

Abstract
Background:
Renin-angiotensin-aldosterone system (RAAS) activation in heart failure with reduced ejection fraction (HFREF) is detrimental through promotion of ventricular remodeling and salt and water retention.
Aims:
The aims of this article are to describe RAAS activity in distinct HFREF populations and to assess its prognostic impact.
Methods:
Venous blood samples were prospectively obtained in 76 healthy volunteers, 72 patients hospitalized for acute decompensated HFREF, and 78 ambulatory chronic HFREF patients without clinical signs of congestion. Sequential measurements were performed in patients with acute decompensated HFREF.
Results:
Plasma renin activity (PRA) was significantly higher in ambulatory chronic HFREF (7.6 ng/ml/h (2.2; 18.1)) compared to patients with acute decompensated HFREF (1.5 ng/ml/h (0.8; 5.7)) or healthy volunteers (1.4 ng/ml/h (0.6; 2.3)) (all p < 0.05). PRA was significantly associated with arterial blood pressure and renin-angiotensin system blocker dose. A progressive rise in PRA (+4 ng/ml/h (0.4; 10.9); p < 0.001) was observed in acute decompensated HFREF patients after three consecutive days of decongestive treatment. Only in acute HFREF were PRA levels associated with increased cardiovascular mortality or HF readmissions (p = 0.035).
Conclusion:
PRA is significantly elevated in ambulatory chronic HFREF patients but is not associated with worse outcome. In contrast, in acute HFREF patients, PRA is associated with cardiovascular mortality or HF readmissions.
Keywords: Aldosterone, biomarkers, renin, renin-angiotensin system, systolic heart failure
Introduction
Renin-angiotensin-aldosterone system (RAAS) activation in heart failure with reduced ejection fraction (HFREF) has detrimental long-term effects such as water and salt retention as well as promoting adverse ventricular remodeling. Outcomes in HFREF patients have drastically improved during the past two decades through strategies that have targeted RAAS activation.1–4 Plasma renin activity (PRA) and plasma aldosterone levels are biomarkers that quantitatively reflect RAAS activation and might be used for risk stratification in HFREF. Indeed, previous studies have linked higher levels of RAAS activation to more advanced disease stages and worse outcomes both in acute and chronic HFREF.5–9 Most of these studies have focused on PRA as renin is the rate-limiting step of the RAAS, and a more reliable reflection of RAAS activation compared to serum aldosterone.10,11 However, these studies largely predate the current era of HFREF treatment in which angiotensin-converting enzyme inhibitors (ACE-i), angiotensin receptor blockers (ARB), beta-blockers and mineralocorticoid receptor antagonists (MRA) are guideline-recommended therapies. Indeed, nowadays cardiologist uptitrate ACE-i/ARB and beta-blockers to the highest achievable dose without intolerable side effects (e.g. systolic blood pressure <90 mmHg, symptomatic orthostatic hypotension, decline in renal function, hyperkalemia).12 Moreover, whether RAAS activation during decongestive therapy has prognostic significance remains unclear.13 Therefore, the objective of this study is to describe the extent of RAAS activation, and its prognostic impact in well-characterized HFREF populations on optimal medical therapy.
Methods
Study design
This prospective cohort study was carried out in a single tertiary care center (Ziekenhuis Oost-Limburg, Genk, Belgium) between September 2011 and October 2015. The study complies with the Declaration of Helsinki and the institutional review board approved the study protocol. All participants provided written informed consent before any study-specific intervention was performed.
Study population
Patients were eligible for study inclusion if ⩾18 years of age and able to give informed consent. Healthy volunteers were recruited through general announcements and had (1) no history of cardiac or renal disease, (2) a normal clinical examination, and (3) normal cardiac function on transthoracic echocardiography.
Patients with acute decompensated HFREF had (1) the presence of ⩾3 signs or symptoms of volume overload (edema, jugular venous distention, orthopnea, rales or pulmonary vascular congestion on chest X-ray); (2) plasma N-terminal of the prohormone of B-type natriuretic peptide (NT-proBNP) levels >1000 ng/l; (3) a left ventricular ejection fraction (LVEF) ⩽45%; and (4) a clinical diagnosis of HF with evidence of impaired LVEF ⩽40% within six months before inclusion (5) on optimal medical therapy according to current guideline recommendations or12,14 (6) were hospitalized with an anticipated treatment strategy of intravenous loop diuretics. Exclusion criteria were (1) administration of intravenous diuretics before study inclusion; (2) mechanical ventilation; (3) inotropic or vasopressor support; (4) concurrent diagnosis of an acute coronary syndrome; (5) renal replacement therapy; or (6) ventricular assist devices, including the use of an intra-aortic balloon pump, at any time during the index hospitalization. During and after index hospitalization neurohumoral blockers were uptitrated to the highest possible dose without side effects according to guideline recommendations (defined as optimal dose) and at the discretion of the treating cardiologist.15
Ambulatory patients with chronic HFREF had (1) a clinical diagnosis of HF with evidence of impaired LVEF ⩽40% within six months before inclusion; (2) no hospital admission for worsening HF signs or symptoms within six months before inclusion; (3) stable New York Heart Association (NYHA) functional class I–III for ⩾3 months; (4) unchanged pharmacological therapy with ACE-i, ARB, beta-blockers, MRA and diuretics during the last three months prior to inclusion; (5) optimal medical therapy according to current guideline recommendations.12,14
Study endpoint
Cardiovascular mortality and HF readmissions (defined as hospitalizations because of signs or symptoms of congestion or low cardiac output that warranted treatment with parenteral drugs) were prospectively registered in all study patients from inclusion up till three years after which they were censored.
