

Abstract
Introduction:
The study aims to confirm the association of acute myocardial infarction (AMI) with serum angiotensin II (AngII), kallikrein1 (KLK1), and ACE/KLK1 polymorphisms.
Materials and methods:
Serum AngII/KLK1 levels and ACE and KLK1 genotypes were determined in 208 patients with AMI and 216 normal controls. Binary logistic regression was used for data analysis.
Results:
The differences in serum AngII levels were statistically significant between the groups. After adjusting for potential confounding factors, high serum levels of AngII and KLK1 significantly increased the risk of AMI. The individuals with ACE DD and KLK1 GG genotypes significantly increased the risk of AMI compared with those harboring the ACE II and KLK1 AA genotypes (OR = 8.77, 95% CI = 1.74–44.16).
Conclusions:
(1) Increasing the serum levels of AngII increased the risk of AMI. (2) The risk of AMI increased significantly when the serum levels of AngII and KLK1 simultaneously increased. (3) Individuals with the combined genotypes of ACE DD and KLK1 GG showed significantly increased risk of AMI compared with those with the combined genotypes of ACE II and KLK1 AA.
Keywords: Coronary artery stenosis, acute myocardial infarction, angiotensin converting enzyme gene, kallikrein1 gene, polymorphism
Introduction
Acute myocardial infarction (AMI) is induced by coronary artery stenosis (CAS) and is one of the most severe types of coronary artery disease (CAD). Clinical studies showed that the serum levels of angiotensin II (AngII) in patients with AMI are higher than those of healthy people.1 Human tissue kallikrein 1 (KLK1) is assumed to be a protective factor for cardiovascular diseases,2 but significantly high levels of KLK1 have been detected in coronary artery atherosclerotic plaques.3 Genetic studies found that renin catalyzes angiotensinogen to angiotensin I (AngI), which is translated into a bioactive octapeptide (AngII) by the action of angiotensin I-converting enzyme (ACE) or other enzymes.4 ACE is a protein expressed by the ACE gene. Previous studies confirmed the association of the polymorphism of ACE with the occurrence of AMI, where the ACE DD genotype increased the risk of AMI.5,6 By contrast, a meta-analysis of abdominal aortic aneurysm revealed that the disease was associated with multiple susceptibility genes but weakly associated with the ACE DD gene (rs4646994) (OR = 1.67, 95%CI 1.09–1.67).7 Another meta-analysis showed that the ACE DD genotype increased the risk of percutaneous transluminal coronary angioplasties-stent (PTCA-stent) among Asians (OR = 2.18, 95%CI 1.08–4.40, p = 0.03).8
Several KLK1 gene polymorphisms have been identified, and a few genetic variants may be associated with KLK1 activity.9 Our previous study showed that the KLK1 AA genotype was negatively correlated with cardiovascular diseases in the Chinese Han population.10
In summary, the ACE and KLK1 genes play an important role in maintaining normal physiological activities and occurrence of cardiovascular diseases. To the best of our knowledge, no studies have reported the association between serum levels of AngII and KLK1 or the polymorphisms of the combined ACE and KLK1 genotypes with AMI caused by CAS. In the present work, we designed a case-control study to prove the association between the serum levels of AngII and KLK1, and the association of ACE and KLK1 polymorphisms as well as their combination effect on AMI.
Materials and methods
Subjects
This study recruited 208 cases with AMI and 216 healthy controls without CAS from January 2012 to January 2015 in the Qilu Hospital of Shandong University. AMI was diagnosed based on the World Health Organization criteria, i.e. patients should have any two of the following three conditions: resting anginal chest pain lasting more than 30 min and/or electrocardiogram changes (e.g. ST-segment elevation ⩾1 mm in two contiguous leads (or ⩾2 mm in V1 to V3 leads)); serum levels of troponin and/or creatine kinase elevation to at least twice the upper limit of the normal range.11 In addition, cases were within 24 hours of AMI, and were identified with AMI by coronary angiography (CAG) (defined by 100% stenosis (an acute occlusion) in any major coronary artery). The exclusion criteria were as follows: clinical evidence of acute inflammation, tumor, rheumatic condition and acute renal failure. CAG were evaluated by experienced researchers blinded to the study plan.
Control subjects (N = 216) were selected based on the following criteria: negative CAG examination results, matched by gender and hospital, and no abnormalities found on electrocardiogram or cardiac ultrasound examinations. In the CAG examination, the patients were found to have normal coronary artery and were eventually diagnosed with non-coronary atherosclerotic diseases, including hypertension, cardiac hypertrophy, and psychological anxiety or depression (neurosis). The research protocol was approved by the Ethics Committee of Qilu Hospital of Shandong University, and patient consent was obtained prior to the study.
Sample size was calculated using Quanto 4.0 software. The following parameters were used to design a case-control study. One exposure factor was the ACE DD gene polymorphism, which amounted to 10%; another exposure factor was the KLK1 GG gene polymorphism, which accounted for 30%; a marginal risk effect (OR = 2.5 and moderate risk effect OR = 1.5; p-value (α) = 0.05, posterior power (1-β) = 0.8). The sample size of cases or controls was 196.
A detailed personal history of each patient was obtained using demographic data and traditional coronary risk factors (the presence of hypertension, diabetes mellitus, obesity, smoking, and alcohol intake), which were collected in a questionnaire following a standard procedure of our research group. Hypertension is defined as a condition where blood pressure exceeds 140/90 mmHg or person with high blood pressure is treated. Diabetes mellitus is defined as a condition where fasting blood glucose exceeds 126 mg/dl on two occasions or being treated. Smoking is defined as daily smoking habit that continues for more than 1 year, and drinking is defined as the daily habit of drinking at least 50 g of liquor for more than 1 year in a row.
CAG examination
A percutaneous CAG test was performed for all subjects with the puncturing path at radial artery (contrast agents: iodine amine (370); Imaging instrument model, Allura FD20/20, the Netherlands).
Measurement of serum levels of KLK1 and AngII
Serum levels of KLK1 and AngII were measured using enzyme-linked immunosorbent assay (ELISA) through a competitive enzyme immunoassay technique.
