

Abstract
Introduction:
Inhaled budesonide is a novel approach to prevent acute mountain sickness (AMS). However, its mechanism is not completely understood. We aimed to investigate the effects of budesonide and dexamethasone on renin–angiotensin–aldosterone system in AMS prevention.
Materials and methods:
Data were obtained from a randomised controlled trial including 138 participants. The participants were randomly assigned to receive budesonide, dexamethasone or placebo as prophylaxis before they travelled to 3450 m altitude from 400 m by car. Their plasma concentrations of renin, angiotensin-converting enzyme (ACE) and aldosterone were measured at both altitudes.
Results:
All parameters were comparable among the three groups at 400 m. After high-altitude exposure of 3450, renin in all groups increased significantly; the ACE, aldosterone concentrations, as well as the aldosterone/renin ratio, rose markedly in the dexamethasone and placebo groups but not in the budesonide group. Moreover, the aldosterone/renin ratio correlated closely with ACE concentration.
Conclusions:
Upon acute high-altitude exposure, budesonide, but not dexamethasone, blunted the response of aldosterone to renin elevation by suppressing angiotensin converting enzyme.
Keywords: Acute mountain sickness, renin–angiotensin–aldosterone system, angiotensin-converting enzyme, budesonide, dexamethasone
Introduction
Acute mountain sickness (AMS) is a common disease affecting lots of people traveling to high altitudes. It can produce extensive discomfort and unpleasant feelings. In some cases, it may even progress to life-threatening high altitude cerebral edema, if not treated appropriately.1,2
In a double-blind randomised controlled trial,3 we demonstrated that inhaled budesonide and oral dexamethasone could reduce the incidence of AMS without serious adverse effects, compared with placebo. Moreover, we found that inhaled budesonide yielded less deterioration of lung function. Despite these findings, the explicit mechanism through which inhaled budesonide prevents AMS remains to be elucidated.
The renin–angiotensin–aldosterone system (RAAS) plays a pivotal role in the development and maintenance of AMS.4 In this system, angiotensin-converting enzyme (ACE) is a key component. ACE converts inactive angiotensin I to active vasopressor angiotensin II, which further stimulates the secretion of aldosterone.5 Previous studies investigating the effects of acute hypoxia on ACE activity have yielded conflicting results. However, reduced transpulmonary angiotensin conversion was consistently reported, regardless of ACE activity.6–9 We speculate that well-balanced responses of angiotensin conversion upon acute hypoxia exposure rather than ACE activity itself are beneficial to the prevention of AMS.
Traditionally, ACE is localized in the luminal surface of vascular endothelial cells and mostly works in the lungs.10 However, there is now a great amount of evidence indicating that angiotensin II can be formed locally in other tissues such as brain, kidney, adrenal gland, and vascular walls, in the presence of circulating ACE.11–13 Since ACE-induced conversion occurs in all of these sites, we infer that peripheral levels of ACE could also exert a regulatory role in the production of angiotensin II and aldosterone, and on the whole RAAS. The classic viewpoint14 is that the peripheral levels of ACE depend on the integrity of the vascular surface in the lungs. An incomplete endothelial cell junction accelerates the drop of ACE molecules into circulation, and increases the ACE levels in the plasma.14 Previously, glucocorticoids have been shown to preserve surface integrity in an earlier study, whereas ACE activity was not assessed.15 We wondered whether inhaled budesonide or oral dexamethasone could cause any change in the peripheral ACE levels by affecting the lung vascular endothelial cells and thereafter affect the risk of AMS in those people rapidly ascending to high altitudes. In this study, we aimed to evaluate the effects of budesonide and dexamethasone on RAAS and their roles in the prevention of AMS.
Materials and methods
Subjects
Data were obtained from a randomised controlled trial,3 in which 138 men rapidly traveled to altitude 3450 m from 400 m by car within two days. The study protocol was approved by our institutional ethics committee. All subjects provided written informed consent. The study was conducted in accordance with the World Medical Association Declaration of Helsinki (trial registration: Chinese Clinical Trial Registry, ChiCTR-PRC-13003296).
The participants were randomly assigned to receive budesonide, dexamethasone, or placebo as prophylaxis before ascent (46 in each group). Medication started one day before high-altitude exposure (>2500 m) and continued until the third day of exposure. During the whole protocol, any other individual medications were not allowed. The subjects were aware of the main side effects of both agents. They were allowed to retreat if there were any adverse reactions.
Data collection
Demographic data were collected during recruitment. Baseline examinations were performed at altitude 400 m.