Laboratory measurements
Venous blood samples were obtained at the moment of study inclusion with the patient in the supine position after an adaptation period of 30 minutes. Plasma NT-proBNP levels were measured by the Roche Diagnostics Assay (Roche, Rotkreuz, Switzerland). PRA was determined using the Gamma-coat*radio immunoassay (DiaSorin, Sallugia, Italy). Plasma aldosterone levels were assessed by the Aldosterone Maia radioimmunoassay (Adaltis, Rome, Italy).
Within the subpopulation of acute decompensated HFREF sequential venous blood samples were obtained before the start of intravenous therapy (baseline), after three days of decongestive therapy, and during ambulatory follow-up approximately six weeks after discharge. Treating physicians were blinded to test results and treatment during hospitalization was at their own discretion.
Statistical analysis
Continuous variables are expressed as mean±standard deviation, if normally distributed, or otherwise by median (interquartile range). Normality was assessed by the Shapiro-Wilk statistic. Categorical data are expressed as percentages and compared with the Pearson χ²-test. One-way analysis of variance (ANOVA) testing or the Kruskal-Wallis H test were used as indicated. Repeated measures within the acute decompensated HFREF group were compared using the paired Student’s t-test or the Wilcoxon signed-rank test as appropriate. To establish associations between PRA and anticipated factors, a multivariable linear regression model was constructed after the logarithmic transformation of PRA to correct for the non-normal distribution. Variables with a p value <0.100 in univariable regression analyses were included in a standard multivariable model. Cumulative survival rates were calculated according to the Kaplan–Meier method with the log-rank test used for comparison among tertiles of PRA. Statistical significance was always set at a two-tailed probability level of <0.05. All statistics were performed using SAS JMP Pro (version 11.2 for Windows).
Results
Study population
Seventy-six healthy volunteers, 72 patients with acute decompensated HFREF and 78 ambulatory chronic HFREF patients were included. Table 1 summarizes their baseline characteristics. Compared to healthy controls, acute and chronic HFREF patients were older and had a severely impaired LVEF (25±10 vs 33±7, respectively). Neurohormonal blocker use was high in both cohorts of HFREF patients. However, compared to chronic ambulatory HFREF patients, fewer patients with acute decompensated HFREF were on maintenance therapy with an ACE-i or ARB (50% vs 87%). Instead, 26% of acute decompensated patients were taking oral vasodilators (hydralazine and/or nitrates). Loop diuretic use was highest in the cohort of acute HFREF patients.
Table 1.
Baseline characteristics of the study population.
| Healthy volunteers n = 76 |
Acute decompensated HFREF n = 72 |
Ambulatory chronic HFREF n = 78 |
|
|---|---|---|---|
| Age (years) | 42 ± 16 | 67 ± 11 | 66 ± 12 |
| Male gender | 51% | 76% | 77% |
| Heart rate (bpm) | 68 ± 11 | 81 ± 19 | 66 ± 10 |
| Systolic blood pressure (mmHg) | 130 ± 17 | 128 ± 23 | 124 ± 17 |
| Diastolic blood pressure (mmHg) | 76 ± 10 | 71 ± 15 | 63 ± 12 |
| Ischemic cardiomyopathy | N/A | 58% | 62% |
| Left ventricular ejection fraction (%) | 65 ± 6 | 25 ± 10 | 33 ± 7 |
| Medical therapy | |||
| ACE-i/ARB use (%) | 0 | 50% | 87% |
| ⩽50% of target dose | 35% | 47% | |
| >50% of target dose | 15% | 40% | |
| (100% of target dose) | 11% | 38% | |
| Beta-blocker use (%) | 0 | 72% | 97% |
| ⩽50% of target dose | 57% | 45% | |
| >50% of target dose | 15% | 52% | |
| (100% of target dose) | 11% | 52% | |
| MRA use (%) | 0 | 49% | 81% |
| Loop diuretic use (%) | 0 | 64% | 49% |
| Hydralazine/nitrate use (%) | 0 | 26% | 8% |
| Laboratory results | |||
| Creatinine (mg/dl) | 0.93 ± 0.21 | 1.43 ± 0.68 | 1.29 ± 0.51 |
| NT-proBNP (ng/l) | 47 (30; 73) | 4011 (2018; 10,608) | 608 (271; 1407) |
| PRA (ng/ml/h) | 1.4 (0.6; 2.3) | 1.5 (0.8; 5.7) | 7.6 (2.2; 18.1) |
| Plasma aldosterone (ng/l) | 247 (165; 346) | 179 (134; 292) | 213 (144; 374) |
ACE-i: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; bpm: beats per minute; HFREF: heart failure with reduced ejection fraction; MRA: mineralocorticoid receptor antagonist; NT-proBNP: N-terminal of the prohormone of B-type natriuretic peptide; NYHA: New York Heart Association; PRA: plasma renin activity.
