

Abstract
Introduction:
LY3045697 is a potent and selective aldosterone synthase (CYP11B2) inhibitor that was developed as a safer alternative to mineralocorticoid receptor antagonists. Effects of LY3045697 on aldosterone and cortisol synthesis, as well as potassium ion homeostasis, were evaluated in two clinical studies in healthy subjects.
Materials and methods:
Two incomplete, placebo-controlled crossover-design clinical studies examined safety, pharmacodynamics, and pharmacokinetics under single and repeated dose conditions in healthy subjects. Pharmacodynamics was assessed following oral potassium challenge and intravenous adrenocorticotropic hormone procedures with spironolactone 25 mg/d as an active comparator.
Results:
A total of 51 subjects participated in the two studies, which included 38 males and 13 females (of non-childbearing potential), from 18–65 years old. LY3045697 caused rapid dose and concentration-dependent unstimulated plasma aldosterone concentration reduction seen as early as 4 h after the first dose at dose levels as low as 1 mg, and reaching near complete suppression at high doses. The potency (IC50) decreased significantly upon multiple dosing. After eight days of dosing, post-adrenocorticotropic hormone challenge plasma aldosterone concentration increase was dose-dependently blunted by LY3045697 with high potency with a dose as low as 0.1 mg resulting in substantial effect, and with an overall IC50 of 0.38 ng/ml. Minor reductions in cortisol were observed only at the top dose of 300 mg. LY3045697 is generally safe and tolerated, and exhibits linear pharmacokinetics.
Conclusions:
LY3045697 is a potent and highly selective aldosterone synthase inhibitor with selectivity for CYP11B2, offering a substantial potential advantage over previous aldosterone synthase inhibitors evaluated in the clinic.
Keywords: Aldosterone synthase inhibitor, LY3045697, aldosterone, cortisol, mineralocorticoid receptor antagonists, potassium regulation, chronic kidney disease
Introduction
Aldosterone, a mineralocorticoid steroid hormone produced by the adrenal glands, is involved in electrolyte and volume homeostasis.1 It is the primary ligand of the mineralocorticoid receptor (MR), a member of the nuclear hormone receptor family. Traditionally, the main target organ of circulating aldosterone is the kidney, where activation of MR in the distal collecting tubule results in increased Na+ re-absorption, leading to volume expansion.1,2 MR is also widely expressed in the cardiovascular system, including cardiac myocytes, vascular endothelial cells and smooth muscle cells, and is also expressed in kidney mesangial cells. Aldosterone exerts genomic and nongenomic MR-mediated effects,2,3 through which pro-inflammatory and pro-fibrotic pathways are activated, leading to tissue damage and remodeling.4,5
Aldosterone has been shown to be elevated in patients with congestive heart failure,6–8 and stable chronic kidney disease.9 Inhibition of aldosterone effects through MR antagonism produces beneficial effects in patients with cardiovascular and renal disease. Two antagonists are commercially available for clinical use. Spironolactone, a nonselective MR antagonist anti-androgenic, demonstrated mortality reduction in patients with systolic heart failure,10 and reduction in proteinuria in patients with chronic kidney disease (CKD).9 Unfortunately, its lack of selectivity against glucocorticoid receptor and estrogen receptor lead to dose limiting adverse effects that have limited its clinical utility. The more selective MR antagonist, eplerenone, reduced cardiovascular mortality or re-hospitalization due to cardiovascular events in patients with congestive heart failure following myocardial infarction.10,11 Both MR antagonists have been shown in a meta-analysis to have renal protective effects in CKD.12
Currently available MR antagonists have several undesirable features. The anti-androgenic activity of spironolactone causes breast pain and symptoms of hypogonadism. Eplerenone has little anti-androgenic effects, but is less efficacious than spironolactone in lowering blood pressure. Both drugs are offset by increased risk of hyperkalemia under certain conditions. Predisposing factors for developing hyperkalemia include use in combination with angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor antagonists (ARBs),12–15 baseline serum potassium (K+)>5.0 mmol/l, or estimated glomerular filtration rate <30 ml/min/1.73 m2. These conditions are not uncommon in patients who otherwise have an indication for an MR antagonist and in turn either curtail the drug’s use or require careful patient monitoring of serum K+. In addition, there is a compensatory increase in aldosterone production during long-term treatment with MR antagonists.16 This could worsen the MR-independent effects of aldosterone in vascular wall and heart.17 Inhibiting the production of aldosterone represents an alternative strategy to MR antagonism at all sites of aldosterone activity in humans.
Aldosterone is synthesized from cholesterol in the outer-most layer of the adrenal cortex (zona glomerulosa) through a cascade of steroid hydroxylase and deoxygenase enzymes.18 Aldosterone synthase (also termed CYP11B2) catalyzes the last and rate-limiting steps in aldosterone synthesis. The major glucocorticoid, cortisol, is synthesized in the zona fasciculata of the adrenal cortex with CYP11B1 (11β-hydroxylase (cytochrome P450 type I)) as the rate-limiting enzyme. Aldosterone and cortisol biosynthesis share many common steps.19,20 In addition, human CYP11B1 and CYP11B2 share 93% homology at the amino acid level.19
Currently, one aldosterone synthase inhibitor (LCI699) has been tested in the clinic,21–27 but seems to lack adequate selectivity against CYP11B1. Clinical development seems focused on inhibition of cortisol production, as the compound is being investigated as a treatment for Cushing’s syndrome.22 Based on these data, aldosterone synthase inhibitors with more selective towards aldosterone synthase are needed.
LY3045697 is a potent and selective AS inhibitor (ASi) that was developed with the intent of establishing a favorable therapeutic index for effects on aldosterone relative to cortisol. LY3045697 inhibits human AS (in vitro CYP11B2 IC50=4.5 nM) with a 39-fold selectivity over cortisol synthase (in vitro CYP11B1 IC50=176 nM). Two clinical studies that evaluate LY3045697 in healthy subjects have recently completed. The goal of these studies was to characterize the safety, tolerability, pharmacokinetics, and pharmacodynamics of LY3045697. The pharmacodynamic (PD) endpoints of interest included the effects of LY3045697 on aldosterone and cortisol synthesis and on K+ homeostasis after single and multiple doses. A novel selective, potent ASi may have therapeutic utility in patients suffering from CKD, diabetic nephropathy (DN), hypertension (HT: salt sensitive, resistant, isolated systolic), primary hyperaldosteronism or cardiac arrhythmias.
Materials and methods
Study designs
Two randomized, placebo-controlled, incomplete crossover design studies were conducted in healthy volunteers using similar enrollment criteria and study endpoints. Subjects remained confined to the Clinical Research Unit during all dosing and evaluation periods in both studies, except for the follow-up visits, which were ambulatory. The first study, ASEA, was a first-in-human single ascending dose study investigating doses across a range of 0.1–300 mg. The second study, ASEB, assessed doses across a range of 0.1–100 mg administered once-daily for eight days. Subjects were randomized to an LY3045697 dose, placebo or 25 mg spironolactone that was included as a positive comparator to benchmark LY3045697-related effects on K+. The studies underwent institutional review board (IRB) approval and subjects signed an informed consent prior to starting any study procedures. In both studies, dosing was conducted after a standardized breakfast, as in vitro testing suggested potentially LY3045697 better bioavailability in the fed state. In study ASEB, subjects consumed a controlled metabolic diet on Days 1–8 in each dosing period. This diet consisted of moderately restricted Na+ (125 mEq/day) and slightly higher than normal K+ (125 mEq/day).