Determination of serum levels of KLK1 through ELISA
Briefly, 96-well plates were coated in advance with anti-human KLK1 IgG. To each well was added serum samples and horseradish peroxidase (HRP) antigen. The samples were subjected to rocking, covered with sealing film, and incubated at 37°C for 1 h. The samples were repeatedly washed with detergent (PBST) five times (antigen without combination washed off). To each well was then added TMB (A, B) substrate solutions and incubated at 37°C for 10 min without light. The reaction was terminated by adding sulfuric acid solution after 10 min. Optical density values were tested at 450 nm in a Bio-Rad ELISA plate reader (Infinite M200Pro, TECAN). Serum KLK1 levels were determined according to the calibrated standard curve.
Determination of serum levels of AngII through ELISA
Briefly, 96-well plates (Corning) were coated in advance with anti-human AngII IgG. To each well was added serum samples and HRP antigen. The rest of the steps were similar to those of the serum KLK1 assays using ELISA kits (Shanghai Lengton Bioscience Co., Ltd.; LOT: 201500506MY). The performance parameters included coefficient of variation of within batch <3%, coefficient of variation between batch <5%, and sensitivity <0.5 ng/ml.
Genetic analysis
DNA was isolated using the human genome extraction kit (TIANGEN Biotechnology Co., Ltd. Shanghai Office).
Genotyping the ACE (Insertion/Deletion I/D) gene
The genotype of ACE (I/D) was tested by polymerase chain reaction (PCR). The primers were as follows: the upstream primer for 5’- CTG GAG ACC ACT CCC ATC CTT CT - 3’ and the downstream primers for 5’- GAT GTG GCC ATC ACA TTC GTC AGA T - 3’; DNA was amplified for 30 cycles, with predegeneration at 94°C, 30 s degeneration at 94°C, 30 s annealing at 55°C and 10 min extension at 72°C. DNA fragments were separated by 1.5% agarose gel electrophoresis and identified by GelRed nucleic acid dye. Three types of ACE gene PCR products were identified: a 490-bp band indicating the II genotype, a 190-bp band indicating the DD genotype, or a combination of 490-bp and 190-bp bands indicating I/D genotype (Figure 1).
Figure 1.
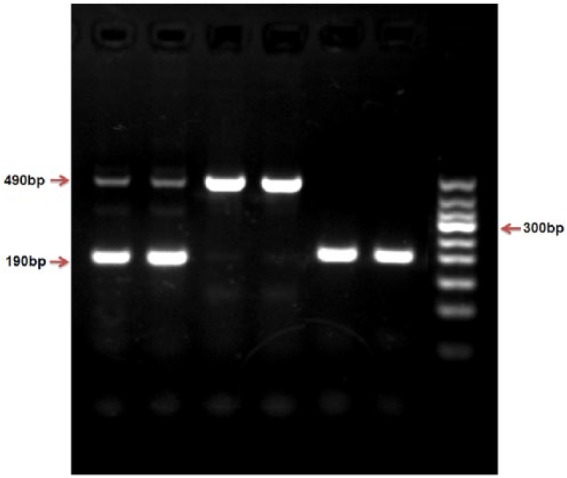
Genotyping of the ACE insertion/deletion (I/D) detected by polymerase chain reaction (PCR).
A combination of 490-bp and 190-bp bands indicate the I/D genotype, a 490-bp band indicates the II genotype, a 190-bp band indicates the DD genotype (Figure 1).
To avoiding overestimation of the ACE DD genotype, all DD genotype samples were confirmed using another specific primer pair for ACE I allele: forward, 5’ - TCG GAC CAC AGC GCC CGC CAC TAC - 3’; and reverse, 5’ - TCG CCA GCC CTC CCA TGC CCA TAA -3’, that produced a 335 bp product only in the presence of the I allele.
Genotyping the KLK1 A1789G (rs5517) gene
The genotype of the KLK1 A1789G (rs5517) gene was tested by PCR-TaqMan-MGB probe. The primers and probes using FAM and VIC fluorescent tagging two report groups were as follows: TaqMan probe using FAM and VIC fluorescent tags two report groups, positive primer: 5’- CAC AGG TGT CTT the TGC CAC CTT - 3’, reverse primer: 5’- CTC CCG GGT TCG TAG TCT CAT - 3’. VIC - 5’- TTT TTC GCA CTC ATC - 3’ - MGB; FAM - 5’- CTT TTTTGC ACT CAT C - 3’ – MGB (all primers, probe and PCR reaction reagent provided by ABI Applied Biosystems by Life Technologies. Assay ID: C - 2932115-20). Dd H2O instead of DNA was used as blank control. The PCR products were determined using Stepone plus Real Time PCR System (Biosystems StepOnePlusTM, USA). A 10% random sample of all samples was repeat tested as quality control. Two kinds of report group were tagged separately by fluorescent FAM and VIC; detecting the fluorescence intensity of FAM or VIC, corresponding to different alleles, enabled us to judge the sample as homozygous genotype of wild type (AA), homozygous mutant (GG) or heterozygote (AG) (Figure 2).
Figure 2.
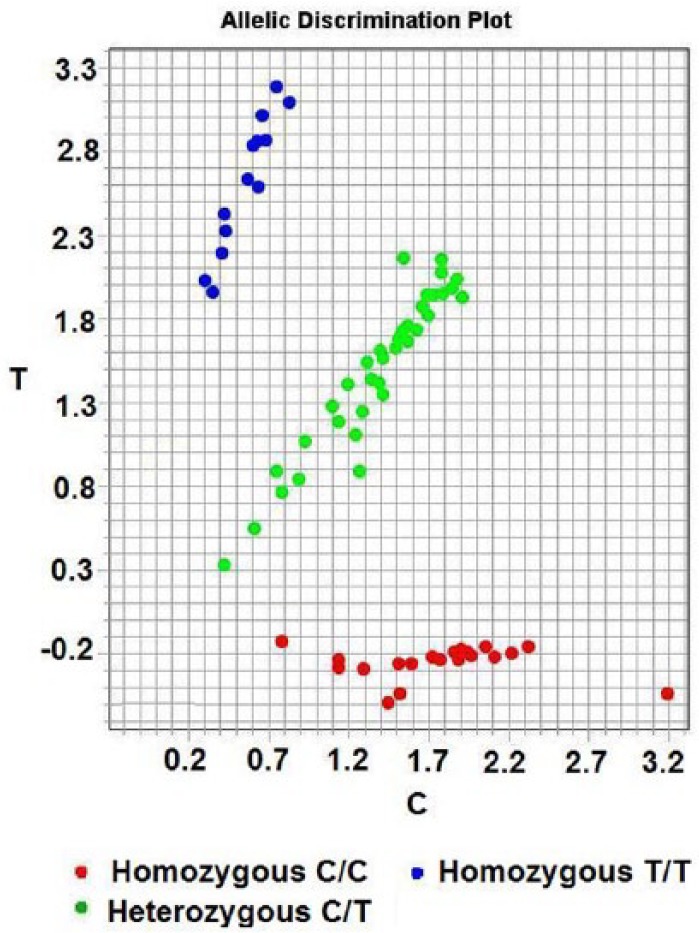
Genotyping of the KLK1 (A/G) detected by PCR-TaqMan-MGB probe SNP genotyping technology.