AMS was diagnosed by the Lake Louise Scoring System (LLS),1 which includes five self-reported symptoms: headache, fatigue/weakness, gastrointestinal symptoms, dizziness/lightheadedness, and difficulty in sleeping. The severity of each symptom is scored on 0–3 points, with 0 indicating none, 1–3 indicating mild, moderate, and severe, respectively. AMS with a score of 3–4 is defined as mild AMS, while severe AMS has a score of ⩾5.
Sample collection
We obtained 10 ml venous blood from each subject at both 400 m and 3450 m. The blood samples were centrifuged at 3000 rpm within 10 min before the plasma was collected. Then the plasma samples were stored in a cryostat filled with liquid nitrogen (N2; –200°C) until analysis. The concentrations of the following substance in the plasma were detected using enzyme-linked immunosorbent assay (ELISA): renin, ACE and aldosterone.
Statistical analyses
Data are expressed as mean±standard deviation (SD). Changes in the detected samples were analyzed within each group by repeated measures analysis of variance (ANOVA). A least significant difference (LSD)-t test was used for intergroup comparison. The relationship between aldosterone/renin ratio and ACE concentration was assessed by Pearson correlation analysis. A two-tailed value of p<0.05 was considered statistically significant. All analyses were performed using SPSS statistical software (version 16.0, SPSS Inc., Chicago, Illinois, USA).
Results
Baseline characteristics
A total of 124 eligible subjects finally completed this study (42, 39, and 43 in the budesonide, dexamethasone, and placebo groups, respectively). At 3450 m, a total of 48 participants were diagnosed with AMS while the other 76 were not. In the AMS group (n=48), 10 received budesonide, 12 dexamethasone and 26 placebo, while in the Non-AMS group (n=76), 32 received budesonide, 27 dexamethasone and 17 placebo. Demographic characteristics, as well as the detected data, were comparable between the three groups (p>0.05).
Associations between AMS and RAAS
We firstly evaluated the differences of renin, ACE and aldosterone between the AMS and non-AMS groups. Upon acute high-altitude exposure, both groups have highly elevated renin (p<0.05); ACE and aldosterone continued to increase in the AMS group (p<0.05) while they increased less in the non-AMS group (Figure 1).
Figure 1.
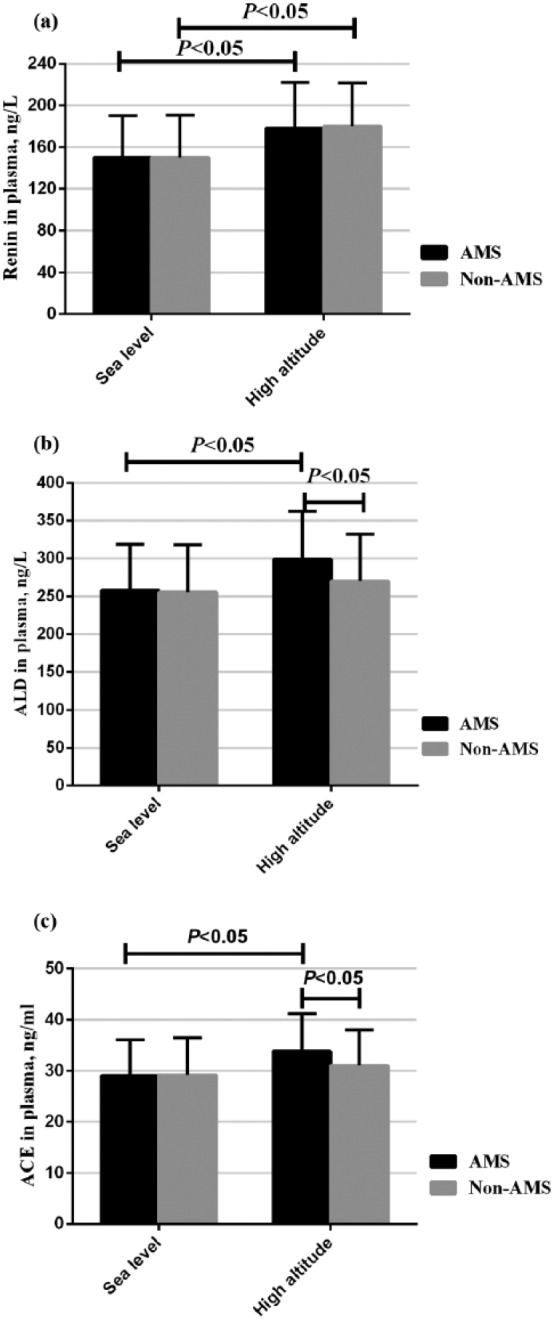
The occurrence of acute mountain sickness (AMS) was associated with an overexpressed renin–angiotensin–aldosterone system (RAAS). ACE: angiotensin-converting enzyme; ALD: aldosterone.
