

Abstract
Levels of angiotensin converting enzyme 2 (ACE2), a cardio and neuro-protective carboxypeptidase, are dynamically altered after stroke in preclinical models. We sought to characterize the previously unexplored changes in serum ACE2 activity of stroke patients and the mechanism of these changes. Serum samples were obtained from patients during acute ischemic stroke (n=39), conditions mimicking stroke (stroke-alert, n=23), or from control participants (n=20). Enzyme activity levels were analyzed by fluorometric assay and correlated with clinical variables by regression analyses. Serum ACE2 activity was significantly lower in acute ischemic stroke as compared to both control and stroke-alert patients, followed by an increase to control levels at three days. Serum ACE2 activity significantly correlated with the presence of ischemic stroke after controlling for other factors (P=0.01). Additional associations with ACE2 activity included a positive correlation with systolic blood pressure at presentation in stroke-alert (R2=0.24, P=0.03), while stroke levels showed no correlation (R2=0.01, P=0.50). ACE2 sheddase activity was unchanged between groups. These dynamic changes in serum ACE2 activity in stroke, which concur with preclinical studies, are not likely to be driven primarily by acute changes in blood pressure or sheddase activity. These findings provide new insight for developing therapies targeting this protective system in ischemic stroke.
Keywords: Angiotensin converting enzyme 2 (ACE2), angiotensin converting enzyme (ACE), renin–angiotensin system, ischemic stroke, angiotensin-(1–7)
Introduction
Despite the important role of the renin–angiotensin system in regulating blood pressure and cardiovascular health, surprisingly little is known regarding its activity during acute ischemic stroke (AIS). The discovery and characterization of a protective pathway within this system, the angiotensin converting enzyme 2/angiotensin-(1–7)/Mas (ACE2/Ang-(1–7)/Mas) axis, has increased the potential impact of an improved understanding of the regulation of this system in AIS.1 By catalyzing the conversion of angiotensin II (Ang II) into Ang-(1–7), ACE2 counteracts the deleterious effects that result from sustained over-activation of the Ang II type 1 receptor. Binding of Ang-(1–7) to its receptor Mas initiates anti-inflammatory, anti-oxidative and vasodilatory effects in a variety of disease states.2 In the context of preclinical studies of AIS, activation of this protective axis by targeted interventions has been proved to induce neuroprotection reproducibly.3 We have demonstrated that ACE2 activation induces significant neuroprotection in a rat model of ischemia when administered via systemic injections starting 4 hours after stroke.4
Studies of the role of ACE2 in human disease, and especially in stroke, are limited. One recent study found that serum ACE2 levels within the first 24 hours post-stroke differ slightly by stroke subtype with lacunar exhibiting lower levels than cardioembolic strokes.5 In rats, expression and activity of ACE2 in ischemic brain tissue is altered,6 and we demonstrated that serum ACE2 activity is initially decreased, followed by rebound increases over the next several days.4 Validating this decrease in human disease will provide further support for targeting ACE2 with neuroprotective interventions.
We assessed the changes in endogenous ACE2 activity in human serum of patients experiencing AIS as compared to patients with symptoms mimicking stroke for whom the early stroke-alert system was activated (stroke-alert) or to control participants. Furthermore, we explored angiotensin converting enzyme (ACE) activity and associations between changes in enzyme activity and clinical measures and the mechanisms underlying these changes.
Methods
Compliance with ethical standards
The collection and use of human samples and clinical data was by written informed consent in a manner approved by the Institutional Review Board of the University of Florida (IRB201200330) and according to the World Medical Association Declaration of Helsinki. This preliminary observational study has been registered with clinicaltrials.gov (NCT02409043).
Subjects and sample collection
Samples of peripheral blood, collected in serum tubes, were obtained between July 2014 and October 2015 at UF Health & Shands hospital. Samples were from 39 acute ischemic stroke patients (AIS group) at an average of 3.6 hours after symptom onset, from 20 control participants consisting primarily of patient spouses and adult children (control group), and from 23 patients presenting with symptoms mimicking stroke for whom the early stroke-alert system was activated prior to determination of non-stroke etiology of symptoms (stroke-alert group). Physiological parameters, including systolic blood pressure, were not collected from control participants. Blood plasma samples from healthy young adults, obtained from a biorepository, were also analyzed.7 Magnetic resonance scans from these stroke-alert patients did not show acute restrictions on diffusion weighted images (DWI). A second phlebotomy was performed in 15 AIS patients who remained hospitalized three days post-stroke, and volumetric analysis was calculated from magnetic resonance DWIs from 17 AIS patients at approximately 24 hours after stroke. Samples were immediately placed on ice, followed by centrifugation for 10 minutes at 3000 rpm. Serum aliquots were frozen and stored at −80°C until analysis. Clinical and demographic variables were obtained from patient interviews and medical records. Modified Rankin score at follow-up one to six months after stroke was available in 28 AIS patients.