Notes: Red, blue and green circles correspond to homozygous mutant GG, homozygous genotype AA and heterozygote AG, respectively. (A and T, C and G are respectively complementary base).
Statistical methods
Mean value ± standard deviation (SD) was used to describe continuous variables with normal distribution, and percentages were used to present categorical variables. Continuous variables that conform to normal distribution were compared using Student’s t-test, whereas categorical data were compared with chi-square test. Hardy–Weinberg equilibrium (HWE) for genotype distribution was performed using chi-square test (p < 0.05). Binary and multinomial logistic regression was performed to adjust the variables that significantly differed in the univariate analysis (p < 0.05). The strength of the association of the serum levels of AngII and KLK1 as well as genetic risk factors with the occurrence of AMI were estimated as ORs with 95% confidence intervals (CI). The two-sided p-value of < 0.05 was considered to be statistically significant. All computations were performed using SPSS version 19.0 (SPSS Co. Chicago, IL, USA) for Windows.
Results
General characteristics of cases and controls
The characteristics of the two groups are shown in Table 1. The univariate analysis showed the continuous variables of ALT, GGT, TP, ALB, GLB, DBIL, AST, TBA, CHO, HDL, GLU, CR, and TT significantly differed between the two groups. Qualitative variables of gender, age, smoking, and diabetes (p < 0.05) also significantly differed between the groups. The other variables were not significantly different between the two groups.
Table 1.
The baseline characteristics of the cases and control groups.
| Variables | Control (N=216) | Cases (N=208) | t/x2 | p |
|---|---|---|---|---|
| Gender(male/female, N) | 120/96 | 148/60 | 11.086+ | 0.001 |
| Age (±S, years) | 58.53 ± 7.00 | 60.63 ± 13.53 | −1.998 | 0.047 |
| BMI | 25.69 ± 3.95 | 26.35 ± 4.01 | −1.691 | 0.092 |
| Smoking (No/Yes, N) | 156/60 | 88/116 | 36.456+ | <0.001* |
| Drinking (No/Yes, N) | 164/52 | 144/64 | 2.390+ | 0.122 |
| Hypertention (No/Yes, N) | 132/84 | 108/100 | 3.642+ | 0.056 |
| Diabetes mellitus (No/Yes, N) | 204/12 | 168/40 | 18.416+ | <0.001* |
| ALT (U/l) | 22.76 ± 10.30 | 46.15 ± 23.30 | −13.283 | <0.001* |
| GGT (U/l) | 30.02 ± 22.39 | 35.29 ± 30.29 | −2.031 | 0.043 |
| AKP (u/l) | 72.89 ± 21.81 | 70.54 ± 20.22 | 1.150 | 0.251 |
| TP (g/l) | 68.78 ± 8.77 | 61.25 ± 4.67 | 11.090 | <0.001* |
| ALB (g/l) | 42.80 ± 3.74 | 39.75 ± 14.87 | 2.876 | 0.004 |
| GLB (g/l) | 26.79 ± 4.08 | 23.09 ± 3.25 | 10.365 | <0.001* |
| IBIL (uM) | 9.70 ± 6.32 | 9.93 ± 4.89 | −0.409 | 0.683 |
| TBIL (uM) | 13.89 ± 7.91 | 14.84 ± 6.04 | −1.391 | 0.165 |
| DBIL (uM) | 4.44 ± 1.61 | 5.40 ± 2.94 | −4.135 | <0.001* |
| AST (u/l) | 22.11 ± 10.65 | 188.92 ± 147.44 | −16.276 | <0.001* |
| A/G | 1.66 ± 0.29 | 1.67 ± 0.28 | −0.516 | 0.606 |
| TBA (uM) | 4.88 ± 3.91 | 3.77 ± 3.78 | 2.954 | 0.003 |
| CHO (mM) | 4.19 ± 0.93 | 4.59 ± 0.98 | −4.261 | <0.001* |
| TGs (mM) | 1.52 ± 0.80 | 1.60 ± 1.17 | −0.854 | 0.394 |
| HDL-c (mM) | 1.324 ± 0.41 | 1.17 ± 0.24 | 4.633 | <0.001 |
| LDL-c (mM) | 3.33 ± 4.25 | 2.90 ± 0.74 | 1.466 | 0.144 |
| GLU (mM) | 5.36 ± 1.03 | 6.34 ± 2.03 | −6.266 | <0.001* |
| BUN (mM) | 4.92 ± 2.19 | 4.99 ± 1.63 | −0.355 | 0.723 |
| CR (uM) | 66.83 ± 15.54 | 78.60 ± 27.96 | −5.327 | <0.001* |
| UA (uM) | 308.46 ± 75.28 | 292.48 ± 107.77 | 1.764 | 0.079 |
| PT (s) | 11.41 ± 4.56 | 11.82 ± 4.05 | −0.962 | 0.336 |
| FIB (g/l) | 3.23 ± 0.73 | 3.23 ± 0.72 | 0.066 | 0.947 |
| TT (s) | 16.58 ± 2.32 | 15.40 ± 3.54 | 4.116 | <0.001* |
BMI: body mass index; ALT: alanine aminotransferase; GGT: gamma-glutamyltranspeptidase; AKP: alkaline phosphatase; TP: total protein; ALB: albumin; GLB: globulin; IBIL: indirect bilirubin; DBIL: detail bilirubin; TBIL: total bilirubin; AST: aspertate aminotransferase; A/G: albumin/globulin; TBA: total bile acid; CHO: cholesterol; TGs: triglycerides; HDL-c: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; GLU: glucose; Cr: creatinine; UA, uric acid; PT: prothrombin; FIB: fibrinogen; TT: thrombin time; x2: chi-square test; t: 2 independent sample Student t-test for quantitative variables.