Subjects in the ASEB study underwent a 40 mEq oral K+ challenge 3 h after dosing on Day 7. Dietary K+ content was reduced by 40 mEq on Day 7 to maintain the total daily K+ intake of 125 mEq. The premise of the oral K+ challenge is that, relative to normal conditions, antagonizing the action of aldosterone in the distal nephron of healthy kidneys should delay the urine excretion of an oral K+ load and produce a transient further rise in serum K+. Application of this challenge in the ASEB study enabled evaluation of the kinetics of serum and urine K+ in the presence or absence of test article or the MR antagonist, spironolactone.
Also in the ASEB study, an intravenous (IV) adrenocorticotropic hormone (ACTH) challenge was performed 1 h after dosing on Day 8. The IV ACTH challenge test is a standard protocol to assess cortisol secretion in humans. In this test, IV administration of ACTH causes a rapid and transient increase in plasma cortisol and aldosterone.28 Application of the IV ACTH challenge in the ASEB study enabled additional assessment of the selectivity of LY3045697 on AS, including any effect on the precursors of aldosterone and cortisol (11-deoxycorticosterone, corticosterone, and 11-deoxycortisol, respectively).
In both studies, LY3045697 or placebo was administered as an oral solution in degassed Sprite. To maintain the blind, a double-dummy design was implemented in the ASEB study in which all subjects received either LY3045697 or placebo oral solution and either spironolactone or placebo capsule on each dosing day.
Evaluations in both studies included clinical safety and tolerability, vital signs (blood pressure and pulse rate, mean arterial pressure (MAP; see Supplementary Material for calculation)), pharmacokinetics and exploratory pharmacodynamics, including Na+ and K+ renal excretion and plasma concentrations of aldosterone (PACs) and cortisol, and their steroidogenic precursors (11-deoxycorticosterone and corticosterone; and 11-deoxycortisol respectively) in both stimulated (post intravenous ACTH challenge in study ASEB) and unstimulated (basal; studies ASEA and ASEB) states. Blood pressure was assessed as part of safety evaluations, as well as multiple times over each of Day 3 and 7 in the ASEB study, from which mean arterial pressure was calculated. Pooled urine was collected from 4-24 h after dosing in the ASEA study and used to quantify the amount of excreted aldosterone as a measure of target engagement (reduction of aldosterone synthesis). In the ASEB study, urine was collected and pooled over 24 h on Days 3 (from pre-dose day 3 to pre-dose day 4) and 7 (from pre-dose Day 7 to pre-dose Day 8) and evaluated for the amount of urine aldosterone excreted. Renal clearance of K+ and Na+ was calculated as the ratio of the amount excreted in the urine divided by the area under the serum concentration curve over the same time frame.
Both the studies were reviewed and approved by the local research ethics committees and internal review boards, and conducted according to the principles expressed in the Declaration of Helsinki. Details of study designs and more details on measured variables can be found in the Supplementary Material (and Supplementary Material, Tables S1a and S1b) and on Clinicaltrials.gov.
PD data analysis
An analysis of variance (ANOVA) model was performed on the PD parameters to compare the active treatment to each of placebo and spironolactone treatment. The PD parameters were natural logarithm transformed prior to analysis. The model included a fixed effect for treatment and period and a random effect for subject. From this model the least squares means (LSMeans) for each treatment were calculated and the p-values were determined from the difference in LSMeans. The ratio of least-squares geometric means between the active treatment and placebo and between the active treatment and spironolactone and the corresponding 90% confidence intervals (CIs) were estimated.
Pharmacokinetic (PK) data analysis
Blood was sampled serially in both studies for the determination of plasma LY3045697 levels. Pharmacokinetics of LY3045697 were characterized using standard noncompartmental methods to calculate PK parameters, using WinNonlin Professional Version 5.0.1 or higher (Pharsight Corp., Mountain View, California, USA).
Dose and concentration-response modeling
The relationship between drug exposure and effects on PAC, urine aldosterone excretion and K+ responses in the different experimental contexts was characterized quantitatively through dose and concentration-response models. All such models used simple hyperbolic functions (Emax, Imax) with dose and observed time-matched plasma LY3045697 concentration as the predictor. Where plasma concentration was not available, an appropriate imputation was applied. The modeling methodology is described in more detail in the Supplementary Material (and Supplementary Material, Figure S1 and Table S3).
All data analysis and visualization was conducted using SAS version 9.1.3 (Cary, North Carolina, USA) and S-PLUS 6.2 Professional edition (SolutionMetrics, Sydney, Australia).
Results
Subject demographics
A total of 51 subjects participated in the two studies, which included 38 males and 13 females (of non-child bearing potential), from 18 to 65 years old.
In the ASEA study, 27 subjects were enrolled, including 20 men and seven women of non-child bearing potential. Eighteen subjects participated in three dosing periods and nine subjects participated in two dosing periods.
In the ASEB study, 24 subjects were enrolled, including 18 men and six women of non-child bearing potential. A total of 23 subjects participated in three periods during the study, and one subject participated in two periods; one subject withdrew consent on Day 5 of Period 3.
Safety and tolerability
In both studies, LY3045697 was well-tolerated across the tested dose range. Incidences of TEAEs were comparable among treatment groups and all TEAEs were mild and reversible. There were no deaths, serious adverse events, or discontinuations for adverse events. Safety labs were unremarkable after a single dose; however a higher incidence of hyperkalemia (serum K+ >5.5 mmol/l) at high doses was observed after multiple dosing in the ASEB study (one subject at 1 mg (Day 3); three subjects at 100 mg (one on Day 3; two on Day 7)). LY3045697 administered as singlet or repeated dosing was not associated with evidence of hepatic or renal toxicity, clinically significant hypertension (systolic blood pressure (SBP)>160 mm Hg) or hypotension (SBP<90 mm Hg), or adverse ECG effects. There were no clear effects on MAP following multiple administration of LY3045697 or spironolactone in the ASEB study (Figure 1).
Figure 1.
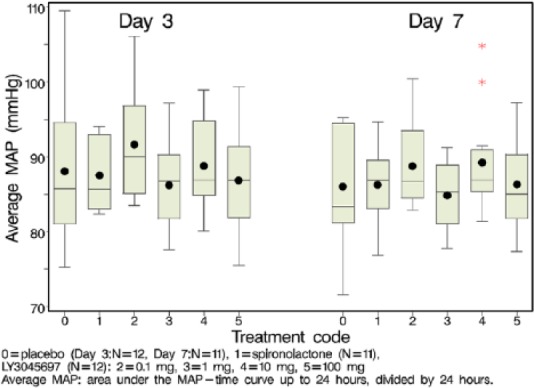
Mean arterial pressure (MAP) at various time points, post-dose, on Day 3 and Day 7. Mean (dot); median (center line within box); 25th and 75th percentiles as edges of the box; whiskers extend as far as the data extend to a distance of at most 1.5 interquartile range (IQR).