Changes in RAAS levels in AMS group
In the AMS group, the concentrations of plasma renin were comparable among the three groups at baseline (400 m). After acute hypoxia exposure (3450 m), the renin levels were all elevated in the three groups (p<0.05). Differences between groups were not statistically significant at 3450 m (p>0.05) (Figure 2(a)).
Figure 2.
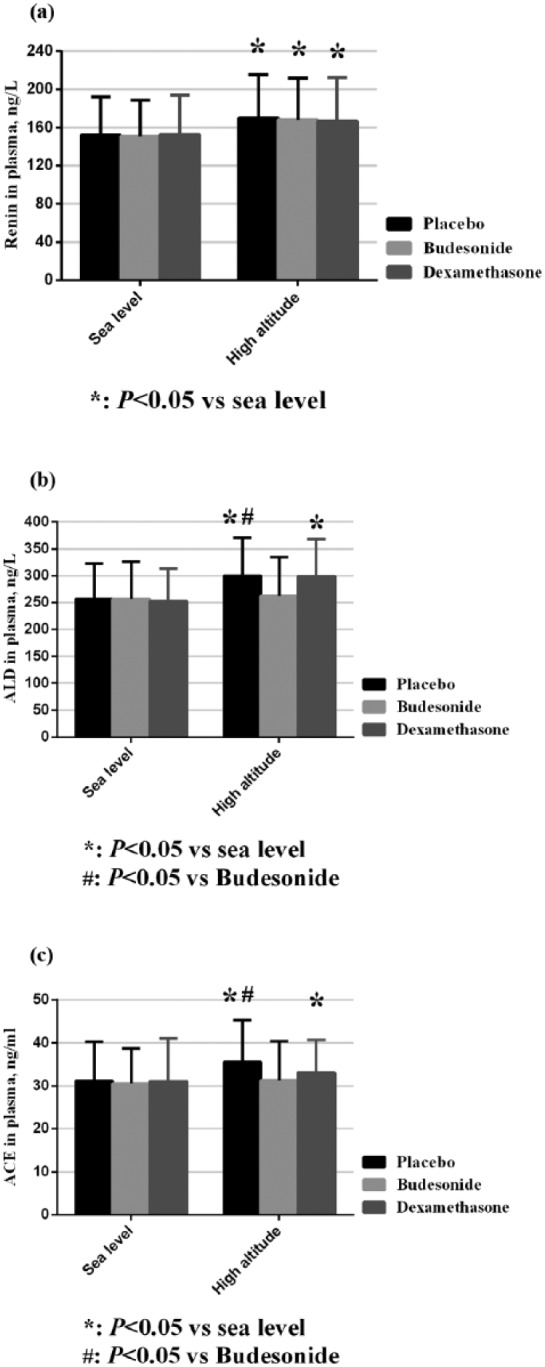
Changes in renin–angiotensin–aldosterone system (RAAS) levels in the acute mountain sickness (AMS) group. ACE: angiotensin-converting enzyme; ALD: aldosterone.
At baseline, the levels of ACE among the three groups were similar (p>0.05). Dexamethasone and placebo groups had markedly increased ACE levels after hypoxia exposure (p<0.05), while the increase in the budesonide group was not statistically significant compared with baseline levels (p>0.05) (Figure 2(b)).
The levels of aldosterone in plasma were also well matched at baseline (p>0.05). Upon acute hypoxia exposure (altitude 3450 m), the concentrations of aldosterone increased markedly (p<0.05) in the dexamethasone and placebo groups, while there was no significant change in the budesonide group (p>0.05). Also, the aldosterone level was much higher in the placebo group than that in the budesonide group (p<0.05) (Figure 2(c)).
Changes in RAAS levels in non-AMS group
In the non-AMS group, renin, ACE, and aldosterone were all comparable at baseline (p>0.05). After ascent to 3450 m, all of the three groups had increased renin levels (p<0.05). However, the ACE and aldosterone concentrations in the budesonide group remained at low levels compared with the other two groups (Figure 3), which was consistent with the levels observed in the AMS group.
Figure 3.