TP, ALB, and GLB were closely associated; similarly, ALT was closely related to AST. Logistic regression analysis indicated that of the above-mentioned variables, 14 variables including gender, age, smoking, diabetes, ALT, GGT, TP, DBIL, TBA, CHO, HDL, GLU, CR and TT were statistically significant. Thus, these 14 variables were potential confounders to be adjusted in the logistic analysis.
Serum levels of AngII and KLK1
Table 2 shows the serum levels of AngII and KLK1 in the two groups. As predicted, serum AngII levels were significantly increased compared with the control group, whereas serum KLK1 level was not significantly different between the two groups. For further statistical analysis, the above serum indicators were divided into low and high classification variables according to the mean of the control group. The two groups significantly differed.
Table 2.
The serum levels of AngII and KLK1 in the case and control groups.
| Variables | Controls | Cases | t/x2 | p |
|---|---|---|---|---|
| AngII (ng/l) | 119.83 ± 52.80 | 185.04 ± 61.55 | −11.69 | <0.001 |
| KLK1 (ng/ml) | 21.68 ± 13.64 | 22.63 ± 8.69 | −0.853 | 0.394 |
| AngII ≦120 | 122 (56.5%) | 42 (20.2%) | 58.833 | <0.001 |
| >120 | 94 (43.5%) | 166 (79.8%) | ||
| KLK1 ≦22 | 146 (67.6%) | 92 (44.2%) | 23.486 | <0.001 |
| >22 | 70 (32.4%) | 116 (55.8%) | ||
| AngII & KLK1 | 71.724 | <0.001 | ||
| ≦120&≦22 | 92 (42.6%) | 20 (9.6%) | ||
| ≦120&>22 | 30 (13.9%) | 22 (10.6%) | ||
| >120&≦22 | 54 (25.0%) | 72 (34.6%) | ||
| >120&>22 | 40 (18.5%) | 94 (45.2%) |
AngII: angiotensin II; KLK1: tissue kallikrein; AngII&KLK1: Angiotensin II and tissue kallikrein.
Table 2 shows the four groupings of serum AngII level combined with serum KLK1 level (AngII ≦120 & KLK1 ≦22, AngII ≦120 & KLK1 >22, AngII >120 & KLK1 ≦22, and AngII >120 & KLK1 >22). The percentages of the four groupings significantly differed between the two groups.
Genotyping ACE and KLK1
The frequencies of the ACE and KLK1 genotypes in the control group were compatible with HWE assessed by chi-square test.
As shown in Table 3, the frequencies of the ACE DD, ID, and II genotypes in the case group were 23.1%, 44.2%, and 32.7%, respectively; whereas those in the control group were 9.3%, 50.0%, and 40.7%, respectively. The frequencies of the KLK1 GG, AG, and AA genotypes in the case group amounted to 40.4%, 50.0%, and 9.6%, respectively; whereas those in the control group amounted to 32.4%, 46.3%, and 21.6%, respectively. For the combined genotype of ACE and KLK1 analysis, 13.5% of the cases possessed ACE DD and KLK1 GG genotypes, whereas only 4.6% possessed these genotypes in the controls. The two groups were significantly different.
Table 3.
The frequency distribution of the ACE and KLK1 genotypes in the cases and controls.
| Variables | Control | Case | x2 | p |
|---|---|---|---|---|
| ACE(I/D)genotype | 15.228 | <0.001 | ||
| II | 88 (40.7%) | 68 (32.7%) | ||
| ID | 108 (50.0%) | 92 (44.2%) | ||
| DD | 20 (9.3%) | 48 (23.1%) | ||
| KLK1(A/G)genotype | 11.447 | 0.003 | ||
| AA | 46 (21.6%) | 20 (9.6%) | ||
| AG | 100 (46.3%) | 104 (50.0%) | ||
| GG | 70 (32.4%) | 84 (40.4%) | ||
| ACE&KLK1 genotypes | 10.026 | 0.006 | ||
| ACE II&KLK1 AA | 20 (9.3%) | 16 (7.7%) | ||
| ACE ID&KLK1 AA/AG/GG | 186 (86.1%) | 164 (78.8%) | ||
| ACE DD&KLK1 GG | 10 (4.6%) | 28 (13.5%) |
x2: chi-square test.
Association of serum levels of AngII, KLK1, and polymorphisms of ACE and KLK1 genotypes with AMI
In the logistic regression model, the dependent variables were (Y) = 1 for case and Y = 0 for control. The independent variables included seven variables (X1 to X14) such as X1 = gender (male = 1, female = 2), X2 = age, X3 = smoking (no = 0, yes = 1), and X4 = history of diabetes (no = 0, yes = 1). X5 to X14 represent ALT, GGT, TP, DBIL, TBA, CHO, HDL, GLU, CR and TT (which are possible confounding factors). Two adjusted logistic regression models were used in the analysis. The first model was adjusted for X1 to X4 variables, and the second model adjusted for X1 to X14 variables.
As shown in Table 4, increasing serum levels of AngII or KLK1 increased the risk for AMI. The OR and 95%CI values were 4.96 (3.08–7.98) and 3.20 (2.04–5.01) in model 1, respectively, and 7.19 (3.20–16.32) and 2.82 (1.36–5.84), respectively, in model 2.
Table 4.