Inclusion and exclusion criteria
AIS and stroke-alert patients presenting within 6 hours of symptom onset were considered for inclusion. Patients with subarachnoid hemorrhage or subdural hematoma were excluded. Stroke was confirmed by the presence of acute lesions on magnetic resonance DWIs.
Fluorometric enzymatic activity assays
Activities of ACE2, ACE, and tumor necrosis factor-alpha converting enzyme (TACE) were assessed by enzyme activity assay involving continuous fluorometric cleavage of specific substrates using a method described previously4 as detailed below. For ACE2 activity assays, human serum samples were first processed to remove endogenous inhibitors of ACE2.8 Testing of serum samples before processing revealed no ACE2 activity. This was done by diluting 250 µL serum into 1.2 mL of low ionic strength buffer (20 mM Tris-HCl, pH 6.5), and mixing for 30 minutes with 200 µl ANX Sepharose 4FastFlow binding resin (GE Healthcare, #17-1287-01) that was prewashed in 1 mL buffer. After spinning for 10 minutes at 1200 rpm, the precipitated resin was washed again with 1.2 mL of buffer, centrifuged again, and protein was eluted by washing with 500 µL of high salt buffer (1 M NaCl, 20 mM Tris-HCl, pH 6.5). Supernatant containing eluted protein was decanted following a final spin-down and stored at −80°C until activity assay. Samples were incubated for one hour in duplicate wells in black flat-bottomed 96-well plates in 100 µL of reaction mixture. For ACE2 activity assays, the reaction mixture contained 75 μL processed serum, 10 µmol/L captopril, and 20 μmol/L fluorogenic Mca-APK(Dnp) ACE2 substrate (Enzo Life Sciences, BML-P163-0001) in ACE2 buffer (1 mol/L NaCl, 75 mmol/L Tris HCl, pH 7.5, and 50 µmol/L ZnCl2). For ACE activity, the reaction mixture contained 0.2 µL of unprocessed serum and 10 μmol/L Mca-RPPGFSAFK(Dnp)-OH ACE substrate (R&D Systems, #ES005) in ACE2 buffer. Due to differences in relative fluorescence levels induced by ACE2 activity found in plasma versus serum samples, human plasma samples (Figure 2(b)) from healthy young adults did not undergo sample processing for inhibitor removal, and fluorometric cleavage assay was performed as described for serum samples with the following changes: the reaction mixture contained 4 μL unprocessed plasma sample and 20 μM ACE2 substrate (Enzo Life Sciences) without captopril in ACE2 buffer. For TACE activity, reaction mixture contained 20 µL of unprocessed serum and 10 µmol/L fluorogenic TACE substrate (Enzo Life Sciences, BML-P132-0500) in TACE buffer (100 mmol/L NaCl, 50 mmol/L Tris HCl, pH 7.5, 100 µmol/L ZnCl2, 10 mmol/L CaCl2). From Synergy Mx Microplate Reader (BioTek Instruments, Inc.) measurements, relative fluorescence unit (RFU) per minute was calculated from minutes 30 to 60. Reaction Km and Vmax were determined using control samples and recombinant human ACE2 (R&D Systems, Inc., #933-ZN-010) as a positive control, and all samples were run in duplicate. Incubation of processed human serum with specific ACE2 inhibitor MLN-4760 (100 µmol/L, Millipore Sigma) completely blocked the observed ACE2 activity. A small number of samples from control (n=1) and mimic (n=3) patients exhibited enzyme activity that was outside of the level of detection.
Figure 2.
