor induced thrombolysis in Wistar rats. Thromb Res 2003; 110: 323–329. [DOI] [PubMed] [Google Scholar]
- 28. Rahman AM, Murrow JR, Ozkor MA, et al. Endothelium-derived hyperpolarizing factor mediates bradykinin-stimulated tissue plasminogen activator release in humans. J Vasc Res 2014; 51: 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ridderstrale W, Ulfhammer E, Jern S, et al. Impaired capacity for stimulated fibrinolysis in primary hypertension is restored by antihypertensive therapy. Hypertension 2006; 47: 686–691. [DOI] [PubMed] [Google Scholar]
- 30. Brown NJ, Gainer JV, Stein CM, et al. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension 1999; 33: 1431–1435. [DOI] [PubMed] [Google Scholar]
- 31. Fleming I, Michaelis UR, Bredenkotter D, et al. Endothelium-derived hyperpolarizing factor synthase (cytochrome p450 2c9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 2001; 88: 44–51. [DOI] [PubMed] [Google Scholar]
- 32. Hori Y, Gabazza EC, Yano Y, et al. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J Clin Endocrinol Metab 2002; 87: 660–665. [DOI] [PubMed] [Google Scholar]
- 33. Aubert H, Frère C, Aillaud MF, et al. Weak and non-independent association between plasma TAFI antigen levels and the insulin resistance syndrome. J Thromb Haemost 2003; 1: 791–797. [DOI] [PubMed] [Google Scholar]
- 34. Chudy P, Kotulicova D, Stasko J, et al. The relationship among TAFI, t-PA, PAI-1 and F1 + 2 in type 2 diabetic patients with normoalbuminuria and microalbuminuria. Blood Coagul Fibrinolysis 2011; 22: 493–498. [DOI] [PubMed] [Google Scholar]
- 35. Chou TC, Yen MH, Li CY, et al. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 1998; 31: 643–648. [DOI] [PubMed] [Google Scholar]
- 36. Bachetti T, Comini L, Pasini E, et al. ACE-inhibition with quinapril modulates the nitric oxide pathway in normotensive rats. J Mol Cell Cardiol 2001; 33: 395–403. [DOI] [PubMed] [Google Scholar]
- 37. Sampaio WO, Henrique de CC, Santos RA, et al. Angiotensin-(1–7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension 2007; 50: 1093–1098. [DOI] [PubMed] [Google Scholar]
- 38. Beaudry F, Guenette SA, Winterborn A, et al. Development of a rapid and sensitive LC-ESI/MS/MS assay for the quantification of propofol using a simple off-line dansyl chloride derivatization reaction to enhance signal intensity. J Pharm Biomed Anal 2005; 39: 411–417. [DOI] [PubMed] [Google Scholar]
- 39. Vasileiou I, Xanthos T, Koudouna E, et al. Propofol: A review of its non-anaesthetic effects. Eur J Pharmacol 2009; 1: 1–8. [DOI] [PubMed] [Google Scholar]
- 40. Lin MC, Lin CF, Li CF, et al. Anesthetic propofol overdose causes vascular hyperpermeability by reducing endothelial glycocalyx and ATP production. Int J Mol Sci 2015; 16: 12092–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reinhart WH, Felix CH. Influence of propofol on erythrocyte morphology, blood viscosity and platelet function. Clin Hemorheol Microcirc 2003; 29: 33–40. [PubMed] [Google Scholar]
- 42. Zhu M, Chen J, Jiang H, et al. Propofol protects against high glucose-induced endothelial adhesion molecules expression in human umbilical vein endothelial cells. Cardiovasc Diabetol 2013; 12: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsai YC, Huang CC, Chu LM, et al. Differential influence of propofol on different cell types in terms of the expression of various oxidative stress-related enzymes in an experimental endotoxemia model. Acta Anaesthesiol Taiwan 2012; 50: 159–166. [DOI] [PubMed] [Google Scholar]
- 44. Kruithof EK, Tran-Thang C, Ransijn A, et al. Demonstration of a fast-acting inhibitor of plasminogen activators in human plasma. Blood 1984; 64: 907–913. [PubMed] [Google Scholar]
- 45. Ko HM, Joo SH, Lee SH, et al. Propofol treatment modulates neurite extension regulated by immunologically challenged rat primary astrocytes: A possible role of PAI-1. Arch Pharm Res 2015; 38: 556–565. [DOI] [PubMed] [Google Scholar]
- 46. Wang DL, Wung BS, Peng YC, et al. Mechanical strain increases endothelin-1 gene expression via protein kinase C pathway in human endothelial cells. J Cell Physiol 1995; 163: 400–406. [DOI] [PubMed] [Google Scholar]
- 47. Petros AJ, Bogle RG, Pearson JD. Propofol stimulates nitric oxide release from cultured porcine aortic endothelial cells. Br J Pharmacol 1993; 109: 6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Z, Yu J, Wu J, et al. The effects of two anesthetics, propofol and sevoflurane, on liver ischemia/reperfusion injury. Cell Physiol Biochem 2016; 38: 1631–1642. [DOI] [PubMed] [Google Scholar]
- 49. Wu F, Dong XJ, Zhang HQ, et al. Role of MnSOD in propofol protection of human umbilical vein endothelial cells injured by heat stress. J Anesth 2016; 30: 410–419. [DOI] [PubMed] [Google Scholar]