The association between the serum levels of AngII/KLK1 and the genotypes of ACE/KLK1 with AMI.
| Variables | AOR(95%CI): Model 1 | AOR(95%CI): Model 2 |
|---|---|---|
| AngII ≦120 | 1.00 | 1.00 |
| >120 | 4.96 (3.08–7.98) | 7.19 (3.20–16.32) |
| KLK1 ≦22 | 1.0 | 1.0 |
| >22 | 3.20 (2.04–5.01) | 2.82 (1.36–5.84) |
| ACE genotype | ||
| II | 1.00 | 1.00 |
| ID | 1.20 (0.74–1.93) | 1.12 (0.45–2.78) |
| DD | 5.13 (2.50–10.50) | 4.89 (1.32–18.08) |
| KLK1 genotype | ||
| AA | 1.00 | 1.00 |
| AG | 2.61 (1.30–5.22) | 4.12 (1.18–14.32) |
| GG | 3.87 (1.86–8.03) | 3.00 (0.88–10.32) |
| AngII&KLK1 | ||
| ≦120&≦22 | 1.00 | 1.00 |
| ≦120&>22 | 3.46 (1.54–7.76) | 3.84 (1.03–14.42) |
| >120&≦22 | 5.21 (2.71–9.99) | 8.75 (2.86–26.75) |
| >120&>22 | 12.21 (6.23–23.94) | 19.52 (5.93–64.28) |
| ACE&KLK1 genotypes | ||
| ACE II&KLK1 AA | 1.00 | 1.00 |
| ACE ID&KLK1 AA/AG/GG | 1.35 (0.85–2.13) | 1.25 (0.55–2.86) |
| ACE DD&KLK1 GG | 9.02 (3.40–23.89) | 4.93 (5.93–64.28) |
Notes: Model 1: 4 variables (gender, age, the history of smoking and diabetes mellitus) were adjusted; Model 2: the above four variables plus 10 variables (ALT, GGT, TP, DBIL, TBA, CHO, HDL, GLU, CR and TT) were adjusted. AOR: adjusted odds ratio; 95% CI: 95% confidence interval.
The combination of serum AngII and KLK1 levels were observed. Compared with the grouping of AngII ≦120 and KLK1 ≦22, the three groupings of AngII ≦120 and KLK1 >22, AngII >120 and KLK1 ≦22, and AngII >120 and KLK1 >22 significantly increased the risk for AMI, with ORs (95%CI) of 3.46 (1.54–7.76), 5.21 (2.71–9.99) and 12.21 (6.23–23.94), respectively, and 3.84 (1.03–14.42), 8.75 (2.86–26.75) and 19.52 (5.93-64.28) in model 2.
The OR and 95%CI values for the ACE DD genotype were 5.13 (2.50–10.50) and 4.89 (1.32–18.08), respectively, compared with those of the ACE II genotype in the association of AMI in the two adjusted models. The OR and 95%CI values for the KLK1 GG genotype were 3.87 (1.86–8.03) and 3.00 (0.88–10.32) respectively, compared with the KLK1 AA genotype in the two adjusted models.
The combination of the two genotypes was compared with people who possessed the ACE II and KLK1 AA combined genotypes. Individuals with the ACE DD and KLK1 GG combined genotype showed significantly increased risk of AMI (OR=9.02, 95%CI 3.40–23.89, and OR=4.93, 95%CI 5.93–64.28) in the two adjusted models, respectively (Table 4).
Analysis of the association of serum levels of AngII, KLK1, and polymorphism of ACE/KLK1 with AMI by using the same logistic regression model
Analysis was performed for the two serum biomarkers combined and for the two genes combined. Hence, three models were used in the analysis. The first model included the above four terms individually and the 14 variables (as mention in Table 4), the second model included the combined serum biomarkers and two genes individually and the 14 variables, and the third model included the combined terms of the two serum biomarkers and the two genes combined and the 14 variables (Table 5).
Table 5.
Association of the serum levels of AngII, KLK1 and ACE/KLK1 polymorphisms with AMI in the same logistic regression model.
| Variables | OR(95%CI)^ Model 1a |
Variables | OR(95%CI)^ Model 2a |
Variables | OR(95%CI)^ Model 3a |
|---|---|---|---|---|---|
| AngII | AngII&KLK1 | AngII&KLK1 | |||
| ≦120 | 1.00 | ≦120&≦22 | 1.00 | ≦120&≦22 | 1.00 |
| >120 | 6.43 (2.77–14.97) | ≦120&>22 | 3.08 (0.77–12.40) | ≦120&>22 | 4.62 (1.11–19.33) |
| KLK1≦22 | 1.00 | >120&≦22 | 7.51 (2.37–23.76) | >120&≦22 | 11.32 (3.47–37.00) |
| >22 | 2.46 (1.08–5.60) | >120&>22 | 16.57 (4.88–56.20) | >120&>22 | 23.85 (6.88–82.65) |
| ACE genotype | ACE genotype | ACE&KLK1 genotypes | |||
| II | 1.00 | II | 1.00 | ACE II&KLK1AA | 1.00 |
| ID | 1.38 (0.51–3.72) | ID | 1.32 (0.48–3.65) | ACEID&KLK1AA/AG/GG | 1.13 (0.45–2.85) |
| DD | 4.66 (1.12–19.29) | DD | 4.46 (1.06–18.66) | ACE DD&KLK1 GG | 8.77 (1.74–44.16) |
| KLK1 genotype | KLK1 genotype | ||||
| AA | 1.00 | AA | 1.00 | ||
| AG | 2.36 (0.58–9.63) | AG | 2.27 (0.55–9.39) | ||
| GG | 2.31 (0.59–9.02) | GG | 2.20 (0.55–8.76) |
OR: adjusted odds ratio; 95%CI: 95% confidence interval.
Model 1: in the regression model included 14 adjusted variables (as mentioned in Table 4), the serum levels of AngII and KLK1, the genotypes of ACE I/D and KLK1 (rs5517) A/G; Model 2: in the regression model included 14 adjusted variables (as mentioned in Table 4), the variable of the serum levels of AngII combined with the serum levels of KLK1, and ACE I/D genotype and KLK1 (rs5517) A/G genotype; Model 3: in the regression model included 14 adjusted variables (as mentioned in Table 4), the variable of the serum levels of AngII combined with the serum levels of KLK1, the variable of the ACE/KLK1 genotype combined.