P = placebo (Day 3; N=12, Day 7; N=11), S = spironolactone (N=11), L = LY3045697 (N=12); Average MAP: area under the MAP – time curve up to 24 hours, divided by 24 hours.
Pharmacokinetics
Following LY3045697 administration after a standard breakfast, drug plasma concentrations increased rapidly and reached peak concentration typically as early as the first post dose sampling time of 0.5 h (Figure 2(a)). After reaching maximum concentration, the LY3045697 plasma concentration-time profiles exhibited a mostly biphasic decline (Figure 2(a)). The PK profile after multiple daily dosing was similar to that observed after the first dose. Plasma PK parameters are reported in the Table 1. Plasma half-life (t1/2) was approximately 10 h, and appeared to be dose-independent and consistent between single and multiple dose administration. Minimal accumulation was observed (Figure 2(b)), and drug exposure appeared to have reached steady state by the third daily dose (data not shown). Both Cmax and AUC generally increased in proportion to dose.
Figure 2.
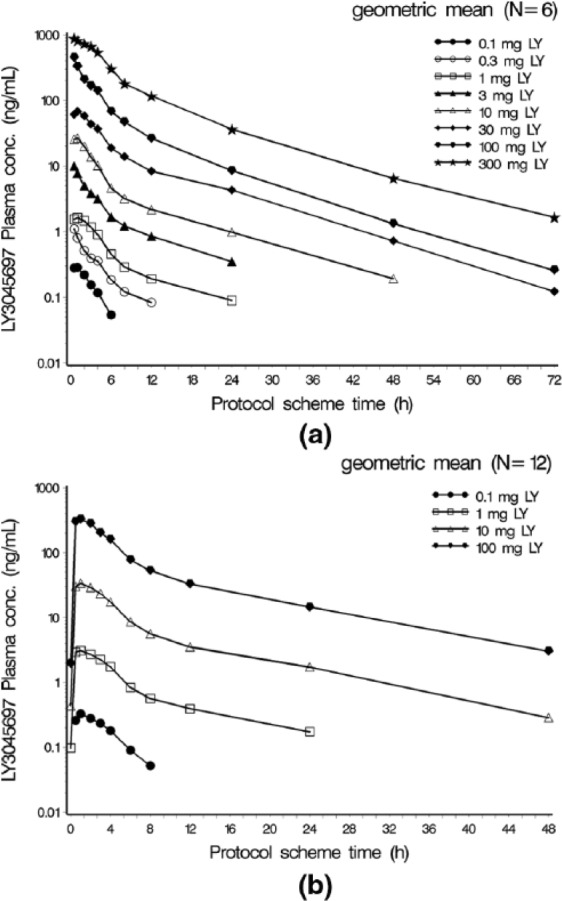
Geometric Mean LY3045697 Plasma Concentration-Time Profiles by Treatment (Semi-Logarithmic Scale) after single (a) and 8 days of daily (b) dosing in healthy subjects.
conc: concentration; h = hour; LY = LY3045697; N = number of subjects exposed.
Table 1.
Summary statistics of LY3045697 plasma pharmacokinetic (PK) parameters after single and multiple-daily dosing in the fed state by dose group (geometric mean (coefficient of variation (CV)%)).
| Variable | 0.1 mg LY3045697 (n=6) |
0.3 mg LY3045697 (n=6) |
1 mg LY3045697 (n=6) |
3 mg LY3045697 (n=6) |
10 mg LY3045697 (n=6) |
30 mg LY3045697 (n=6) |
100 mg LY3045697 (n=6) |
300 mg LY3045697 (n=6) |
|---|---|---|---|---|---|---|---|---|
| After single daily dose of LY3045697 in the fed state | ||||||||
| Cmax (ng/ml) | 0.32 (28) | 1.10 (33) | 1.96 (49) | 9.95 (37) | 27.9 (24) | 83.7 (25) | 458 (27) | 951 (20) |
| tmax (h)a | 0.75 (0.50–1.00) | 0.50 (0.50–1.00) | 1.00 (0.50–2.00) | 0.50 (0.50–0.50) | 0.75 (0.50–2.00) | 1.00 (0.50–2.00) | 0.50 (0.50–1.00) | 0.50 (0.50–1.00) |
| AUC0–inf (h*ng/ml) | nrb | nrb | 12.1 (51)c | 49.1 (31)d | 138 (19) | 482 (32) | 1726 (15) | 6226 (19) |
| t1/2 (h) | nrb | nrb | 9.11 (43)c | 8.73 (32)d | 9.99 (20) | 9.57 (20) | 9.62 (28) | 10.8 (30) |
| After 8 once-daily doses of LY3045697 in the fed state | ||||||||
| (n=12) | (n=12) | (n=12) | (n=12) | |||||
| Cmax (ng/ml) | 0.368 (30) | 3.45 (36) | 37.4 (29) | 371 (37) | ||||
| tmax (h) a | 0.95 (0.50–3.00) | 0.95 (0.50–3.00) | 0.95 (0.50–2.00) | 0.95 (0.50–0.95) | ||||
| AUC0–tau (h*ng/ml) | NC | 19.2 (29) | 195 (22) | 1818 (28) | ||||
| t1/2 (h) | e | e | 9.84 (18) | 10.8 (26) | ||||
AUC0–inf : area under the plasma concentration-time curve from time 0 to infinity; AUC0–tau: area under the plasma concentration-time curve during the dosing interval of 24 h; Cmax: maximum observed plasma concentration; CI: confidence interval; h: hour; n: number of subjects exposed; NC: if no quantifiable observations were reported until 24 h post-dose than AUC0–tau was not calculated; nr: not reported; t1/2: terminal elimination half-life; tmax: time to attain Cmax.
Median (range); bhalf-life and other PK parameters that rely on the terminal phase elimination rate constant were not calculable due to inadequate quantifiable observation interval; cn=3; dn=5; enot reported due to inadequate quantifiable terminal phase.
Urine aldosterone
After a single LY3045697 dose, there was a dose-dependent reduction in urine aldosterone excretion, starting at 1 mg (−24%) and becoming statistically significantly lower compared to placebo starting at 3 mg (Supplementary Material, Figure S2), which was associated with a peak LY3045697 plasma concentration of 9.95 ng/ml. The percent change in urinary aldosterone relative to placebo was −62%, −76%, −85%, −88%, and −95%, respectively, for the 3 mg, 10 mg, 30 mg, 100 mg, and 300 mg LY3045697 doses.
Consistent with the single dose results, there was a dose-dependent reduction in 24-hour urine aldosterone excretion after multiple dosing with LY3045697 (Figure 3(a)). Urine aldosterone excretion observed during dosing with 10 and 100 mg LY3045697 were significantly lower than placebo on Day 3: (11,461 and 3172 pmol, respectively, versus 31,656 pmol; p<0.0001 for both treatments) and Day 7: (13,081 and 7638 pmol, respectively, versus 26,512 pmol; p=0.0001 and p<0.0001, respectively). The 10 and 100 mg doses were associated with mean peak LY3045697 plasma concentrations of 37.4 and 371 ng/ml, and time-averaged plasma concentrations at steady state (calculated as the area under the curve divided by the dosing interval of 24 hours) of 8.13 and 75.8 ng/ml, respectively (Table 1).