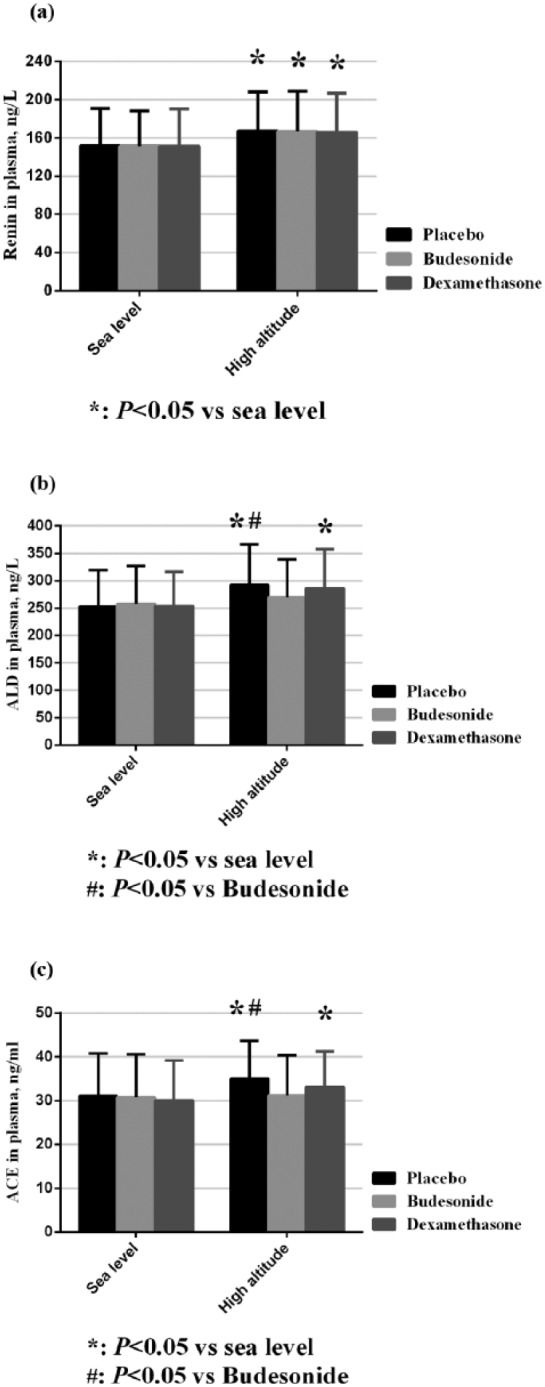
Changes in renin–angiotensin–aldosterone system (RAAS) levels in the non-acute mountain sickness (AMS) group. ACE: angiotensin-converting enzyme; ALD: aldosterone.
Aldosterone response to renin
To investigate the changes of aldosterone response to renin, we also assessed the ratio of aldosterone/renin during the course of high altitude. As shown in Table 1, upon acute high-altitude exposure, the aldosterone/renin ratio remained unchanged in the budesonide group (p>0.05) while it rose significantly in the dexamethasone and placebo groups (p<0.05). In addition, at 3450 m, the aldosterone /renin ratio in the budesonide group was much lower than that in the placebo group (p<0.05).
Table 1.
Ratio of aldosterone/renin in three groups during the course of high altitude exposure.
| Altitude levels | Budesonide | Dexamethasone | Placebo |
|---|---|---|---|
| Altitude 400 m Altitude 3450 m |
1.72±0.42 1.72±0.45 |
1.69±0.49 1.78±0.51a,b |
1.70±0.50 1.82±0.54a,b |
p<0.05 vs altitude 400 m; bp<0.05 vs budesonide group.
Relationship between the aldosterone/renin ratio and ACE concentration
The changes in the aldosterone/renin ratio were consistent with ACE changes from 400 m to 3450 m. Following Pearson correlation analysis, we found that the aldosterone/renin ratio (1.65±0.83) correlated with ACE concentration (32.21±9.49, ng/ml) (r=0.516, p<0.001) (Figure 4).
Figure 4.
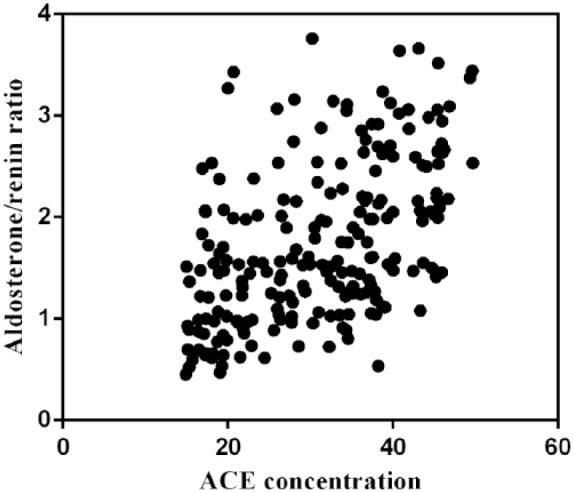
The aldosterone/renin ratio correlated with angiotensin-converting enzyme (ACE) concentration.