In the first model, increasing serum levels of AngII and KLK1 biomarkers after adjusting the above variables were significantly associated with AMI. Compared with the ACE II genotype, the ACE DD genotype increased the risk of AMI (OR=4.66, 95%CI 1.12–19.29); however, the KLK1 GG genotype did not show a significant correlation with AMI as compared with the KLK1 AA genotype.
In the second model, the two groupings of AngII >120 and KLK1 ≦22, and AngII >120 and KLK1 >22 increased the risk of AMI compared with the grouping of AngII ≦ 120 and KLK1 ≦ 22 after adjusting for the above variables for the two combined serum biomarkers. The ORs (95%CI) for AngII >120 and KLK1 ≦22, and AngII >120 and KLK1 >22 were 7.51 (2.37–23.26), 16.57 (4.88–56.20) respectively. Only the ACE DD genotype was a risk factor in AMI (OR=4.46, 95%CI 1.06–18.66); this finding is similar to that obtained using model 1.
In the third model, the last three groupings were significantly associated with AMI compared with the first (AngII ≦120 and KLK1 ≦22) after adjusting for the above-mentioned variables for the two combined serum biomarkers. Compared with individuals exhibiting the ACE II and KLK1 AA combined genotype, those with the ACE DD and KLK1 GG combined genotype exhibited significantly increased risk of AMI (OR=8.77; 95%CI 1.74–44.16).
Discussion
This study showed two novel findings. First, the levels of the two serum biomarkers AngII and KLK1 increased at the same time, thereby increasing the risk of AMI. Second, a synergistic interaction was observed between the ACE DD and KLK1 GG genotypes in the risk of AMI for Chinese people compared with the ACE II and KLK1 AA genotypes. Therefore, the ACE DD and KLK1 GG combined genotype increases the risk of AMI.
Inactive AngI is converted into the bioactive octapeptide (AngII) through the action of ACE or other enzymes.4 AngII generated by ACE elicits a vasoconstrictive effect, promotes thrombosis, and enhances the expression of platelet-derived growth factor, proliferation of smooth muscle cells, and inhibition of plasmin activity.12 Patients with AMI exhibited significantly higher AngII levels than the normal controls.1 Some reports suggested that KLK1 is a unique key enzyme of the kallikrein–kinin system (KKS), which catalyzes inactive kininogen and converts it to the bioactive bradykinin (BK).3,13,14 KLK1 and BK confer a series of cardiovascular protective effects. However, some studies have suggested that a high serum KLK1 level is positively associated with the presence of CAD and negatively associated with the severity of CAD; moreover, plasma-elevated KLK1 levels are a useful predictor for the presence and extent of CAD.3,15
This study found that simultaneously increases in serum levels of AngII and KLK1 were significantly associated with AMI. This finding could be due to acute coronary occlusion of patients with AMI. The KKS is activated earlier than the renin–angiotensin–aldosterone system (RAAS) and plays a key role in protecting against acute-phase ischemia. In severe myocardial ischemia, the RAAS is also over activated as a stress response.16
The human ACE gene is located on chromosome 17q23 and is a typical insertion (I)/deletion(D) polymorphism because of the absence of a 287bp Alu repeat sequence at intron 16.17 The polymorphism produces three genotypes including a heterozygous genotype (ID) and homozygous genotypes (DD and II). ACE catalyzes the conversion of AngI into bioactive AngII, as well as the inactivation of BK via the KKS.4 The levels of circulating, intracellular, and heart tissue activities of ACE in subjects with the ACE DD genotype were higher than those with the II genotype.18 The ACE DD genotype is related to the risk of CAS.5,19 Moreover, the ACE DD genotype may play a key role in the onset of AMI17,18 by altering serum ACE level or activity and increasing the instability of atherosclerotic plaques.20 In the present study, the ACE DD genotype was significantly associated with AMI in Chinese patients. The results are in accordance with those of previous reports, in which the ACE DD genotype increased the risk of PTCA-stent among Asians (OR = 2.18, 95%CI 1.08–4.40, p = 0.03).8
KLK1 A1789G (rs5517) is a unique polymorphism of the KLK1 gene exon 4 coupled with the single nucleotide substitution Lys→Glu. Functional analysis has suggested that different haplotype alleles significantly contribute to kallikrein expression.21,22 In one animal study, increasing BK outflow of transgenic rats harboring KLK1 gene-modified endothelial progenitor cells were investigated under basal and ischemic conditions to promote angiogenesis in rats with ischemic hindlimb.23 Some reports have shown that KLK1GG may be associated with human essential hypertension.24,25 Our previous results suggested that the KLK1 GG genotype of KLK1 A1789G (rs5517) is associated with CAS in the Chinese Han population.8
Our results suggest that patients with the ACE DD and KLK1 GG combined genotype had a significantly increased in the risk of AMI (p < 0.05) compared with those with the ACE II and KLK1 AA combined genotype, showing their synergistic effect on the occurrence of AMI. However, we did not find a significant correlation between the serum levels of AngII or KLK1 and the genotypes of ACE and (or) KLK1, indicating they have independent effects in the risk of AMI.
The mechanism for the observed results remains unclear. We have established the following views based on literature review. AngII is believed to be a potential pro-inflammatory factor. AngII can stimulate the secretion of cytokines, such as IL-6 and tumor necrosis factor alpha (TNFα) as well as serum C-reactive protein (CRP), which induces the inflammation and instability of atherosclerosis (AS) plaques.26 A recent study identified the lack of association between six different genetic polymorphisms (ACE (I/D), A-240T and A2350G, angiotensinogen M235T, AT1 receptor A1166C, and AT2 receptor C3123A polymorphisms) of RAS with increase in serum CRP level.27 These reports provide evidence that supports the present findings.