RAAS activation in distinct populations of HFREF
PRA was significantly higher in ambulatory chronic HFREF patients (7.6 ng/ml/h (2.2; 18.1)) compared to acute decompensated HFREF patients (1.5 ng/ml/h (0.8; 5.7)) or healthy volunteers (1.4 ng/ml/h (0.6; 2.3)) (both p <0.0001). There was no significant difference in PRA levels between acute decompensated HFREF patients and healthy volunteers (p = 0.13) (Figure 1). Plasma concentrations of aldosterone were the lowest in acute decompensated HFREF but not significantly different from healthy individuals (p = 0.08) or ambulatory chronic HFREF patients (p = 0.19) and comparable among the two other groups (p = 0.76). Overall, PRA was significantly associated with blood pressure, ACE-i/ARB dose and MRA dose after multivariable regression analysis. There was no significant association between PRA and either loop diuretic dose or NT-proBNP level (Table 2).
Figure 1.
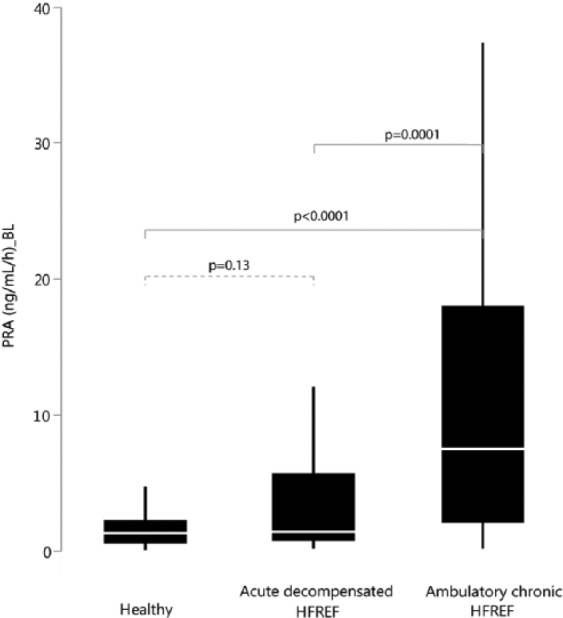
Plasma renin activity (PRA) in healthy volunteers, patients with acute decompensated heart failure with reduced ejection fraction (HFREF) and ambulatory chronic HFREF.
Table 2.
Uni- and multivariable regression analysis for significant determinants of plasma renin activity in the total study population.
| Univariable |
Multivariable |
|||||
|---|---|---|---|---|---|---|
| Beta | S.E. | p | Beta | S.E. | p | |
| Age (years) | 0.12 | 0.10 | 0.199 | |||
| Male gender | ||||||
| Left ventricular ejection fraction (%) | −0.31 | 0.07 | <0.001 | −0.13 | 0.08 | 0.12 |
| Heart rate (bpm) | −0.06 | 0.10 | 0.544 | |||
| Mean arterial pressure (mmHg) | −0.51 | 0.11 | <0.001 | −0.44 | 0.09 | <0.001 |
| ACE-i/ARB dose (% of guideline recommended dose) | 0.54 | 0.10 | <0.001 | 0.40 | 0.12 | 0.001 |
| Beta-blocker dose (% of guideline recommended dose) | 0.37 | 0.10 | <0.001 | −0.25 | 0.13 | 0.058 |
| MRA dose (% of guideline recommended dose) | 0.50 | 0.09 | <0.001 | 0.24 | 0.12 | 0.041 |
| Loop diuretic dose (mg bumetanide equivalents) | 0.16 | 0.10 | 0.107 | |||
| Creatinine (mg/dl) | 0.19 | 0.09 | 0.047 | 0.10 | 0.09 | 0.288 |
| NT-proBNP (ng/l) | −0.05 | 0.10 | 0.576 | |||
To correct for non-normal distribution, PRA was logarithmically transformed. Beta and standard error (S.E.) for continuous variables reported per standard deviation change. ACE-i: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; bpm: beats per minute; MRA: mineralocorticoid receptor antagonist; NT-proBNP: N-terminal of the prohormone of B-type natriuretic peptide.