Discussion
Inhaled budesonide, mainly used for the treatment of asthma and chronic obstructive pulmonary disease, was proved effective for the prevention of AMS.3,16 In this study, we firstly validated the early opinion that subjects who have overexpressed RAAS are likely to develop AMS.2 Secondly, we investigated the effect of inhaled budesonide, as well as oral dexamethasone, on RAAS expression. More importantly, we demonstrated for the first time that in the setting of acute high-altitude exposure, inhaled budesonide could alleviate the excessive rise of circulating ACE, regardless of AMS status.
The explicit etiology of AMS remains unclear. Neurohumoral regulation upon acute hypoxia contributes greatly. During the present high-altitude exposure (from 400 m to 3450 m), renin in all subjects increased significantly. Plasma renin concentration or activity was found to be elevated in some studies,17,18 but unchanged in some others.19,20 Actually, the response of renin to acute hypoxia depends on the altitude reached and the time spent on high altitude.21 We speculate that the present altitude level or ascent speed probably accounts for this elevation in our study. In addition, hypoxia has a direct inhibitory effect on aldosterone release,22–24 so that ascent to high altitude reduces aldosterone levels.25 However, exercise, always accompanied with mountaineering, rescue work or military task, stimulates renin release, which in turn enhances aldosterone generation via angiotensin pathway.26,27 An inappropriate rise in aldosterone is associated with fluid retention, electrolyte disturbances, and concomitant hypoxemia, which predisposes people to AMS.28 AMS-susceptible subjects were found to have elevated aldosterone levels compared with AMS-resistant ones.27 Upon acute high-altitude exposure, the aldosterone concentration remained at a low level in the budesonide group but continued to increase in the dexamethasone and placebo groups. Similarly, the aldosterone responses to the elevation of renin levels also varied, as reflected by the differences in the aldosterone/renin ratio among the three groups. Compared with the other two groups, the response of aldosterone to renin in the budesonide group was greatly blunted, which means that highly elevated renin resulting from acute hypoxia exposure did not correspondingly lead to high levels of aldosterone. The observed dissociation of aldosterone from renin was in accordance with previous results,17,29–32 and supported the opinion that the ability to dissociate aldosterone in presence of elevated renin is supposed to be protective from AMS.23
ACE, which is localized and generated in the luminal surface of vascular endothelial cells in the lungs, is a potent component of RAAS and plays a critical role in conversion of inactive angiotensin I to vasoactive angiotensin II.5 Effects of acute hypoxia on ACE activity have yielded conflicting results. In some early studies, reduced ACE activity was reported in response to acute hypoxia.17,29,31,33 This reduction was supposed to decrease excessive production of downstream angiotensin II and aldosterone, and thereafter maintain an internal homeostasis. However, subsequent studies found that on some occasions the enzyme was not affected by acute hypoxia in experimental models.7,9,34 The lack of effects of acute hypoxia on ACE activity was further confirmed in human.21,30,35 Despite apparent preservation of pulmonary ACE activity, Oparil et al. demonstrated that the conversion of angiotensin I to angiotensin II was still significantly reduced in hypoxia-exposed lungs.8 This suggested that hypoxia-induced suppression of angiotensin I conversion may be attributed to the endothelial membrane levels that regulate enzyme-substrate interactions rather than altered ACE activity or synthesis. Indeed, reduction in transpulmonary angiotensin conversion was consistently reported regardless of ACE levels.6–9 These observations indicated that some other factors, such as altered contact time between the enzyme and substrate (e.g. as a result of pulmonary hemodynamic changes) could also regulate ACE function and aldosterone levels.8,36
Taken together, upon acute hypoxia exposure, a decline in ACE function (via either suppressed ACE activity or reduced contact time) is supposed to preserve the ability to dissociate aldosterone from renin. On the contrary, overexcited ACE (via activated enzyme or prolonged contact time) would probably increase the risk of AMS by promoting angiotensin conversion and raising the aldosterone level.
In our study, the ACE level was reduced in the budesonide group, but not in the dexamethasone and placebo groups. A possible explanation is that inhaled budesonide, as a glucocorticoid agent, preserved the integrity of pulmonary endothelial cell membrane37 and thereafter prevented the drop of ACE molecules from the lungs to the circulation. As peripheral ACE function has been demonstrated to be an important regulatory step in the control of angiotensin II and aldosterone,11–13 inhaled budesonide may exert beneficial effects by blocking the cascade above. Clinically, long-term use of exogenous glucocorticoid may result in adrenal insufficiency by negative feedback. High-dose inhaled budesonide was not associated with reduced cortisol concentration in plasma.38 As for dexamethasone, the medication duration in this study was strictly restricted to only four days, thus the side-effects were likely to be minimal.