Figure 3.

(a) Box and whisker plot of the amount of aldosterone excreted in urine after 24 h collection on Day 3 and Day 7 by treatment (after a single dose). Mean (dot); median (center line within box); 25th and 75th percentiles as edges of the box; whiskers extend as far as the data extend to a distance of at most 1.5 interquartile range (IQR); (b) Mean percent (%) change from baseline for plasma aldosterone versus time from dosing by treatment after a single dose administration; (c) Arithmetic mean percentage change from Baseline versus time profiles on Day 7 for plasma aldosterone after multiple daily dose administration.
As expected, administration of the MR antagonist spironolactone produced an elevated urine aldosterone excretion (Figure 3(a)), consistent with a compensatory increase in PAC (Figure 3(c)).
Plasma aldosterone concentration
Basal concentration
Basal (i.e. not stimulated by exogenous ACTH challenge) PAC was measured prior to dosing and at 4, 12 and 24 h post dose in the ASEA study and after seven days of dosing in the ASEB study. After single dose administration (ASEA study), there was a dose-dependent reduction in PAC that was observed at the first post-dose sampling point (4 h) (Figure 3(b)). Mean reductions in PAC of greater than 75% were observed for doses of LY3045697 between 3–300 mg at this timepoint (mean plasma LY3045697 concentrations of 3.28–553 ng/ml). By 24 h after dosing, PAC levels trended back toward baseline for doses of LY30456097 less than 30 mg; however, PAC remained at peak reduction throughout the 24-hour observation period at doses of 30 mg and above. Mean LY30456097 plasma concentrations at 24 h at the 30 mg dose was 4.59 ng/ml, and the dynamic PAC changes appear to follow changes in drug levels.
After multiple dosing (ASEB study), PAC reductions were pronounced at 10 and 100 mg (Figure 3(c)), with peak reductions observed at 4 h post dose, with associate mean plasma LY3045697 concentrations of 18.2 and 168 ng/ml, respectively, similar to the phenotype after single dose administration. However, in contrast to the phenotype observed after single dosing, PAC levels after seven days of multiple dosing approached baseline by 24 h post dose across the entire dose range evaluated (0.1–100 mg), with associated mean plasma LY3045697 concentrations up to 15.7 ng/ml. Mean 24-hour PAC at steady state was significantly lower after treatment with 10 mg and 100 mg LY3045697 versus placebo (p=0036 and p<0.0005 respectively).
After ACTH challenge
ACTH administered IV on Day 8 (Study ASEB) produced the anticipated increase in PAC in placebo treated subjects, with a mean peak increase from pre-challenge concentration of 104 pg/ml occurring 30 min post-challenge, and an average increase of 66.4 pg/ml (average of 30 and 60 min post-challenge). The excursions from pre-challenge PAC were dose-dependently blunted by LY3045697, with mean change±standard deviation (SD) (average of 30 and 60 min post-challenge) of 41±23 pg/ml after 0.1 mg dose, –11±20, –8±6 and −7±7 pg/ml after 1, 10 and 100 mg respectively, indicating complete blunting of the ACTH stimulated increase in aldosterone synthesis at LY3045697 doses of 1 mg and above (Figure 4(a)), which was associated with mean peak plasma LY3045697 concentrations of 3.45 ng/ml. Spironolactone had no effect on PAC following the ACTH challenge.
Figure 4.

Mean change from pre-challenge plasma aldosterone (a), corticosterone (b) and 11-deoxycorticosterone (c) after IV ACTH challenge. IV ACTH challenge was conducted 1 hr post-dose on Day 8. Steroids measured at -15 mins (baseline), 30 mins and 60 mins post-challenge. Cave calculated as baseline subtracted AUC(0-1hr) divided by 1hr.
Box and whiskers plots: mean(dot); median (center line within box); 25th and 75th percentiles as edges of the box; whiskers extend as far as the data extend to a distance of at most 1.5 interquartile range (IQR); Outliers (data points outside of ±1.5 IQR) are presented as stars (*).
The effect of LY3045697 on plasma corticosterone concentrations was similar to that of the placebo after the IV ACTH challenge, i.e. expected increase in plasma corticosterone concentrations due to the ACTH stimulus and no obvious further increase by administration of LY3045697 (Figure 4(b)). Similarly, plasma 11-deoxycorticosterone levels were elevated by the ACTH challenge in placebo-treated subjects and after administration of LY3045697. Additional slight elevation in plasma 11-deoxycorticosterone concentrations were observed after administration of 10 mg and more substantially after administration of 100 mg LY3045697 compared to placebo (Figure 4(c)). Collectively, the basal and ACTH-stimulated aldosterone results indicate that LY3045697 inhibited AS (CYB11B2) resulting in reduced PAC via reduced aldosterone synthesis and increased levels of its precursors corticosterone and 11-deoxycorticorsterone.
Plasma cortisol and 11-deoxycortisol
Basal concentrations
To determine if the effects of LY3045697 were selective for CYP11B2 (and therefore aldosterone reduction), plasma 11-deoxycortisol and cortisol, the respective substrate and product of CYP11B1, were also measured. After single dosing with LY3045697, there were no measured decreases in plasma cortisol concentrations at doses up to 300 mg, with mean peak plasma LY3045697 concentration of 951 ng/ml; however, there was a trend for increased concentrations of 11-deoxycortisol observed at doses beyond 30 mg, potentially indicating an inhibitory effect on the CYP11B1 enzyme at the higher plasma LY3045697 concentrations (Supplementary Material, Figure S4).
After multiple dosing with LY3045697 (ASEB study), there was no effect on mean concentrations (over 24 h) of cortisol at steady state at doses between 0.1–10 mg, similar to the phenotype after placebo or spironolactone (Figure 5(a)), with associated mean peak and time-averaged plasma LY3045697 concentrations up to 37.4 and 8.13 ng/ml, respectively. However, paradoxically, an increase in mean plasma cortisol concentration at steady state was observed at 100 mg LY3045697 dose compared to placebo (Figure 5(a); mean concentrations±SD: 272±43 versus 212±78.0 nmol/l, respectively; p<0.0001). This increase was not indicative of an inhibitory effect of LY3045697 on CYP11B1. A similar pattern was observed for plasma 11-deoxycortisol as for plasma cortisol, with no effect after dosing with LY3045697 at doses between 0.1–10 mg versus placebo (p=0.85, p=0.72 and p=0.89 for 0.1, 1, 10 mg respectively as compared to placebo). There was a significantly higher average concentration of 11-deoxycortisol at steady state observed after multiple dosing with 100 mg LY3045697 when compared to placebo (average plasma concentrations of 836±465 pg/ml versus 222±113 pg/ml, respectively; p<0.0001; Figure 5(b)).
Figure 5.