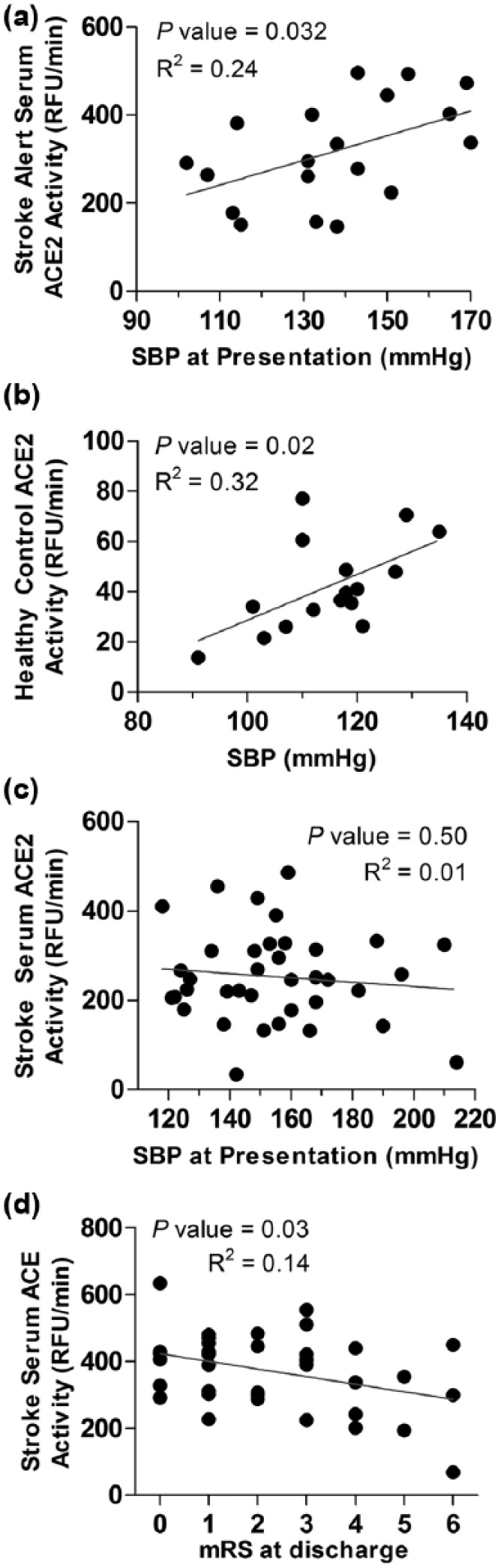
Serum ACE2 activity is significantly correlated with SBP in stroke-alert patients and healthy young adults, but not AIS patients. Correlation graphs of ACE2 activity and SBP among stroke-alert patients (a) and healthy young adults (b) as compared to stroke patients (c). Young adult blood plasma samples in panel (b) were from a biorepository established by Wegman et al.,7 which were obtained from research participants undergoing baseline measurements. (d) Correlation graph of ACE activity and mRS at discharge from hospital among AIS patients. ACE2: angiotensin converting enzyme 2; AIS: acute ischemic stroke; mRS: modified Rankin score; RFU: relative fluorescence unit; SBP: systolic blood pressure.
Statistics
Enzyme activity was assessed by blinded investigators and is presented as mean±SEM. Analysis assuming a 20% reduction in serum ACE2 activity in AIS and SD of 30% as observed in our preclinical studies, with power of 0.8 and α<0.05, calculated a sample size of 28. Significance was assessed by one-way analysis of variance with post-hoc t-test with Welch’s correction for comparisons of enzyme activity level by group, by two-way Mann–Whitney tests for comparison of baseline characteristics, and by two-way Wilcoxon matched-pairs signed rank test to compare acute versus convalescent enzyme activity levels. Enzyme activity levels were normally distributed. Multiple linear and logistic regression analyses were performed using SAS version 9.4 (SAS Institute) with the designation of the dependent variable as the presence of ischemic stroke and independent variables as level of ACE2 activity, age, race, sex, and history of diabetes and hypertension. These variables were selected due to their relevance as potential stroke risk factors. One outlier from the acute time point for each enzyme activity assay was identified as ⩾2.5 times the absolute deviation around the median9 and removed. For correlation analyses of systolic blood pressure (SBP) with activity levels, one outlier ⩾2.5 times the standard deviation around the mean was removed from each group. Statistical significance was assumed at α<0.05.