Unlike budesonide, dexamethasone did not exhibit sufficient effects on ACE and aldosterone. One possible explanation for this divergence is that budesonide produced more potent local effects (e.g. preserving the integrity of airway epithelia barrier) in the lungs than dexamethasone. As inhaled budesonide was delivered directly to the site of action, peak concentration of inhaled budesonide could be much higher in the lung than oral dexamethasone. Since dexamethasone was given orally and acted systemically, it might not be abundant enough to exhibit its complete actions in the lungs. On the other hand, glucocorticoids may have intricate impacts on pulmonary physiology, including preserving the integrity of airway epithelial barriers,37 stimulating surfactant secretion,39 and preventing transvascular protein escape.40 Due to the nature of this field study and lack of detailed information, we could not determine to what extent any of these mechanisms were involved in the regulation of pulmonary and systemic angiotensin conversion. Besides, some other factors remain to be uncovered, for instance, the molecular conformation may also affect the interaction between ACE and substrate. Future research is required to better address these issues.
It is also known that the occurrence of AMS is multifactorial. Both blunted ventilation and ACE response play important roles. Another explanation for AMS and its prevention is that there is an association between the ACE level and the ventilation response to hypoxia.28 With our colleagues, we recently found that inhaled budesonide alleviated the impairment of high altitude on forced vital capacity.3 Based on the current findings in this study, we consider that the observed protective effect on lung function may be related to anti-inflammatory impacts by suppressed ACE. Angiotensin II is generated by the action of ACE and present in high concentrations in lung tissues. Angiotensin II, known primarily as a potent vasopressor, is also a pro-inflammatory molecule.41 Its pro-inflammatory profile may recruit some characteristic inflammatory and immune cells into the lungs and thereby contribute to lung function decline, especially in those individuals exposed to hypoxia.42 Blocking ACE-induced angiotensin II effects may attenuate the rapid decline of lung function.
Our study has several limitations. Firstly, our subjects were all healthy young men, so that it is uncertain whether our results would be applicable to a broader population. Secondly, the Renin and ACE activity may be more accurate but absent here, i.e., these could be measured in other ways, such as high performance liquid chromatography (HPLC), but these methods require strict conditions. It is impossible to conduct these tests due to the field-study nature, so we did not obtain the data here. Their concentrations in plasma may give an alternative but less specific index of RAAS expression during high-altitude exposure. Besides, what we detected was peripheral ACE concentration and we do not think it valid to use the plasma concentration of ACE to reflect its pulmonary level. Since pulmonary angiotensin conversion constitutes a great part in overall ACE function, whether inhaled budesonide has any effects on regional ACE activity, conformation, or contact with angiotensin I is to be explored.
Conclusion
In this trial, we demonstrated for the first time that in the setting of acute high-altitude exposure, budesonide, but not dexamethasone, blunted the response of aldosterone to renin elevation by suppressing ACE. These effects were expected to be beneficial to the prevention of AMS. Our findings help understand the mechanisms through which budesonide and dexamethasone prevent AMS, as well as the difference in their contributions to AMS incidence reduction. Also, our data indicate that aldosterone-targeted approach may represent a new direction for AMS management.
Acknowledgments
The authors wish to thank all of the individuals who participated in this study. They also express sincere gratitude to Xu-Bin Gao, Shi-Zhu Bian, Can Chen, Xu-Gang Tang, Lian-You Wang, Li Guo, Jie Yang, Xiao Liang, and You-Zhu Qiu for data collection and valuable discussion.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article :This work was supported by the Special Health Research Project, Ministry of Health of P.R. China (grant number 201002012); and the Military Special Project (grant number BWS14J040).