Mean cortisol (a) and 11-deoxycortisol (b) concentrations (over 24 hr) at steady state. Plasma samples collected at pre-dose, 4 hr, 12 hr, 24 hr post-dose on Day 7. Cave calculated as AUC(0-24) divided by 24hr. Mean change from pre-challenge plasma cortisol (c) and 11-deoxycortisol (d) after IV ACTH challenge. IV ACTH challenge was conducted 1 hr post-dose on Day 8. Steroids measured at -15 mins (baseline), 30 mins and 60 mins post-challenge. Cave calculated as baseline subtracted AUC(0-1hr) divided by 1hr. Box and whiskers plots: mean(dot); median (center line within box); 25th and 75th percentiles as edges of the box; whiskers extends as far as the data extend to a distance of at most 1.5 interquartile range (IQR); Outliers (data points outside of ± 1.5 IQR) are presented as stars (*).
After ACTH challenge
ACTH administered IV on Day 8 (Study ASEB) resulted in increased plasma cortisol concentrations in placebo-treated subjects as expected (Figure 5(c); average increased plasma concentration over the one-hour evaluation period). Mean peak increase from pre-challenge concentration was 466±102 nmol/l at 60 min post-challenge. No differences in plasma cortisol concentrations were seen for the 0.1, 1, and 10 mg LY3045697 doses (with associated mean peak plasma LY3045697 concentrations up to 37.4 ng/ml) or spironolactone compared to placebo (Figure 5(c)). In contrast, ACTH-induced increase in plasma cortisol concentration was substantially lower for the 100 mg LY3045697 dose as compared to placebo (150.3±60.4 nmol/l versus 299.1±78.2 nmol/l for average post-challenge plasma concentration, respectively; Figure 5(c); mean change from pre-challenge values), with associated mean peak LY3045697 concentrations of 371 ng/ml, indicating an inhibitory effect of LY3045697 on CYP11B1 at a daily dose of 100 mg.
ACTH administration increased 11-deoxycortisol concentrations relative to pre-challenge values in placebo-treated subjects (mean peak increase from pre-challenge value of 1403 pg/ml). Treatment with LY3045697 at 0.1 and 1 mg doses showed a similar response to placebo, indicating no effect of LY3045697 at these doses on ACTH-induced plasma 11-deoxycortisol concentrations. After treatment with 10 mg LY3045697 (mean peak plasma concentrations 37.4 ng/ml), the magnitude of increase in plasma 11-deoxycortisol was slightly higher than at the lower doses and placebo, with a substantial further increase after treatment with 100 mg LY3045697 (Figure 5(d); mean change from pre-challenge values of 3443±1,491 pg/ml after 100 mg LY3045697 versus 982±467 pg/ml after placebo). This phenotype was consistent with that of cortisol, further indicating an inhibitory effect of LY3045697 on CYP11B1, with initial signal of effect on CYP11B1 at 10 mg (11-deoxycortisol response) and a more prominent effect at 100 mg.
Collectively, the cortisol and 11-deoxycortisol results after IV ACTH challenge suggest an inhibitory effect of LY3045697 on CYP11B1 occurring between a daily dose of 10–100 mg.
Potassium effects
Basal serum potassium
After a single dose of LY3045697, serum K+ did not change. After multiple dose administration, there was a dose-related increase in serum K+ concentration over time beginning with 1 mg LY3045697 compared to placebo (Figure 6(a)). The effect of 1 mg LY3045697 on serum K+ was comparable to that of 25 mg spironolactone, with larger effects observed at 10 mg and 100 mg. Effects on serum K+ appeared to plateau around 72 h after initiating dosing (Figure 6(a)).
Figure 6.

Mean serum K+ over time by treatment after multiple dosing (a) x-axis timing reflects hours since first dosing; Mean serum potassium after oral potassium challenge (b); Box and whisker plot of K+ clearance on Day 3 and Day 7 by treatment (c). Mean (dot); median (center line within box); 25th and 75th percentiles as edges of the box; whiskers extend as far as the data extend to a distance of at most 1.5 interquartile range (IQR); Outliers (data points outside of ± 1.5 IQR) are presented as stars (*).
After oral K+ challenge
The oral K+ challenge was conducted on Day 7 approximately 3 h after dosing study drug and results are shown in Figures 6(b) and (c). Due to the increase in serum K+ from daily LY3045697 dosing (Figure 6(a)), the serum K+ concentrations prior to the oral K+ challenge on Day 7 were higher for the 10 mg and 100 mg doses versus placebo (see pre-challenge values, Figure 6(b)). As expected, there was an increase in serum K+ in response to the oral K+ challenge. This was observed after all treatments, with peak increase observed at 1 h post-challenge with a progression back toward pre-challenge values over the 5 h post-challenge evaluation period. After placebo treatment, where the average serum K+ concentration over 5 h post-challenge (Cavg, mean±SD) was 4.4±0.2 mmol/l (Figure 6(b)). LY3045697 dose-dependently increased serum K+ concentrations after the oral K+ challenge. Compared to placebo treatment, no statistically significant difference in serum K+ was observed after 0.1 mg LY3045697 treatment (4.5±0.9 mmol/l, p=0.31). However, statistically significant increases in serum K+ were observed after all other active treatments relative to placebo, with average serum K+ after spironolactone treatment of 4.5±0.2 mmol/l (p=0.04); and 4.5±0.2 mmol/l (p=0.008), 4.8±0.2 mmol/l (p<0.0001) and 4.9±0.3 mmol/l (p<0.0001) after 1, 10 and 100 mg of LY3045697 treatment, respectively. Comparing serum K+ response of LY3045697 treatment to spironolactone, there was no clear separation for the 0.1 and 1 mg LY3045697 doses after oral K+ challenge (p>0.05); while serum K+ concentrations were significantly higher after 10 and 100 mg LY3045697 treatment (p=0.02 and p<0.0001, respectively).
Urine potassium
Unstimulated
For K+ renal clearance after single dosing, the majority of the LY3045697 dose groups (0.1–30 mg) had similar mean clearance values, with a subtle trend for reduction at doses between 100 mg and 300 mg. Potassium clearance was lower at the 100 mg and 300 mg doses (0.0092 and 0.0082 l/min, respectively) compared to the other LY3045697dose groups and placebo (Supplementary Material, Table S2).
Renal clearance of K+ was assessed on Days 3 and 7 of the ASEB study using 24-hour urine collection (Figure 6(c)). Compared to urinary K+ excretion in placebo-treated subjects (mean±SD values of 99±19 and 110±23 mmol for Days 3 and 7, respectively), urinary K+ excretion was similar after spironolactone treatment (97±20 and 117±17 mmol for Days 3 and 7, respectively) and after LY3045697 treatment up to 10 mg (urinary potassium excretion ranging from 103±19 to 94±17 mmol on Day 3, and 134±15 to 116±16 mmol on Day 7). A subtle trend for reduction in urinary K+ excretion was observed after 100 mg LY3045697 treatment (80±15 mmol) on Day 3, but it seemed to normalize on Day 7 (117±23 mmol). As a result, no trends were noted for urine excretion of K+ on Day 7.