Results
Ischemic stroke and serum activity of ACE2 and ACE
Baseline participant characteristics are presented in Table 1. The AIS group included 24 large vessel/cardioembolic and 15 small vessel/lacunar strokes, and intravenous tissue plasminogen activator (IV tPA) was administered in 25 AIS patients. The stroke-alert group, five of whom received IV tPA, included patients presenting with symptoms later determined to be due to transient ischemic attack (n=4), atypical migraine (n=7), hypertensive encephalopathy (n=1), seizure (n=3), altered mental status (n=3), or idiopathic or other causes (n=5). Of the participants in the control group made up primarily of patient spouses and family members, 50% indicated a history of hypertension (see Table 1). Variables that were significantly higher in the AIS group are identified in Table 1 and included age and SBP at presentation, although they were not different in risk factors such as history of hypertension and diabetes.
Table 1.
Characteristics of research participants.
| CONT (n=20) | ALERT (n=23) | AIS (n=39) | |
|---|---|---|---|
| Age, years, mean ± SD | 58.2 ± 13 | 56.8 ± 15.4 | 70.7 ± 14.8a,b |
| Female/male | 10/10 | 12/11 | 15/24 |
| Race, n (%) | |||
| Caucasian | 15 (75%) | 16 (70%) | 29 (74%) |
| African American | 2 (10%) | 4 (17%) | 8 (21%) |
| Hispanic | 2 (10%) | 3 (13%) | 2 (5%) |
| Indian | 1 (5%) | 0 (0%) | 0 (0%) |
| Hypertension, n (%) | 10 (50%) | 18 (78%) | 29 (74%) |
| Diabetes, n (%) | 5 (25%) | 11 (48%) | 20 (51%) |
| Hyperlipidemia, n (%) | – | 13 (57%) | 27 (69%) |
| AFib, n (%) | – | 2 (9%) | 8 (21%) |
| Smoking, n (%) | – | 9 (39%) | 8 (21%) |
| Prior stroke, n (%) | – | 9 (39%) | 11 (28%) |
| BMI, kg/m2, mean ± SD | – | 29.8 ± 5.1 | 28.9 ± 6.5 |
| SBPp, mmHg, mean ± SD | – | 135.8 ± 19.8 | 156.7 ± 26.6c |
| DBPp, mmHg, mean ± SD | – | 75.9 ± 14.4 | 79.4 ± 16.6 |
| WBC, count*109/L, mean ± SD | – | 8.2 ± 3.2 | 9.4 ± 2.9 |
| Hemoglobin, g/dL, mean ± SD | – | 13.2 ± 2.1 | 13.3 ± 2.1 |
| Hematocrit, %, mean ± SD | – | 39.2 ± 6.2 | 39.9 ± 6.2 |
| Sodium, mmol/L, mean ± SD | – | 139.6 ± 3.3 | 139.6 ± 3.1 |
| Potassium, mmol/L, mean ± SD | – | 3.9 ± 0.6 | 4.2 ± 0.5 |
| Creatinine, mg/dL, mean ± SD | – | 1.4 ± 1.4 | 1.3 ± 0.6 |
| Glucose, mg/dL, mean ± SD | – | 151 ± 90.5 | 132.3 ± 43.0 |
| ProTime, seconds, mean ± SD | – | 13.6 ± 1.4 | 13.6 ± 1.1 |
| INR, ratio, mean ± SD | – | 1.04 ± 0.14 | 1.05 ± 0.10 |
| ACEip, n (%) | – | 4 (17%) | 7 (18%) |
| ARBp, n (%) | – | 5 (22%) | 4 (10%) |
| tPA treatment, n (%) | – | 5 (22%) | 25 (64%)b |
| % ejection fraction, median (IQR) | – | 60 (55–60) | 55 (55–60) |
| NIHSSp, median (IQR) | – | 4 (3–6) | 6 (3–12) |
| NIHSSd score, median (IQR) | – | – | 2 (1–5) |
| mRSd score, median (IQR) | – | – | 2 (1–3.25) |
| SBP72h, mmHg, mean ± SD | – | – | 146.2 ± 20.06 |
ACEip: angiotensin converting enzyme inhibitor previously prescribed; AFib: atrial fibrillation; AIS: acute ischemic stroke group; ALERT: stroke-alert group; ARBp: angiotensin receptor blocker previously prescribed; BMI: body mass index; DBPp: diastolic blood pressure at presentation; CONT: control group; Mimic: stroke mimic and transient ischemic attack group; INR: international normalized ratio; IQR: interquartile range; mRSd: modified Rankin scale; NIHSSp or d: national institutes of health stroke scale at presentation (p) or discharge (d); Protime: prothrombin time; SBPp: systolic blood pressure at presentation; SD: standard deviation; tPA: tissue plasminogen activator; WBC: white blood cell count.