The protective effect of the KKS against cardiovascular diseases involves several signaling pathways, such as nitric oxide (NO)-cyclic guanosine monophosphate (Akt-GSK-3β) which is activated by binding KLK1 to their respective receptors to protect against apoptosis, ischemia, inflammation, and ventricular remodeling.28 Nuclear factor-related factor 2 (Nrf2) is a master regulator of redox homeostasis that protects cells in adapting to oxidative stress and accelerates cell proliferation. The phosphorylation of Akt–GSK–3β active Nrf2 with non-phosphorylated DSGIS motif within the Neh6 domain activates the Nrf2/ARE (antioxidant responses elements) pathway, increasing Nrf2 stability and ARE-driven gene expression, such as antioxidant and anti-inflammatory protein gene expression.29 The phosphorylation of Akt-GSK-3β also inhibits thrombus growth to increase plaque stability.30 Furthermore, KLK1 inhibits nuclear factor-kappa-binding (NF-κB) activation by inhibiting the phosphorylation and degradation of IκB-α. These events lead to the inhibition of NF-κ B release from the I3B-3/NF-3B complex and NF-κ B-driven gene expression, such as oxidant and inflammatory protein gene expression.28
ACE and AngII play a key role in the development of cardiovascular diseases and can be modulated by some components of ACE gene abnormalities and disorders.31 Several signaling pathways are linked with angiogenesis, including EGFR-PI3K-PTEN-AKT, KRAS-BRAF-MEK-MAPK, and external stimulating factor ROS-GST-NF-κ B.32,33 The expression of ACE is directly related to the NF-κ B pathway through 20 hydroxyeicosatetraenoicacid (20-HETE) and promotes myocardial injury. 20-HETE is identified based on the transcription of ACE via NF-κB translocation and promoter binding by the distinct EGFR-MAPK-IKK-NF-κB pathway.34
NF-κB is related to inflammation and cardiovascular complications because of the capacity of ACE gene transcription factor.35 The present study suggested that the combined ACE DD and KLK1 GG genotypes increased the risk of AMI, which may have been associated with the above-mentioned common joint points (NF-κB) of biological information pathways.
Overall, 147 gene variants have been detected, which comprises only a small proportion of the heritability (20%) and is weakly associated with AS, CAS, and AMI (OR = 1.1–1.5) by using the genome-wide association study (GWAS) method.36 The pathogenesis of AMI involves multiple genetic and environmental factors as well as their interaction.37,38 Structural gene variants, such as insertion or deletion as well as the interactions of gene–gene or gene–environment, are poorly captured by GWAS methodology.36,39 Notable, the GWAS study did not indicate that ACE and KLK1 genes are associated with AS. The reason may be related to the GWAS itself; detecting a powerful effect on a locus of the gene made others genes’ effects indistinguishable.40 The risk or protective factors and pathogenesis are different for AS, CAD, CAS and AMI.39 Furthermore, the traditional epidemiology heritability of CAS and in-stent restenosis has not been established. Phenotypically well-defined cases and controls must be analyzed to overcome the limitations of the GWAS method.36-40
The conclusion should be proven by a larger and a more precise selection of homogenous cases and controls, because of insufficient sample size used in the study.
In conclusion, this study presented the following novel results. (1) The risk of AMI will increase significantly if the serum AngII and KLK1 levels simultaneously increase. (2) Individuals with the ACE DD and KLK1 5517GG combined genotype had significantly increased risk of AMI compared with those with the ACE II and KLK1 5517AA combined genotypes. The results of the study may lay a foundation for further research on the etiology of AMI.
Footnotes
Authors’ contributions: DQL and SHD designed the experiments; DQL, SHD, JFL and JBF carried out all experiments; RJL, CBL, ZL, and YZ provided clinical specimens; DQL and SHD analyzed the data; RJL, CBL, ZL, and YZ contributed reagents and materials; DQL and SHD drafted the manuscript, edited and revised the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Shandong Province (grant numbers ZR2015HM012 and NO.BS2011YY024) and National Natural Science Foundation of China Grant (grant number 21576206).
References
- 1. Oyamada S, Bianchi C, Takai S, et al. Impact of acute myocardial ischemia reperfusion on the tissue and blood-borne renin-angiotensin system. Basic Res Cardiol 2010; 105: 513–522. [DOI] [PubMed] [Google Scholar]
- 2. Rhaleb NE, Yang XP, Carretero OA.The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr Physiol 2011; 1: 971–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yao Y-Y, Fu C, Ma G-s, et al. Tissue kallikrein is related to the severity of coronary artery disease. Clinica Chimica Acta 2013; 423: 90–98. [DOI] [PubMed] [Google Scholar]
- 4. Fildes JE, Walker AH, Keevil B, et al. The effects of ACE inhibition on serum angiotensin II concentration following cardiac transplantation. Transplant Proc 2005; 37: 4525–4527. [DOI] [PubMed] [Google Scholar]
- 5. Chen Y, Dong S, He M, et al. Angiotensin-converting enzyme insertion/deletion polymorphism and risk of myocardial infarction in an updated meta-analysis based on 34993 participants. Gene 2013; 522: 196–205. [DOI] [PubMed] [Google Scholar]
- 6. Mehri S, Baudin B, Mahjoub S, et al. Angiotensin-converting enzyme insertion/deletion gene polymorphism in a Tunisian healthy and acute myocardial infarction population. Genet Test Mol Biomarkers 2010; 14: 85–91. [DOI] [PubMed] [Google Scholar]
- 7. Bradley DT, Badger SA, McFarland M, et al. Abdominal aortic aneurysm genetic associations: Mostly false? A systematic review and meta-analysis. Eur J Vasc Endovasc Surg 2016; 51: 64–75. [DOI] [PubMed] [Google Scholar]
- 8. Pan Y, Wang F, Qiu Q, et al. Influence of the angiotensin converting enzyme insertion or deletion genetic variant and coronary restenosis risk: Evidence based on 11,193 subjects. PLOS One 2013; 8: e83415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chao J, Chao L. Kallikrein-kinin in stroke, cardiovascular and renal disease. Exp Physiol 2005; 90: 291–298. [DOI] [PubMed] [Google Scholar]