After oral K+ challenge
Renal clearance of K+ (CL0–5h) was also assessed after the oral K+ challenge on Day 7. After oral K+ challenge, K+ clearance in subjects randomized to placebo (mean±SD values of 1.78±0.39 l/h) and spironolactone (1.7±0.3 l/h) was similar (p=0.50), as were the clearance values in subjects randomized to LY3045697 at 0.1 mg (1.8±0.1 l/h, p=0.42) and 1 mg (1.6±0.3 l/h, p=0.15) doses. Renal clearance of K+ was significantly lower in subjects randomized to LY3045697 at 10 mg (1.5±0.2, p=0.03) and 100 mg (1.5±0.3 l/h, p=0.004). Subjects who received 100 mg LY3045697 also had significantly lower K+ clearance compared to spironolactone (p=0.03).
Sodium effects
There were no significant changes in serum sodium ion (Na+) concentration or Na+ renal clearance after a single dose of LY3045697. After multiple daily dosing with LY3045697, there was a dose-related reduction in serum Na+ beginning at 1 mg (Supplementary Material, Figure S3 (a)). An increase in urine excretion of Na+ was seen beginning at 10 mg on Day 3, which was on track to normalize on Day 7. However, within the first 5 h of oral K+ challenge (on Day 7), urine excretion of Na+ increased at doses as low as at 1 mg LY3045697. In addition, a reduction on body weight over time after administration of 100 mg LY3045697 was seen which is most likely due to its natriuretic effect. The effects of spironolactone on renal Na+ clearance followed a similar trend in that the Na+ clearance in spironolactone-treated subjects was comparable to that in placebo-treated subjects on Days 3 and 7 (24-h urine collections), while greater natriuresis was observed within 5 h of oral K+ challenge (Supplementary Material, Figure S3 (b)). It is well known that the natriuretic effects of diuretics are most prominent within days of initiation of treatment, and renal Na+ clearance commonly normalize within one week. Our observation was consistent with the literature, and implies that the natriuretic effects of LY3045697 may be clinically relevant at doses at low as 1 mg.
Dose and concentration-response relationships
Models were established for basal (unstimulated) plasma aldosterone and serum K+ after single and multiple dose administrations, as well as post-challenge plasma aldosterone and K+ after multiple dose administration (Figures 7(a) and 7(b)). The key model parameter estimates are summarized in Supplementary Material, Table S3.
Figure 7.

Model-fitted (with 90% Prediction Intervals) relationships between LY3045697 plasma concentration and basal aldosterone and potassium after a single LY3045697 dose administration (a) and after multiple LY3045697 dose administration (b).
The effects of LY3045697 on basal (unstimulated plasma aldosterone and serum K+) aldosterone and K+ show that multiple daily dosing resulted in a different profile than after a single dose (Figures 7(a) and 7(b)). Upon repeat dosing, the effect of LY3045697 on PAC appears to lose potency (higher IC50) without a substantial difference in the maximum inhibition possible (Imax), whereas there is an increase in maximal serum K+ effect (Emax) effect with no evidence of change to potency (EC50), albeit a poor estimate of EC50 after a single dose administration due to a low Emax.
Similarly, the effects of LY3045697 and spironolactone on PAC following stimulated production of aldosterone (via IV ACTH administration on Day 8) and serum K+ (following an oral K+ challenge on Day 7) are illustrated in Figures 8(a) and 8(b) respectively. These relationships suggest that the potency (IC50) of LY3045697 to inhibit post-challenge PAC remains very high after multiple daily dosing (0.38 ng/ml), similar to the potency of inhibiting basal (unstimulated) PAC after the first dose (0.60 ng/ml). No direct comparison for post-challenge potency can be made between single and multiple dose administrations, as the challenge test was administered only after multiple daily administration.
Figure 8.

Model-fitted (with 90% Prediction Intervals) relationships between LY3045697 and stimulated aldosterone (after IV ACTH challenge) and potassium (after oral K+ challenge) after multiple LY3045697 dose administration based on LY3045697 dose (a) and based on LY3045697 plasma concentration (b).
Discussion
Aldosterone synthesis inhibition is a promising new mode of MR antagonism. It offers the potential for added efficacy through reduction of non-MR related aldosterone effects, and can circumvent anti-androgenic selectivity issues associated with current MR antagonists. However, this mode also comes with its own challenges, including selectivity for CYP11B2 (aldosterone synthesis route) relative to CYP11B1 (cortisol synthesis route), and uncertainty regarding the magnitude of hyperkalemic effect. The studies discussed herein comprehensively probed the pharmacology of the potent AS inhibitor LY3045697, its selectivity for CYP11B2 relative to CYP11B1, and its impact on serum and urine K+, uncovering interesting facets of the dynamics of AS inhibition.
LY3045697 is a potent and highly effective AS inhibitor, as evident by suppression of PAC to below the limit of quantitation in both ACTH-stimulated and unstimulated states at doses as low as 0.1 mg. These results confirm that AS is the primary, if not the only, de novo synthesis route of circulating aldosterone.
Selectivity of AS (CYP11B2) inhibitors against the highly homologous CYP11B1 enzyme is of key importance to therapeutic utility, due to the detrimental effect on the body’s natural response to stress via inhibition of cortisol synthesis. By design, LY3045697 is a potent inhibitor of AS with 39-fold less in vitro potency for CYP11B1 (data on file at Lilly). The results from these studies clearly demonstrate selectivity for AS in humans, as LY3045697 resulted in some blunting of cortisol response to ACTH challenge at doses that are 2–3 orders of magnitude higher than those demonstrating significant aldosterone blunting. Since the inhibition of cortisol production did not reach a maximum, precise estimation of the IC50 for CYP11B1 is not possible from these data. Therefore, the relative in vivo selectivity (i.e. IC50 ratio) cannot be estimated. Nonetheless, it is clear that LY3045697 demonstrates a CYP11B2/CYP11B1 selectivity in vivo that is consistent with the in vitro data. Thus, a wide range of LY3045697 doses with high AS inhibition can be administered with little risk of blunting cortisol production.
This level of selectivity far exceeds that of LCI699. In Na+-depleted healthy volunteers, LCI699 decreased both plasma and 24-h urinary aldosterone in a dose-dependent manner. In these subjects, the adrenocorticotropic hormone (ACTH)-challenge test inhibited ACTH-stimulated aldosterone elevation at all three doses (0.5, 1 and 3 mg) of LCI699. However, the compound also significantly inhibited ACTH-stimulated cortisol production and induced accumulation of the cortisol precursor, 11-deoxycortisol, at the higher dose of 3 mg.23 In a study of patients with primary hypertension, LCI699 was dosed for eight weeks at 0.25 mg, 0.5 mg, and 1 mg once daily, as well as 0.5 mg twice daily (BID). All doses of LCI699 significantly reduced ambulatory blood pressure compared to placebo.24 At 1 mg once daily dosing, the reduction of blood pressure was comparable to that achieved by eplerenone at 50 mg BID. However, LCI699 significantly inhibited ACTH-stimulated cortisol production at all doses tested25 LY3045697 has other desirable drug properties, as it was also safe and well-tolerated in healthy volunteers over the range and duration of dosing, and demonstrated dose-proportional pharmacokinetics. In this population, we found no evidence of reduction in blood pressure by LY3045697 relative to placebo, which is likely due to the fact that blood pressure is normal to begin with. Spironolactone also failed to show a reduction.