P<0.01 versus control bP<0.01 versus mimic cP<0.05 versus mimic.
Our studies from rats revealed that AIS results in initial decreases in serum ACE2 activity when measured 4 hours after focal ischemia followed by rebound increases over three days.4 Consistent with these findings, levels of serum ACE2 activity among patients with AIS measured at an average of 3.6 hours after symptom onset were significantly lower than levels from control participants and from stroke-alert patients (Figure 1(a)). This decrease in ACE2 activity was reversed by three days post-stroke (Figure 1(b)), with the average serum ACE2 activity levels increasing by more than 40% during that time (<6 hours: 219.9 ± 17.5 RFU/minute vs. 3 days: 314.6 ± 38.95 RFU/minute, P<0.04). Although serum levels of ACE activity were not lower than control groups at the <6 hours acute time point (Figure 1(c)), they were significantly decreased by nearly 15% compared to acute levels at three days after stroke (Figure 1(d); <6 hours: 400.4 ± 26.3 vs. 3 days: 341.6 ± 19.5 RFU/minute, P<0.01). Activity of serum TACE, which exhibits sheddase activity by cleaving the ACE2 membrane bound to its soluble form, was not significantly affected as compared to levels from control or stroke-alert patients among AIS patients at either time point (data not shown).
Figure 1.
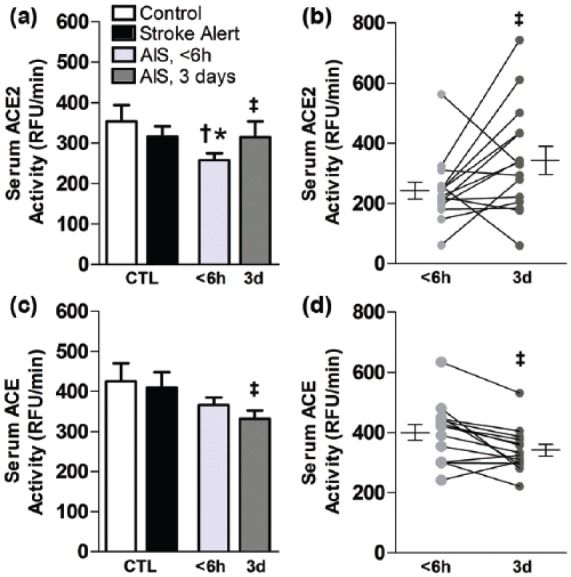
Activity of ACE2 and ACE in serum is altered following stroke. For human serum, bar graphs are means ± SEM and represent enzyme activity levels of ACE2 (a) and ACE (c) from control, stroke-alert, or AIS patients at an average of 3.6 hours and again at 3 days after stroke. Individual differences and means ± SEM in ACE2 (b) and ACE (d) are shown. *P<0.05 versus control and †P<0.05 versus stroke-alert. ‡P<0.05 versus AIS <6 hours. ACE: angiotensin converting enzyme; ACE2: angiotensin converting enzyme 2; AIS: acute ischemic stroke; RFU: relative fluorescence unit.
Among control, stroke-alert, and AIS patients, variables that were significantly correlated with the presence of AIS by multiple linear regression included increased age and lower levels of ACE2 activity when controlling for sex, race, and history of type II diabetes or hypertension (R2=0.29, P<0.001, Table 2). In addition, when controlling for these variables among these patients by multiple logistic regression analysis, those with serum ACE2 activity in the lowest two quartiles had four-fold increased odds ratio (95% confidence interval (CI) 1.02–15.3) of having suffered AIS as compared to those with levels in the top quartile. After controlling for ACE2 activity levels, along with age, sex, and history of diabetes and hypertension, it was found that among all participants, African American race was associated with 11.3 times increased odds ratio of having suffered an AIS as compared to Caucasian race (95% CI 1.3–99.0).
Table 2.