PRA during decongestive therapy and uptitration of neurohormonal blockers in patients with acute decompensated HF
During decongestive treatment (from baseline to day 3) in patients with acute decompensated HFREF, the absolute and relative increase in PRA was +2.4 ng/ml/h (−0.2; 6.9) and +107% (−12%; 553%), respectively (both p < 0.001). Seventy-one percent of patients showed an increase in PRA during decongestion and uptitration of neurohormonal blockers (Figure 2, Table 3). Additionally, from day 3 until six weeks after discharge, neurohormonal blockers were further uptitrated, and 58% of patients showed an additional increase in PRA, which was however non-significant (+0.5 ng/ml/h (−2.3; 7.8) (p = 0.35)) (Figure 2). Taken together, from admission until six weeks’ follow-up 79% of patients showed an increase in PRA (+4 (0.4; 10.9) ng/ml/h or +238% (30; 654) compared to baseline; p < 0.001)). Similar trends were observed with regards to serum aldosterone concentration.
Figure 2.
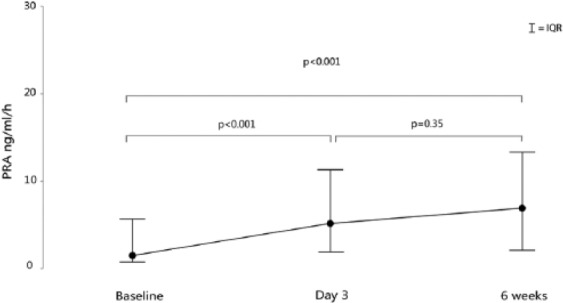
Plasma renin activity (PRA) from admission until six weeks after discharge in patients with acute heart failure and reduced ejection fraction. PRA levels rise during decongestive therapy and neurohumoral uptitration.
Table 3.
Use of neurohormonal blockers and loop diuretics in patients presenting with acute decompensated heart failure and reduced ejection fraction at baseline (BL), after three days (D3) of decongestive treatment, and at six weeks (6W) after discharge.
| Baseline (BL) | Day 3
(D3) |
Six weeks
(6W) |
p
BL–D3 |
p
BL–6W |
p
D3–6W |
|
|---|---|---|---|---|---|---|
| ACE-I or ARB use | 50% | 68% | 68% | <0.0001 | <0.0001 | <0.0001 |
| ○ ⩽50% of target dose | 35% | 55% | 38% | |||
| ○ >50% of target dose | 15% | 13% | 30% | |||
| ○ (100% of target dose) | 11% | 6% | 22% | |||
| Beta-blocker use | 72% | 88% | 97% | 0.0038 | 0.0038 | <0.0001 |
| ○ ⩽50% of target dose | 57% | 66% | 68% | |||
| ○ >50% of target dose | 15% | 22% | 29% | |||
| ○ (100% of target dose) | 11% | 14% | 23% | |||
| MRA use | 49% | 90% | 86% | <0.001 | 0.006 | <0.0001 |
| Loop diuretic use | 64% | 100% | 70% | |||
| ○ Loop diuretic dose (mg bumetanide equivalent) | 1 (0; 2) | N/A | 0.5 (0; 1) | 0.0005 | ||
| ○ Cumulative loop diuretic dose used during D1–D3 (mg bumetanide equivalent) | N/A | 4 (2; 5) | N/A |
ACE-i: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; MRA: mineralocorticoid receptor antagonist.
PRA and clinical outcome
During the entire follow-up period, 36 events occurred in patients with acute decompensated HFREF (22 patients died from cardiovascular causes and 14 patients were readmitted for worsening HF). PRA levels at admission in the highest tertile were associated with a significantly increased event rate (log rank=0.035) (Figure 3). There was no difference in outcome between patients with a PRA rise vs decline during recompensation (from baseline until day 3) (log rank=0.96) (Figure 4, Table 3 Supplementary Appendix). In the subgroup with ambulatory chronic HFREF patients, one death and six HF readmissions occurred. PRA levels were not associated with clinical outcome (log rank = 0.99) (Figure 3). Baseline characteristics of acute decompensated and chronic stable HFREF patients per tertile are presented in the supplementary appendices.
Figure 3.

Kaplan–Meier curves for the combined endpoint of heart failure-associated hospitalization and cardiovascular mortality in patients with acute decompensated heart failure with reduced ejection fraction (HFREF) (upper panel) and ambulatory chronic HFREF (lower panel) according to tertiles of plasma renin activity (PRA).
Figure 4.
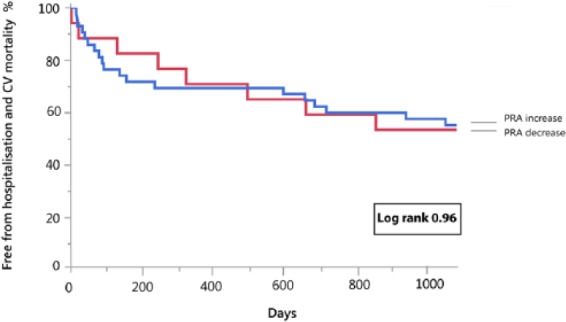
Kaplan–Meier curve for the combined endpoint of heart failure-associated hospitalization and cardiovascular mortality in patients hospitalized for acute heart failure with plasma renin activity (PRA) increase vs PRA decrease during decongestive therapy.