LY3045697 actions result in perturbation in the K+ homeostasis, likely secondary to its aldosterone reducing effect. LY3045697 increases basal K+ in a time and dose/concentration dependent manner, with a negligible effect after the first dose, reaching up to a maximum of 5.3 mmol/l after seven days of treatment at the top dose of 100 mg. Upon repeat dosing, the magnitude (Emax) of K+ rise increases. The PK/PD model predicts a maximum drug effect of 0.8 mmol/l increase in basal K+ on Day 8, and an LY3045697 EC50 (achieving half maximum response) of 4.3 ng/ml. The steady state for serum K+ effect of LY3045697 cannot be deduced due to relatively short treatment duration of the ASEB study. Average K+ levels after the oral K+ challenge (Day 7) were also higher for LY3045697-treated subjects in a dose and concentration-dependent fashion. The maximum magnitude of difference from placebo was estimated to be 0.58 mmol/l, which is similar to the magnitude of the increase in basal K+ predicted by the model (0.7 mmol/l). The potency (EC50) by which LY3045697 increases basal and post- challenge serum K+ is also similar. Collectively, this suggests that LY3045697 increases basal K+ while having a negligible effect on the acute response to K+ ingestion. This effect of LY3045697 on K+ is to be expected given the normal inter-regulatory relationship between aldosterone and K+. An LY3045697 dose of 1 mg is roughly equipotent to 25 mg of spironolactone with respect to the effect on K+ after the oral K+ challenge, which was administered close to the expected peak levels of both drugs. In contrast, 50 mg spironolactone showed a marginal effect on basal (unstimulated) K+ measured at trough drug levels prior to dosing. Of note, the 25 mg dose of spironolactone is considered low, as doses as high as 200 mg or more are used in practice. A comparison of LY3045697 effect on K+ relative to LCI699 cannot be made, due to the different populations and experimental conditions of the studies in which this has been assessed for the two compounds.
The dynamics of aldosterone reduction in single vs repeated dosing and in the presence and absence of ACTH stimulation provides interesting insights into the regulatory interrelationship between aldosterone and K+. The reduction in unstimulated PAC was rapid and observed at the first post-dose time point (4 h). The relationship between LY3045697 and PAC showed no clear evidence of hysteresis, indicating a nearly direct PK/PD relationship. The within-day time course of PAC was not measured with great frequency, hindering the ability to discern a rapid hysteresis. However, if a hysteresis exists, its magnitude is minor. In a system where drug effect is on the input rate of aldosterone production, a near direct PK/PD relationship is consistent with rapid equilibration of PAC upon inhibition of production, which suggests a rather fast aldosterone clearance rate, consistent with previously reported rapid disappearance half-life of radiolabeled aldosterone of only 35 min.28
The analyses provided herein uncovers an interesting phenomenon, where the concentration of LY3045697 needed to reduce PAC in the unstimulated state are much smaller after first LY3045697 administration compared to repetitive daily dosing, as illustrated in Figure 7. The IC50 is 47 times higher (28 vs 0.60 ng/ml) after multiple dosing than after single dosing (Table 2). Thus, LY3045697 aldosterone-reducing potency appears to drop precipitously upon daily dosing. This large potency shift cannot be explained based on the pharmacokinetics of LY3045697, as the potency estimates are already based on plasma concentrations. Furthermore, the pharmacokinetics of LY3045697 after single and multiple dose administration are comparable, with only minor accumulation of drug concentrations. The potency of LY3045697 in blunting the IV ACTH-stimulated aldosterone production on Day 8 remained quite high, with slightly smaller IC50 compared to the IC50 for inhibition of basal aldosterone on Day 1 of dosing. The IV ACTH test was only conducted on the last day of dosing, precluding a comparison to LY3045697 response to this test in the acute setting.
Table 2.
Key parameter estimates (coefficient of variation (CV)%) of LY3045697 plasma concentration-response models for various basal and stimulated pharmacodynamic (PD) endpoints.
| Basal |
Stimulated |
|||||
|---|---|---|---|---|---|---|
| Single dose |
Multiple dose |
Post-ACTH |
Post-K+ challenge |
|||
| PAC | K+ | PAC | K+ | PAC | K+ | |
| Placebo response (pg/ml for PAC; mM for K+) |
52 (5.9%) | 4.5 (0.8%) | 98 (8.3%) | 4.3 (1.2%) | 73 (18%) | 4.4 (1.1%) |
| Maximum LY3045697 effecta
(fractional reduction for PAC; mM for K+) |
0.90 (0.8%) | 0.12 (47%) | 0.94 (1.4%) | 0.70 (14%) | 1.1 (4.1%) | 0.58 (12%) |
| LY3045697 C50b
(ng/ml) |
0.60 (18%) | 1.3 (180%)c | 28 (20%) | 4.3 (46%) | 0.38 (32%) | 10 (50%) |
| Spironolactone (50 mg) effect (pg/ml for PAC; mM for K+)a |
NA | NA | 49 (19%) | 0.02 (232%) | 18 (89%) | 0.09 (68%) |
ACTH: adrenocorticotropic hormone; K+: potassium ion; PAC: plasma concentration of aldosterone.
Maximum drug effect for PAC (Imax) was modeled as a fractional reduction (e.g. 0.9 means 90% reduction) relative to placebo response, while for K+ (Emax) as an additive effect above placebo response (e.g. 0.12 mM means 0.12 mM above that observed for placebo).
The plasma concentration at 50% of the effect (i.e. IC50 for inhibitory effect on PAC or EC50 for stimulatory effect on K+).
Poorly estimated due to low maximum response.
Aldosterone precursor 11-deoxycorticosterone was predictably dose-dependently increased upon LY3045697 administration, as its formation is independent of AS (CYP11B2) or CYP11B1, while it is metabolized to corticosterone via both enzymes.18 Corticosterone, on the other hand, is formed from 11-deoxycorticosterone by both AS and CYP11B1, and is converted to aldosterone by AS. Thus, our expectation was that corticosterone levels would drop as a result of LY3045697 inhibition of AS (and CYP11B1 at much higher concentrations). However, the opposite was inexplicably observed.
The phenomenon of time-dependent potency reduction (IC50 increase) may be explained by the fact that serum K+ levels slowly increased upon daily dosing. Potassium is a known potent stimulator of aldosterone production, independent of its effects on the renin-angiotensin system.29,30 An increase in the amount of active AS or its substrate would naturally require a higher LY3045697 concentration to inhibit aldosterone production. We observed a few-fold increase in the aldosterone precursors, corticosterone and 11-deoxycortecosterone on Day 7 just prior to LY3045697 dosing, as would be expected from the inhibition of their conversion to aldosterone by AS, but only at the 10 and 100 mg doses. However, the increase would not seem adequate to explain the drop in potency also observed at lower doses of LY3045697 where precursor levels were not elevated. Therefore, it is more likely that a significant increase in AS enzyme activity occurs over days of dosing with the AS inhibitor LY3045697.