- 10. Li QD, Li FJ, Liu XC, et al. KLK1 A1789G gene polymorphism and the risk of coronary artery stenosis in the Chinese population. Genet Mol Res 2013; 12: 1636–1645. [DOI] [PubMed] [Google Scholar]
- 11. Langorgen J, Ebbing M, Igland J, et al. Implications of changing definitions of myocardial infarction on number of events and all-cause mortality: The WHO 1979, ESC/ACC 2000, AHA 2003, and Universal 2007 definitions revisited. Eur J Prev Cardiol 2014; 21: 1349–1357. [DOI] [PubMed] [Google Scholar]
- 12. Malik FS, Lavie CJ, Mehra MR, et al. Renin-angiotensin system: Genes to beside. Am Heart J 1997; 134: 514–526. [DOI] [PubMed] [Google Scholar]
- 13. Sato M, Engelman RM, Otani H, et al. Myocardial protection by preconditioning of heart with losartan, an angiotensin II type 1-receptor blocker – Implication of bradykinin-dependent and bradykinin-independent mechanisms. Circulation 2000; 102: 346–351. [DOI] [PubMed] [Google Scholar]
- 14. Ren YL, Garvin J, Carretero OA. Mechanism involved in bradykinin-induced efferent arteriole dilation. Kidney Int 2002; 62: 544–549. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Q, Ran X, Wang DW. Relation of plasma tissue kallikrein levels to presence and severity of coronary artery disease in a Chinese population Plos One 2014; 9: e91780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Heuzey J-Y, Marijon E, Chachoua K, et al. Pathophysiology of atrial fibrillation: Insights into the renin-angiotensin system. Arch Cardiovasc Dis 2008; 101: 787–791. [DOI] [PubMed] [Google Scholar]
- 17. Cambien F, Costerousse O, Tiret L, et al. Plasma level and gene polymorphism of angiotensin-converting enzyme in relation to myocardial infarction. Circulation 1994; 90: 669–676. [DOI] [PubMed] [Google Scholar]
- 18. Rigat B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 1990; 86: 1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vaisi-Raygani A, Ghaneialvar H, Rahimi Z, et al. The angiotensin converting enzyme D allele is an independent risk factor for early onset coronary artery disease. Clin Biochem 2010; 43: 1189–1194. [DOI] [PubMed] [Google Scholar]
- 20. Igic R, Behnia R. Properties and distribution of angiotensin I converting enzyme. Curr Pharm Des 2003; 9: 697–706. [DOI] [PubMed] [Google Scholar]
- 21. Slim R, Torremocha F, Moreau T, et al. Loss-of-function polymorphism of the human kallikrein gene with reduced urinary kallikrein activity. J Am Soc Nephrol 2002; 13: 968–976. [DOI] [PubMed] [Google Scholar]
- 22. Song Q, Chao J ad, Chao L. DNA polymorphisms in the 5’-flanking region of the human tissue kallikrein gene. Hum Genet 1997; 99 :727–734. [DOI] [PubMed] [Google Scholar]
- 23. Fu SS, Li FJ, Wang YY, et al. Kallikrein gene-modified EPCs induce angiogenesis in rats with ischemic hindlimb and correlate with integrin alpha v beta 3 expression. Plos One. 2013; 8: e73035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev 1992; 44: 1–80. [PubMed] [Google Scholar]
- 25. Zhao WY, Wang LY, Lu XF, et al. A coding polymorphism of the kallikrein 1 gene is associated with essential hypertension: A tagging SNP-based association study in a Chinese Han population. J Hypertens 2007; 25: 1821–1827. [DOI] [PubMed] [Google Scholar]
- 26. Sattler KJE, Woodrum JE, Galili O, et al. Concurrent treatment with renin-angiotensin system blockers and acetylsalicylic acid reduces nuclear factor kappa B activation and C-reactive protein expression in human carotid artery plaques. Stroke 2005; 36: 14–20. [DOI] [PubMed] [Google Scholar]
- 27. Bahramali E, Firouzabadi N, Jonaidi-Jafari N, et al. Renin-angiotensin system genetic polymorphisms: Lack of association with CRP levels in patients with coronary artery disease. J Renin Angiotensin Aldosterone Syst 2014; 15: 559–565. [DOI] [PubMed] [Google Scholar]
- 28. Yao Y-Y, Yin H, Shen B, et al. Tissue kallikrein infusion prevents cardiornyocyte apoptosis, inflammation and ventricular remodeling after myocardial infarction. Regul Pept 2007; 140: 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayes JD, Chowdhry S, Dinkova-Kostova AT, et al. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta-TrCP and GSK-3. Biochem Soc Trans 2015; 43: 611–620. [DOI] [PubMed] [Google Scholar]
- 30. Laurent PA, Severin S, Gratacap MP, et al. Class I PI 3-kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv Biol Regul 2014; 54: 162–174. [DOI] [PubMed] [Google Scholar]
- 31. Dzida G, Sobstyl J, Puzniak A, et al. Polymorphisms of angiotensin-converting enzyme and angiotensin II receptor type 1 genes in essential hypertension in a Polish population. Med Sci Monit 2001; 7: 1236–1241. [PubMed] [Google Scholar]
- 32. Michaelis UR, Fisslthaler B, Medhora M, et al. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor. FASEB J 2003; 17: 770–2. [DOI] [PubMed] [Google Scholar]
- 33. Yan G, Chen S, You B, et al. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovasc Res 2008; 78: 308–314. [DOI] [PubMed] [Google Scholar]
- 34. Garcia V, Brian S, Milhau L, et al. 20-HETE activates the transcription of angiotensin converting enzyme (ACE) via NF-kB translocation and promoter binding. J Pharmacol Exp Ther 2016; 356: 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harrison DG, Guzik TJ, Lob HE, et al. Inflammation, immunity, and hypertension. Hypertension 2011; 57: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dai X, Wiernek S, Evans JP, et al. Genetics of coronary artery disease and myocardial infarction. World J Cardiol 2016; 8: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Topol EJ. The genomic basis of myocardial infarction. J Am Coll Cardiol 2005; 46: 1456–1465. [DOI] [PubMed] [Google Scholar]
- 38. Gibson G. Hints of hidden heritability in GWAS. Nat Genet 2010; 42: 558–560. [DOI] [PubMed] [Google Scholar]
- 39. Dorn GW, Cresci S.Genome-wide association studies of coronary artery disease and heart failure: Where are we going? Pharmacogenomics 2009; 10: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kitsios GD, Dahabreh IJ, Trikalinos TA, et al. Heterogeneity of the phenotypic definition of coronary artery disease and its impact on genetic association studies. Circ Cardiovasc Genet 2011; 4: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]