Discussion
Longitudinal data from distinct well-characterized HFREF populations in the current era of treatment with neurohormonal blockers provide a unique opportunity to examine RAAS activation. The primary findings of this study are (1) significant PRA is present in ambulatory chronic HFREF patients without signs and symptoms of congestion, while PRA seems depressed during episodes of acute decompensated HFREF with clear signs of volume overload; (2) PRA levels correlate significantly to worse outcomes only in patients with acute decompensated HFREF before initiation of decongestive treatment; and (3) treatment with neurohormonal blockers significantly influence neurohormonal levels.
The RAAS
To better appreciate the study results, a brief review of the RAAS is useful. Renin, an enzyme released by juxtaglomerular cells of the renal afferent arteriole, starts a cascade in which angiotensinogen is cleaved first into angiotensin I, which is further metabolized to angiotensin II (Ang II) by ACE. Ang II causes systemic and renal arteriolar vasoconstriction, promotes renal tubular sodium and water reabsorption, and is a potent stimulator of aldosterone release from the adrenal glands. Upon an acute drop of the cardiac output, RAAS activation helps to preserve organ perfusion in general and the glomerular filtration rate in particular.16–18 Renin is released from the afferent arteriole in response to three main stimuli: (1) decreased arterial blood pressure sensed by baroreceptor cells in the afferent arteriolar vessel wall, (2) decreased chloride concentrations in macula densa cells lining the renal tubules at the end of Henle’s loop, and (3) sympathetic nerve system activation.19,20 As a result, renin release is physiologically inhibited by normal or elevated systemic blood pressure and a diet high in salt.21–23 Persistent and excessive RAAS activation causes adverse ventricular remodeling and contributes to fluid retention with signs and symptoms of congestion.24–30
High PRA in ambulatory chronic HFREF
More than two decades ago, before the standard use of neurohormonal blockers in HFREF, Francis et al. compared neurohormonal activation—including PRA—in healthy volunteers vs asymptomatic HFREF patients vs HFREF patients with signs and symptoms of congestion. The authors concluded that neurohormonal activation already occurred in patients with left ventricular dysfunction before the onset of symptoms, which was further exaggerated as overt HF ensued and diuretics were added to therapy.7 Remarkably, important increases in neurohormonal activation were mainly seen in the patients with symptomatic HFREF, while the PRA increases in asymptomatic patients were modest. This is in contrast to our findings in a contemporary cohort of HFREF patients, where the most pronounced PRA rise was observed in ambulatory chronic HFREF patients without signs and symptoms of congestion but well treated with ACE-i, ARB, beta-blockers and MRA.
Indeed, ACE-i and ARB tend to lower aldosterone concentrations, but increase PRA, while beta-blockers might lower both, and MRA increase both.31–37 Although none of the patients included in this study were taking an angiotensin receptor blocker-neprilysin inhibitor (ARNI), in light of the new HF guidelines it may be interesting to point out that previous studies have demonstrated that the association of sacubitril, a neprilysin inhibitor, does not affect PRA or serum aldosterone concentration.38,39 Hypertensive patients treated with neurohormonal blockers demonstrate increased PRA and plasma aldosterone levels.40–42 Yet, the individual response to medication varies greatly because of genetic polymorphisms.6,43–47 Our data corroborate this as we observed a wide spread both in PRA and plasma aldosterone levels among patients with ambulatory chronic HFREF. Overall, patients with ambulatory chronic HF had significantly higher levels of PRA compared to acute decompensated HF patients while exhibiting a much lower risk. Thus, the association between PRA and outcome seems to apply only to the higher-risk groups of HF patients. Therefore, most probably the PRA and serum aldosterone levels do not reflect disease-related (harmful) RAAS activation and are not a reliable surrogate for downstream receptor activation.48,49 Interestingly, the RAAS consists of two main axes: the classical ACE/Ang II and the counteracting ACE2/angiotensin 1 to 7 (Ang 1–7) axis.50 ACE2, a homolog of ACE, degrades Ang II into Ang 1–7. Ang 1–7 exerts a wide array of actions, many of which are opposite to those attributed to Ang II (vasodilation, decrease in fibrosis and cardiomyocyte hypertrophy, inhibition of aldosterone secretion etc.).50,51 In our study we measured PRA, the rate-limiting step of the RAAS system and common to both axes and serum aldosterone. However, we did not measure Ang II, Ang 1–7 or other products of intermediate steps of the complex RAAS. It has been shown that in response to ACE-i, PRA rises but plasma levels of Ang 1–7 also increase. A high percentage of ambulatory chronic HF patients, but also acute decompensated patients were on ACE-i as well as MRA. Therefore, it may be a logical hypothesis that the increased renin activity due to efficient blockade of the classical ACE/Ang II axis leads to stimulation of the counteracting and beneficial axis of the RAAS (ACE2/Ang 1–7), which may be partly responsible for the beneficial effects of this therapy in HF patients. This could be an interesting topic for further research for the development of new targets for HF therapies.