Of note, the studies and conclusions reported herein are limited by two factors. First, the studies are conducted in healthy volunteers, and the translation of the conclusion to therapeutic contexts in patient populations should be made with caution. Second, the studies were small in size, typical of phase one studies.
In summary, LY3045697 demonstrated potent inhibition of AS at doses that are highly selective relative to activity toward CYP11B1 and, therefore, LY3045697 is a cortisol sparing AS inhibitor. Doses of LY3045697 in the 0.1–1 mg range would be expected to provide complete blunting of the aldosterone response with a K+ elevation response significantly lower than that of low dose spironolactone. This relationship between the effects on aldosterone and K+ is likely inherent to the AS inhibition regardless of the inhibitor, as the K+ response is secondary to changes in aldosterone levels. In contrast to spironolactone, LY3045697 has potential to potently reduce both MR-mediated and non-MR mediated aldosterone effects, without anti-androgenic adverse effects.
Supplementary Material
Footnotes
Declaration of conflicting interests: The authors declare that there is no conflict of interest.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- 1. Funder JW. Aldosterone and mineralocorticoid receptors: Past, present, and future. Endocrinology 2010; 151: 5098–5102. [DOI] [PubMed] [Google Scholar]
- 2. Briet M, Schiffrin EL. Aldosterone: Effects on the kidney and cardiovascular system. Nat Rev Nephrol 2010; 6: 261–273. [DOI] [PubMed] [Google Scholar]
- 3. Gilbert KC, Brown NJ. Aldosterone and inflammation. Curr Opin Endocrinol Diabetes Obes 2010; 17: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grossmann C, Gekle M. New aspects of rapid aldosterone signaling. Mol Cell Endocrinol 2009; 308: 53–62. [DOI] [PubMed] [Google Scholar]
- 5. Gros R, Ding Q, Sklar LA, et al. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 2011; 57: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345: 1689–1697. [DOI] [PubMed] [Google Scholar]
- 7. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure: Randomized Aldactone Evaluation Study Investigators. N Eng J Med 1999; 341: 709–717. [DOI] [PubMed] [Google Scholar]
- 8. Maron BA, Opotowsky AR, Landzberg MJ, et al. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: A pilot study. Eur J Heart Fail 2013; 15: 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hene RJ, Boer P, Koomans HA, et al. Plasma aldosterone concentrations in chronic renal disease. Kidney Intern 1982; 21: 98–101. [DOI] [PubMed] [Google Scholar]
- 10. Pitt B, Remme W, Zannad F, et al. ; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309–1321. [DOI] [PubMed] [Google Scholar]
- 11. Zannad F, McMurray JJ, Krum H, et al. ; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11–21. [DOI] [PubMed] [Google Scholar]
- 12. Navaneethan SD, Nigwekar SU, Sehgal AR, et al. Aldosterone antagonist for preventing the progression of chronic kidney diseases: A systemic review and meta-analysis. Clin J Am Soc Nephrol 2009; 4: 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351: 543–551. [DOI] [PubMed] [Google Scholar]
- 14. Masoudi FA, Gross CP, Wang Y, et al. Adoption of spironolactone therapy for older patients with heart failure and left ventricular dysfunction in the United States, 1998–2001. Circulation 2005; 112: 39–47. [DOI] [PubMed] [Google Scholar]
- 15. Pitt B, Bakris G, Ruilope LM, et al. ; EPHESUS Investigators. Serum potassium and clinical outcomes in the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS). Circulation 2008; 118: 1643–1650. [DOI] [PubMed] [Google Scholar]
- 16. Rousseau MF, Gurné O, Duprez D, et al. ; Belgian RALES Investigators. Beneficial neurohormonal profile of spironolactone in severe congestive heart failure: Results from the RALES neurohormonal substudy. J Am Coll Cardiol 2002; 40: 1596–1601. [DOI] [PubMed] [Google Scholar]
- 17. Fujita M1, Minamino T, Asanuma H, et al. Aldosterone nongenomically worsens ischemia via protein kinase C-dependent pathways in hypoperfused canine hearts. Hypertension 2005; 46: 113–117. [DOI] [PubMed] [Google Scholar]
- 18. Williams GH. Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart Fail Rev 2005; 10: 7–13. [DOI] [PubMed] [Google Scholar]
- 19. Han TS, Walker BR, Arlt W, et al. Treatment and health outcomes in adults with congenital adrenal hyperplasia. Nat Rev Endocrinol 2014; 10: 115–124. [DOI] [PubMed] [Google Scholar]
- 20. Mornet E, Dupont J, Vitek A, et al. Characterization of two genes encoding human steroid 11 beta-hydroxylase (P-450(11) beta). J Biol Chem 1989; 264: 20961–20967. [PubMed] [Google Scholar]
- 21. Wang HZ, Tian JB, Yang KH. Efficacy and safety of LCI699 for hypertension: A meta-analysis of randomized controlled trials and systematic review. Eur Rev Med Pharmacol Sci 2015; 19: 296–304. [PubMed] [Google Scholar]
- 22. Bertagna X1, Pivonello R, Fleseriu M, et al. LCI699, a potent 11β-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: Results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab 2014; 99: 1375–1383. [DOI] [PubMed] [Google Scholar]
- 23. Karns AD, Bral JM, Hartman D, et al. Study of aldosterone synthase inhibition as an add-on therapy in resistant hypertension. J Clin Hypertens (Greenwich) 2013; 15: 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amar L, Azizi M, Menard J, et al. Sequential comparison of aldosterone synthase inhibition and mineralocorticoid blockade in patients with primary aldosteronism. J Hypertens 2013; 31: 624–629; discussion 629. [DOI] [PubMed] [Google Scholar]
- 25. Andersen K, Hartman D, Peppard T, et al. The effects of aldosterone synthase inhibition on aldosterone and cortisol in patients with hypertension: A phase II, randomized, double-blind, placebo-controlled, multicenter study. J Clin Hypertens (Greenwich) 2012; 14: 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calhoun DA1, White WB, Krum H, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: Results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation 2011; 124: 1945–1955. [DOI] [PubMed] [Google Scholar]
- 27. Amar L, Azizi M, Menard J, et al. Aldosterone synthase inhibition with LCI699: A proof-of-concept study in patients with primary aldosteronism. Hypertension 2010; 56: 831–838. [DOI] [PubMed] [Google Scholar]
- 28. Greig WR, Maxwell JD, Boyle JA, et al. Criteria for distinguishing normal from subnormal adrenocortical function using the Synacthen test. Postgrad Med J 1969; 45: 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mortensen RM, Williams GH. Aldosterone action. In: DeGroot LJ, Jameson JL, Burger HG, et al. (eds) Endocrinology, 4th ed Philadelphia: WB Saunders, 2001, pp.1783–1790. [Google Scholar]
- 30. Dluhy RG, Williams GH. Adrenal cortex. In: Kasper DL, Braunwald E, Fauci AS, et al. (eds) Harrison’s principles of internal medicine, 16th ed McGraw Hill: New York, 2005, p.321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.