Predictors of acute ischemic stroke by multiple linear regression analysis.
| R2 model | Variable | β coefficient | 95% CI of β | t | P value |
|---|---|---|---|---|---|
| R2=0.29 | ACE2 activity | −0.00105 | −0.00187 to −0.00024 | −2.57 | 0.01 |
| Age | 0.01162 | 0.00485 to 0.01839 | 3.42 | 0.001 | |
| Sex | −0.13479 | −0.34323 to 0.07275 | −1.30 | 0.20 | |
| Race | −0.01457 | −0.16683 to 0.13769 | −0.19 | 0.85 | |
| Type II diabetes | 0.16093 | −0.07677 to 0.39862 | 1.35 | 0.18 | |
| Hypertension | −0.07220 | 0.5703 to −0.32473 | −0.57 | 0.57 |
ACE2: angiotensin converting enzyme 2; CI: confidence interval.
Values for groups were assigned as follows: acute ischemic stroke=1, control or stroke-alert=0; female sex=1, male sex=0; race: Indian=3, Hispanic=2, African American=1, Caucasian=0; history of type II diabetes, or hypertension=1, negative history=0.
Correlations of serum ACE2 and ACE activity with clinical measures
To evaluate the relationship of serum ACE2 activity and blood pressure, we performed correlation analyses among several groups. Activity of serum ACE2 among stroke-alert patients showed a significant positive correlation with SBP at presentation (Figure 2(a)). This was also true for ACE2 activity among a separate population of healthy young adults (Figure 2(b)). By contrast, SBP and ACE2 activity levels showed no correlation among patients experiencing AIS (Figure 2(c)). Analysis of other clinical variables – including measures of stroke severity, laboratory results, cardioembolic versus small vessel lacunar stroke, magnetic resonance image infarct volume, other baseline demographics – did not reveal additional associations with acute or three day post-stroke ACE2 activity among stroke-alert or AIS patients. Although differences were not significant, the average ACE2 activity at three days post-stroke among patients treated with tPA was 30% lower (3 days: 292.6 ± 50.42 RFU/minute) than levels from those who did not receive tPA treatment (3 days: 419.1 ± 83.03 RFU/minute, P=0.11). Finally, ACE activity was negatively correlated with modified Rankin scores at discharge from the hospital (Figure 2(d)), indicating that lower levels of ACE activity during AIS may be associated with worse functional outcomes, at least initially.
Discussion
The novel findings from this observational study of decreased ACE2 activity in human serum following stroke, followed by a return to control levels, and of associations with SBP add to an increasingly complex picture of ACE2 activity and cardiovascular health. In light of increasing preclinical evidence for a protective role of the ACE2–Ang-(1-7)-Mas axis in stroke,10 the decreased ACE2 activity in AIS may represent a promising target for future studies of experimental neuroprotective therapies in humans.
In considering the mechanism(s) that may be responsible for the observed changes in ACE2, we assessed for differences in the activity of TACE, which cleaves membrane bound ACE2 to form active soluble ACE2 contributing to altered serum ACE2 activity in hypertension,11 which may also play a role in stroke.4 Although we did not observe a significant change in TACE activity in the serum (data not shown), a change in vascular or brain TACE activity with resulting alterations in shedding of ACE2 into interstitial and vascular spaces may regulate the initially decreased serum ACE2 in stroke. We also explored the potential for a blood-pressure dependent mechanism of control over serum ACE2 activity in ischemic stroke. As we observed among stroke-alert patients and healthy young adults (Figure 2), others have reported positive correlations between soluble ACE2 activity and blood pressure.12 In AIS, however, we observed no correlation between SBP and ACE2 activity (Figure 2(c)). The wider range and higher pressures among the stroke patients as compared to other groups may be an important factor in this difference. The mechanisms responsible for these dynamic changes are unknown and are likely to be influenced differently during periods of rapid alterations or very high levels of blood pressure, such as during stroke, as compared to chronically altered pressures. The absence of a correlation between ACE2 activity and SBP in AIS does not rule out a contribution of blood pressure to the mechanism of ACE2 activity alterations, but suggests that other mechanisms are also likely to be involved. Interestingly, no correlation of ACE2 activity and SBP was observed 72 hours after stroke (data not shown), and SBP among AIS patients was not significantly lower by 72 hours as compared to pressures at presentation (Table 1).
Our analysis did not indicate a correlation between acute serum ACE2 activity and infarct volume, a finding that supports the interpretation of the observed ACE2 activity changes as being regulated physiologically by stroke-specific mechanisms, rather than by the magnitude of injury or as a dose-dependent/spill-over effect from damaged brain tissue. The increased odds ratio of AIS among African Americans after controlling for relevant factors, including ACE2 activity, highlights potentially important questions about the cause and effect relationship between these elevated ACE2 levels by race and other related stroke risk factors.