References
- 1. Luks AM, McIntosh SE, Grissom CK, et al. Wilderness Medical Society practice guidelines for the prevention and treatment of acute altitude illness: 2014 Update. Wilderness Environ Med 2014; 25: S4–S14. [DOI] [PubMed] [Google Scholar]
- 2. Bartsch P, Swenson ER. Acute high-altitude illnesses. N Engl J Med 2013; 369: 1666–1667. [DOI] [PubMed] [Google Scholar]
- 3. Zheng CR, Chen GZ, Yu J, et al. Inhaled budesonide and oral dexamethasone prevent acute mountain sickness. Am J Med 2014; 127: 1001–1009. [DOI] [PubMed] [Google Scholar]
- 4. Goldfarb-Rumyantzev AS, Alper SL. Short-term responses of the kidney to high altitude in mountain climbers. Nephrol Dial Transplant 2014; 29: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ucar G, Yildirim Z, Ataol E, et al. Serum angiotensin converting enzyme activity in pulmonary diseases: Correlation with lung function parameters. Life Sci 1997; 61: 1075–1082. [DOI] [PubMed] [Google Scholar]
- 6. Peacock AJ, Matthews A. Transpulmonary angiotensin II formation and pulmonary haemodynamics in stable hypoxic lung disease: The effect of captopril. Respir Med 1992; 86: 21–26. [DOI] [PubMed] [Google Scholar]
- 7. Jackson RM, Narkates AJ, Oparil S. Impaired pulmonary conversion of angiotensin I to angiotensin II in rats exposed to chronic hypoxia. J Appl Physiol (1985) 1986; 60: 1121–1127. [DOI] [PubMed] [Google Scholar]
- 8. Oparil S, Winternitz S, Gould V, et al. Effect of hypoxia on the conversion of angiotensin I to II in the isolated perfused rat lung. Biochem Pharmacol 1982; 31: 1375–1379. [DOI] [PubMed] [Google Scholar]
- 9. Szidon P, Oparil S, Osikowicz G, et al. Effect of hypoxia on the conversion of angiotensin I to II in cultured porcine pulmonary endothelial cells. Biochem Pharmacol 1983; 32: 1201–1205. [DOI] [PubMed] [Google Scholar]
- 10. Caldwell PR, Seegal KC, Hsu M, et al. Angiotensin-converting enzyme: Vascular endothelial localization. Science (Wash., DC) 1976; 191: 1050–1051. [DOI] [PubMed] [Google Scholar]
- 11. Oshima G, Gecse A, Erdös EG. Angiotensin I-converting enzyme of the kidney cortex. Biochim Biophys Acta 1974; 350: 26–37. [DOI] [PubMed] [Google Scholar]
- 12. Yang H, Neff NH. Distribution and properties of angiotensin converting enzyme of rat brain. J Neurochem 1972; 19: 2443–2450. [DOI] [PubMed] [Google Scholar]
- 13. Ward PE, Sheridan MA, Hammon KJ, et al. Angiotensin I converting enzyme (kininaseII) of the brush border of human and swine intestine. Biochem Pharmacol 1980; 29: 1525–1529. [DOI] [PubMed] [Google Scholar]
- 14. Orfanos SE, Kotanidou A, Roussos C. Pulmonary endothelial angiotensin converting enzyme in lung injury. In: Vincent JL. (ed.) Yearbook of intensive care and emergency medicine 2002. 1st ed Berlin: Springer, 2002, pp. 100–110. [Google Scholar]
- 15. Maca RD, Fry GL, Hoak JC. The effects of glucocorticoids on cultured human endothelial cells. Br J Haematol 1978; 38: 501–509. [DOI] [PubMed] [Google Scholar]
- 16. Chen GZ, Zheng CR, Qin J, et al. Inhaled budesonide prevents acute mountain sickness in young Chinese men. J Emerg Med 2015; 48: 197–206. [DOI] [PubMed] [Google Scholar]
- 17. Milledge JS, Catley DM, Ward MP, et al. Renin-aldosterone and angiotensin-converting enzyme during prolonged altitude exposure. J Appl Physiol Respir Environ Exerc Physiol 1983; 55: 699–702. [DOI] [PubMed] [Google Scholar]
- 18. Frayser R, Rennie ID, Gray GW, et al. Hormonal and electrolyte response to exposure to 17,500 ft. J Appl Physiol 1975; 38: 636–642. [DOI] [PubMed] [Google Scholar]
- 19. Hogan RP, 3rd, Kotchen TA, Boyd AE, 3rd, et al. Effect of altitude on renin-aldosterone system and metabolism of water and electrolytes. J Appl Physiol 1973; 35: 385–390. [DOI] [PubMed] [Google Scholar]
- 20. Keynes RJ, Smith GW, Slater JD, et al. Renin and aldosterone at high altitude in man. J Endocrinol 1982; 92: 131–140. [DOI] [PubMed] [Google Scholar]
- 21. Millar EA, Angus RM, Nally JE, et al. Effect of hypoxia and beta 2-agonists on the activity of the renin–angiotensin system in normal subjects. Clin Sci (Lond) 1995; 89: 273–276. [DOI] [PubMed] [Google Scholar]