PRA in acute HFREF
As neurohormonal activation is often perceived as the key driver in HFREF disease progression, it may seem odd that PRA and serum aldosterone levels are significantly lower in patients with signs and symptoms of congestion. Furthermore, this contradicts former observations in medication-naive HFREF patients.7 Yet, most chronic HFREF patients in the current study were on maximally tolerated dosages of neurohormonal blockers and also have rather low blood pressure. Both are powerful predictors of PRA levels in our overall population (Table 2). In contrast, most patients with acute decompensated HFREF present with elevated rather than low arterial blood pressure, a finding also present in the current study.52 Intriguingly, this might indicate that the RAAS in advanced HFREF treated with neurohormonal blockers remains appropriately responsive to hemodynamic changes including blood pressure and volume overload. Also, the most important increase in RAAS activation during the treatment of acute HFREF is seen in the first days of hospitalization, which seem to be linked to decongestive therapy (reduction in plasma volume as well as intensified diuretic therapy) and introduction and/or uptitration of neurohormonal blockers. Therefore, one might speculate that PRA could be a potential surrogate for effective circulatory volume assessment.
Prognostic value of PRA
PRA levels correlate significantly to worse outcomes only in patients with acute decompensated HFREF before decongestive treatment. High levels of neurohormones in stable HFREF patients are not predictive for rehospitalization due to water and salt retention or death. Also, the relation of high PRA levels and negative outcome in acute decompensated HFREF might not be driven by higher neurohormonal activation but rather reflect more advanced disease in this subgroup, reflecting volume overload and low pressure, both known to be related to worse outcome.50,51 In conclusion, it seems that the association between an RAAS biomarker and adverse outcomes applies only to the setting and not to its absolute value.
Study limitations
First, we recruited and compared two groups of HFREF patients. Although we were able to characterize these groups in detail, it is uncertain to what extent observed differences in RAAS activation were due to heterogeneity between groups. The fact that all patients were recruited from a single institution, the limited sample size and event rates makes findings hypothesis-generating and ask for separate confirmation. Second, patients with urgent need for administration of loop diuretics, inotropic/vasodilator or mechanical support were not included in this study. Thus, our data do not apply to severely decompensated HF patients or patients in cardiogenic shock. Third, all patients were put in a semi-supine position for an adaptation period before a venous blood sample was drawn. However, besides physical activity, PRA also depends on many other variables such as circadian rhythm, sodium intake, and presence/absence of disease states that could not be accounted for in this cohort study.53
Conclusion
PRA is decreased in a state of acute decompensation compared to ambulatory chronic HFREF. An increase in PRA activity is observed in the majority of patients during decongestive treatment and neurohormonal blocker uptitration. However, increased PRA is associated with adverse outcomes only in the setting of acute decompensated HFREF before initiation of decongestive treatment.
Supplementary Material
Footnotes
Declaration of conflicting interests: None declared.
Funding: P. N., F.H.V., P.M. and W.M. are researchers for the Limburg Clinical Research Program (LCRP) UHasselt–ZOL–Jessa, supported by the foundation Limburg Sterk Merk (LSM), Hasselt University, Ziekenhuis Oost-Limburg and Jessa Hospital. F.H.V. and P.B.B. are supported by a PhD fellowship of the Research Foundation–Flanders (FWO). P.N. and M.D. are supported by a research grant provided by Vision4Life-Sciences.