This preliminary study has several limitations. First, the use of enzymatic activity assays to measure system components provides accurate data regarding the levels of functional serum enzymes, but does not allow for a determination of the tissue and cellular sources of these enzymes, or the total protein levels. ACE2 functions in both membrane-bound and soluble states, and it is unclear which might provide the primary source of serum ACE2 activity in the setting of stroke, where ischemic cell death may be contributing to increased release of both types. Future studies to evaluate the role and effect of ACE2 shedding from various tissues in stroke are needed. Second, our study population was limited in number and exhibited substantial variability in risk factors and stroke mechanisms. The discovery of significant physiological differences within this small heterogeneous population may indicate potential for more substantial findings to be made from larger, more homogeneous populations. Finally, an evaluation of serum levels of Ang II and Ang-(1–7) as downstream effects or of renin activity as an upstream signal in the cascade could provide potentially valuable information regarding the downstream effects of the enzyme activity changes we have observed. The sample collection and preparation protocol in this study was found in our hands to be inadequate to assess these inherently less stable peptides reliably.
Conclusion
A clearer understanding of the alterations in activity of the renin–angiotensin system in ischemic stroke, and the consequences of these changes, has great potential to augment the development and testing of clinically viable therapies and diagnostic or prognostic tools. In this observational human study, we report the novel finding that stroke induces dynamic changes in the serum activity of ACE2, a key regulatory enzyme of the protective arm of the renin–angiotensin system, with initially decreased ACE2 activity during acute ischemic stroke, and convalescent increases by three days post-stroke. The novel evidence that ACE2 is dynamically altered in humans in a potentially detrimental way during stroke is consistent with observations from preclinical studies in which activation of ACE2 induces stroke neuroprotection, which suggests ACE2 as an important clinical target for future studies of stroke therapies.
Acknowledgments
The authors gratefully acknowledge the assistance of JinDong Xu in participant screening and sample collection.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Heart Lung and Blood Institute (2T32HL083810-06A1); the American Heart Association Greater Southeast Affiliate (12PRE11940010); and the McKnight Brain Institute of the University of Florida.
References
- 1. Pena-Silva RA, Heistad DD. Stages in discovery: angiotensin-converting enzyme type 2 and stroke. Hypertension 2015; 66: 15–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jiang F, Yang J, Zhang Y, et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol 2014; 11: 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennion DM, Haltigan E, Regenhardt RW, et al. Neuroprotective mechanisms of the ACE2-angiotensin-(1–7)-mas axis in stroke. Curr Hypertens Rep 2015; 17: 3–014–0512–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bennion DM, Haltigan EA, Irwin AJ, et al. Activation of the neuroprotective angiotensin-converting enzyme 2 in rat ischemic stroke. Hypertension 2015; 66: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mogi M, Kawajiri M, Tsukuda K, et al. Serum levels of renin–angiotensin system components in acute stroke patients. Geriatr Gerontol Int 2014; 14: 793–798. [DOI] [PubMed] [Google Scholar]
- 6. Lu J, Jiang T, Wu L, et al. The expression of angiotensin-converting enzyme 2-angiotensin-(1–7)-mas receptor axis are upregulated after acute cerebral ischemic stroke in rats. Neuropeptides 2013; 47: 289–295. [DOI] [PubMed] [Google Scholar]
- 7. Wegman MP, Guo MH, Bennion DM, et al. Practicality of intermittent fasting in humans and its effect on oxidative stress and genes related to aging and metabolism. Rejuvenation Res 2015; 18: 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lew RA, Warner FJ, Hanchapola I, et al. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp Physiol 2008; 93: 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leys C, Ley C, Klein O, et al. Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J Exp Soc Psychol 2013; 49: 764–766. [Google Scholar]
- 10. Fouda A, Artham S, El-Remessy A, et al. Renin–angiotensin system as a potential therapeutic target in stroke and retinopathy: experimental and clinical evidence. Clin Sci 2016; 130: 221–238. [DOI] [PubMed] [Google Scholar]
- 11. Xia H, Sriramula S, Chhabra KH, et al. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res 2013; 113: 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soro-Paavonen A, Gordin D, Forsblom C, et al. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J Hypertens 2012; 30: 375–383. [DOI] [PubMed] [Google Scholar]