- 22. Raff H, Ball DL, Goodfriend TL. Low oxygen selectively inhibits aldosterone secretion from bovine adrenocortical cells in vitro. Am J Physiol 1989; 256: E640–E644. [DOI] [PubMed] [Google Scholar]
- 23. Raff H, Jankowski BM, Engeland WC, et al. Hypoxia in vivo inhibits aldosterone synthesis and aldosterone synthase mRNA in rats. J Appl Physiol (1985) 1996; 81: 604–610. [DOI] [PubMed] [Google Scholar]
- 24. Zaccaria M, Rocco S, Noventa D, et al. Sodium regulating hormones at high altitude: Basal and post-exercise levels. J Clin Endocrinol Metab 1998; 83: 570–574. [DOI] [PubMed] [Google Scholar]
- 25. Sutton JR, Viol GW, Gray GW, et al. Renin, aldosterone, electrolyte, and cortisol responses to hypoxic decompression. J Appl Physiol Respir Environ Exerc Physiol 1977; 43: 421–424. [DOI] [PubMed] [Google Scholar]
- 26. Maher JT, Jones LG, Hartley LH, et al. Aldosterone dynamics during graded exercise at sea level and high altitude. J Appl Physiol 1975; 39: 18–22. [DOI] [PubMed] [Google Scholar]
- 27. Bärtsch P, Maggiorini M, Schobersberger W, et al. Enhanced exercise-induced rise of aldosterone and vasopressin preceding mountain sickness. J Appl Physiol (1985) 1991; 71: 136–143. [DOI] [PubMed] [Google Scholar]
- 28. Loeppky JA, Icenogle MV, Maes D, et al. Early fluid retention and severe acute mountain sickness. J Appl Physiol (1985) 2005; 98: 591–597. [DOI] [PubMed] [Google Scholar]
- 29. Milledge JS, Catley DM. Renin, aldosterone, and converting enzyme during exercise and acute hypoxia in humans. J Appl Physiol Respir Environ Exerc Physiol 1982; 52: 320–323. [DOI] [PubMed] [Google Scholar]
- 30. Colice GL, Ramirez G. Effect of hypoxemia on the renin–angiotensin–aldosterone system in humans. J Appl Physiol (1985) 1985; 58: 724–730. [DOI] [PubMed] [Google Scholar]
- 31. Milledge JS, Catley DM, Williams ES, et al. Effect of prolonged exercise at altitude on the renin-aldosterone system. J Appl Physiol Respir Environ Exerc Physiol 1983; 55: 413–418. [DOI] [PubMed] [Google Scholar]
- 32. Bouissou P, Guezennec CY, Galen FX, et al. Dissociated response of aldosterone from plasma renin activity during prolonged exercise under hypoxia. Horm Metab Res 1988; 20: 517–521. [DOI] [PubMed] [Google Scholar]
- 33. Jin H, Oparil S, Ann HS, et al. Hypoxia-induced inhibition of converting enzyme activity: Role in vascular regulation. J Appl Physiol (1985) 1987; 63: 1012–1018. [DOI] [PubMed] [Google Scholar]
- 34. Brecher P, Tercyak A, Chobanian AV. Properties of angiotensin-converting enzyme in intact cerebral microvessels. Hypertension 1981; 3: 198–204. [DOI] [PubMed] [Google Scholar]
- 35. Milledge JS, Catley DM. Angiotensin converting enzyme activity and hypoxia. Clin Sci (Lond) 1987; 72: 149. [DOI] [PubMed] [Google Scholar]
- 36. Catravas JD, Gillis CN. Metabolism of [3H]benzoyl-phenylalanyl-alanyl-proline by pulmonary angiotensin converting enzyme in vivo: Effects of bradykinin, SQ 14225 or acute hypoxia. J Pharmacol Exp Ther 1981; 217: 263–270. [PubMed] [Google Scholar]
- 37. Sekiyama A, Gon Y, Terakado M, et al. Glucocorticoids enhance airway epithelial barrier integrity. Int Immunopharmacol 2012; 12: 350–357. [DOI] [PubMed] [Google Scholar]
- 38. Volovitz B, Kauschansky A, Nussinovitch M, et al. Normal diurnal variation in serum cortisol concentration in asthmatic children treated with inhaled budesonide. J Allergy Clin Immunol 1995; 96: 874–878. [DOI] [PubMed] [Google Scholar]
- 39. Young SL, Ho YS, Silbajoris RA. Surfactant apoprotein in adult rat lung compartments is increased by dexamethasone. Am J Physiol 1991; 260: L161–L167. [DOI] [PubMed] [Google Scholar]
- 40. Stelzner TJ, O’Brien RF, Sato K, et al. Hypoxia-induced increases in pulmonary transvascular protein escape in rats. Modulation by glucocorticoids. J Clin Invest 1988; 82: 1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamazato Y, Ferreira AJ, Hong KH, et al. Prevention of pulmonary hypertension by Angiotensin-converting enzyme 2 gene transfer. Hypertension 2009; 54: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petersen H, Sood A, Meek PM, et al. Rapid lung function decline in smokers is a risk factor for COPD and is attenuated by angiotensin-converting enzyme inhibitor use. Chest 2014; 145: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]