References
- 1. CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 1987; 316: 1429–1435. [DOI] [PubMed] [Google Scholar]
- 2. Cohn JN, Tognoni G. Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 2001; 345: 1667–1675. [DOI] [PubMed] [Google Scholar]
- 3. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341: 709–717. [DOI] [PubMed] [Google Scholar]
- 4. Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11–21. [DOI] [PubMed] [Google Scholar]
- 5. Brown MJ. Renin: Friend or foe? Heart 2007; 93: 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castrop H, Höcherl K, Kurtz A, et al. Physiology of kidney renin. Physiol Rev 2010; 90: 607–673. [DOI] [PubMed] [Google Scholar]
- 7. Francis GS, Benedict C, Johnstone DE, et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 1990; 82: 1724–1729. [DOI] [PubMed] [Google Scholar]
- 8. Vergaro G, Emdin M, Iervasi A, et al. Prognostic value of plasma renin activity in heart failure. Am J Cardiol 2011; 108: 246–251. [DOI] [PubMed] [Google Scholar]
- 9. Girerd N, Pang PS, Swedberg K, et al. Serum aldosterone is associated with mortality and re-hospitalization in patients with reduced ejection fraction hospitalized for acute heart failure: Analysis from the EVEREST trial. Eur J Heart Fail 2013; 15: 1228–1235. [DOI] [PubMed] [Google Scholar]
- 10. Atlas SA. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J Manag Care Pharm 2007; 13: 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skeggs LT, Jr, Kahn JR, Lentz K, et al. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med 1957; 106: 439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012; 33: 1787–1847. [DOI] [PubMed] [Google Scholar]
- 13. Verbrugge FH, Tang WH, Mullens W. Renin-angiotensin-aldosterone system activation during decongestion in acute heart failure: Friend or foe? JACC Heart Fail 2015; 3: 108–111. [DOI] [PubMed] [Google Scholar]
- 14. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62: e147–e239. [DOI] [PubMed] [Google Scholar]
- 15. Verbrugge FH, Duchenne J, Bertrand PB, et al. Uptitration of renin-angiotensin system blocker and beta-blocker therapy in patients hospitalized for heart failure with reduced versus preserved left ventricular ejection fractions. Am J Cardiol 2013; 112: 1913–1920. [DOI] [PubMed] [Google Scholar]
- 16. Seikaly MG, Arant BS, Jr, Seney FD., Jr Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clinical Invest 1990; 86: 1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Kats JP, Danser AH, van Meegen JR, et al. Angiotensin production by the heart: A quantitative study in pigs with the use of radiolabeled angiotensin infusions. Circulation 1998; 98: 73–81. [DOI] [PubMed] [Google Scholar]
- 18. Vallotton MB, Gerber-Wicht C, Dolci W, et al. Interaction of vasopressin and angiotensin II in stimulation of prostacyclin synthesis in vascular smooth muscle cells. Am J Physiol 1989; 257 (5 Pt 1): E617–E624. [DOI] [PubMed] [Google Scholar]
- 19. Schnermann J. Juxtaglomerular cell complex in the regulation of renal salt excretion. Am J Physiol 1998; 274 (2 Pt 2): R263–R279. [DOI] [PubMed] [Google Scholar]
- 20. Verbrugge FH, Dupont M, Steels P, et al. The kidney in congestive heart failure: ‘Are natriuresis, sodium, and diuretics really the good, the bad and the ugly?’ Eur J Heart Fail 2014; 16: 133–142. [DOI] [PubMed] [Google Scholar]
- 21. Shin SJ, Lim C, Oh SW, et al. The unique response of renin and aldosterone to dietary sodium intervention in sodium sensitivity. J Renin Angiotensin Aldosterone Syst 2014; 15: 117–123. [DOI] [PubMed] [Google Scholar]
- 22. Lu H, Wu C, Howatt DA, et al. Differential effects of dietary sodium intake on blood pressure and atherosclerosis in hypercholesterolemic mice. J Nutr Biochem 2013; 24: 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pimenta E, Gaddam KK, Oparil S, et al. Effects of dietary sodium reduction on blood pressure in subjects with resistant hypertension: Results from a randomized trial. Hypertension 2009; 54: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baker KM, Aceto JF. Angiotensin II stimulation of protein synthesis and cell growth in chick heart cells. Am J Physiol 1990; 259 (2 Pt 2): H610–H618. [DOI] [PubMed] [Google Scholar]
- 25. Harrap SB, Dominiczak AF, Fraser R, et al. Plasma angiotensin II, predisposition to hypertension, and left ventricular size in healthy young adults. Circulation 1996; 93: 1148–1154. [DOI] [PubMed] [Google Scholar]
- 26. Peng J, Gurantz D, Tran V, et al. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res 2002; 91: 1119–1126. [DOI] [PubMed] [Google Scholar]
- 27. Weber KT. Extracellular matrix remodeling in heart failure: A role for de novo angiotensin II generation. Circulation 1997; 96: 4065–4082. [DOI] [PubMed] [Google Scholar]
- 28. Brilla CG, Zhou G, Matsubara L, et al. Collagen metabolism in cultured adult rat cardiac fibroblasts: Response to angiotensin II and aldosterone. J Mol Cell Cardiol 1994; 26: 809–820. [DOI] [PubMed] [Google Scholar]
- 29. Harada E, Yoshimura M, Yasue H, et al. Aldosterone induces angiotensin-converting-enzyme gene expression in cultured neonatal rat cardiocytes. Circulation 2001; 104: 137–139. [DOI] [PubMed] [Google Scholar]
- 30. Mizuno Y, Yoshimura M, Yasue H, et al. Aldosterone production is activated in failing ventricle in humans. Circulation 2001; 103: 72–77. [DOI] [PubMed] [Google Scholar]
- 31. Waeber B, Nussberger J, Perret L, et al. Experience with perindopril in normal volunteers. Arch Mal Coeur Vaiss 1989; 82 Spec No 1: 35–41. [PubMed] [Google Scholar]
- 32. Yasumura Y, Miyatake K, Okamoto H, et al. Rationale for the use of combination angiotensin-converting enzyme inhibitor and angiotensin II receptor blocker therapy in heart failure. Circ J 2004; 68: 361–366. [DOI] [PubMed] [Google Scholar]
- 33. Lees KR, Reid JL. Haemodynamic and humoral effects of oral perindopril, an angiotensin converting enzyme inhibitor, in man. Br J Clin Pharmacol 1987; 23: 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jorde UP, Vittorio T, Katz SD, et al. Elevated plasma aldosterone levels despite complete inhibition of the vascular angiotensin-converting enzyme in chronic heart failure. Circulation 2002; 106: 1055–1057. [DOI] [PubMed] [Google Scholar]
- 35. Sato A, Suzuki Y, Shibata H, et al. Plasma aldosterone concentrations are not related to the degree of angiotensin-converting enzyme inhibition in essential hypertensive patients. Hypertens Res 2000; 23: 25–31. [DOI] [PubMed] [Google Scholar]
- 36. Azizi M, Chatellier G, Guyene TT, et al. Additive effects of combined angiotensin-converting enzyme inhibition and angiotensin II antagonism on blood pressure and renin release in sodium-depleted normotensives. Circulation 1995; 92: 825–834. [DOI] [PubMed] [Google Scholar]
- 37. Giles TD, Bakris G, Oparil S, et al. Correlations of plasma renin activity and aldosterone concentration with ambulatory blood pressure responses to nebivolol and valsartan, alone and in combination, in hypertension. J Am Soc Hypertens 2015; 9: 845–854. [DOI] [PubMed] [Google Scholar]
- 38. Hubers SA, Brown NJ. Combined angiotensin receptor antagonism and neprilysin inhibition. Circulation 2016; 133: 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37: 2129–2200. [DOI] [PubMed] [Google Scholar]
- 40. Tiryaki O, Usalan C, Buyukhatipoglu H. Effect of combined angiotensin-converting enzyme and aldosterone inhibition on plasma plasminogen activator inhibitor type 1 levels in chronic hypertensive patients. Nephrology (Carlton) 2010; 15: 211–215. [DOI] [PubMed] [Google Scholar]
- 41. Sullivan JM, Ginsburg BA, Ratts TE, et al. Hemodynamic and antihypertensive effects of captopril, an orally active angiotensin converting enzyme inhibitor. Hypertension 1979; 1: 397–401. [DOI] [PubMed] [Google Scholar]
- 42. Grossman E, Peleg E, Carroll J, et al. Hemodynamic and humoral effects of the angiotensin II antagonist losartan in essential hypertension. Am J Hypertens 1994; 7: 1041–1044. [DOI] [PubMed] [Google Scholar]
- 43. Tang WH, Vagelos RH, Yee YG, et al. Impact of angiotensin-converting enzyme gene polymorphism on neurohormonal responses to high- versus low-dose enalapril in advanced heart failure. Am Heart J 2004; 148: 889–894. [DOI] [PubMed] [Google Scholar]
- 44. Hannila-Handelberg T, Kontula KK, Paukku K, et al. Common genetic variations of the renin-angiotensin-aldosterone system and response to acute angiotensin I-converting enzyme inhibition in essential hypertension. J Hypertens 2010; 28: 771–779. [DOI] [PubMed] [Google Scholar]
- 45. Sato A, Saruta T. Aldosterone breakthrough during angiotensin-converting enzyme inhibitor therapy. Am J Hypertens 2003; 16: 781–788. [DOI] [PubMed] [Google Scholar]
- 46. van de Wal RM, Plokker HW, Lok DJ, et al. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol 2006; 106: 367–372. [DOI] [PubMed] [Google Scholar]
- 47. de Boer RA, Schroten NF, Bakker SJ, et al. Plasma renin and outcome in the community: Data from PREVEND. Eur Heart J 2012; 33: 2351–2359. [DOI] [PubMed] [Google Scholar]
- 48. Dzau VJ, Re R. Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation 1994; 89: 493–498. [DOI] [PubMed] [Google Scholar]
- 49. Asano K, Dutcher DL, Port JD, et al. Selective downregulation of the angiotensin II AT1-receptor subtype in failing human ventricular myocardium. Circulation 1997; 95: 1193–1200. [DOI] [PubMed] [Google Scholar]
- 50. Santos RA. Angiotensin-(1–7). Hypertension 2014; 63: 1138–1147. [DOI] [PubMed] [Google Scholar]
- 51. Patel VB, Lezutekong JN, Chen X, et al. Recombinant human ACE2 and the Angiotensin 1–7 axis as potential new therapies for heart failure. Can J Cardiol 2017; 33: 943–946. [DOI] [PubMed] [Google Scholar]
- 52. Adams KF, Jr, Fonarow GC, Emerman CL, et al. Characteristics and outcomes of patients hospitalized for heart failure in the United States: Rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 2005; 149: 209–216. [DOI] [PubMed] [Google Scholar]
- 53. Cugini P, Lucia P. Circadian rhythm of the renin-angiotensin-aldosterone system: A summary of our research studies [article in Italian]. Clinica Ter 2004; 155: 287–291. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.