

Abstract
Background and objective:
Renin–angiotensin system (RAS) inhibitors reduce glomerular injury and proteinuria, indicating that angiotensin II (Ang II) is involved in glomerular diseases. Although the local RAS is reported to play an essential role in maintaining local tissue functions, the role of the local RAS in regulating glomerular function is not well evaluated. In this study, we analyzed the glomerular expression of RAS components in nephrotic models and the effect of Ang II receptor blockers (ARB) on the expression of angiotensinogen (AGT).
Methods:
The levels of glomerular expression of RAS components were analyzed in two nephrotic models: anti-nephrin antibody-induced nephropathy and PAN nephropathy, a mimic of human minimal change nephrotic syndrome. The effect of the ARB irbesartan on the expression of AGT in the nephrotic model was analyzed.
Results:
Glomerular expression of AGT and the receptors for Ang II was clearly increased in the nephrotic models, while the expression levels of renin, ACE and ACE2 were decreased. ARB treatment suppressed the increase of glomerular expression of AGT in the nephrotic model.
Conclusion:
It is conceivable that the promoted local RAS action participated in the glomerular dysfunction, and that ARB treatment ameliorated slit diaphragm injury by inhibiting the positive feedback loop of the activated local Ang II action.
Keywords: Angiotensinogen, podocyte, slit diaphragm, nephrin
Introduction
A number of clinical1–4 and experimental5–9 studies have demonstrated that renin–angiotensin system (RAS) inhibitors, such as an angiotensin-converting enzyme inhibitor and an angiotensin II (Ang II) type 1 receptor (AT1R) blocker (ARB) reduce renal tissue damage and proteinuria. These reports indicated that Ang II is involved in the development of kidney diseases and proteinuria. Although the action of Ang II is basically explained as being on systemic blood pressure and/or intra-glomerular pressure-dependent mechanisms, some studies have focused on the pressure-independent mechanism of Ang II action in kidney.10,11 Although the RAS was originally viewed solely as an endocrine system, some recent studies have emphasized that the local RAS plays an essential role in maintaining local tissue functions.12,13 However, the role of the local RAS in regulating glomerular function is not yet well evaluated.
In the field of nephrology, clarification of the mechanism of proteinuria is one of the most important themes. The filtration barrier of the kidney glomerulus, preventing the leak of plasma proteins into primary urine, comprises three layers: the endothelial cells, the glomerular basement membrane, and the visceral epithelial cells (podocytes). It is now accepted that the slit diaphragm (SD), located between adjacent foot processes of podocytes, functions as the final barrier of the glomerular capillary wall, and that dysfunction of the SD is involved in the development of proteinuria in several common diseases .14–16
We have previously reported that the ARB ameliorates proteinuria in rat nephropathy models in which proteinuria results from SD dysfunction.17,18 In these reports, we showed that ARB reduced proteinuria by preventing the dis-localization of the functional molecules of the SD. These observations indicated that the increase in Ang II action participates in the development of SD dysfunction. However, the differential roles of the systemic and local RAS in regulating the barrier function of the SD remain to be clarified.
In this study, we showed that glomerular expression of angiotensinogen (AGT) and the receptors for Ang II were clearly increased in the nephrotic models. We also showed that ARB treatment rescued the decreased expression of nephrin, a key molecule of the SD, and suppressed the increase of the expression of AGT in nephrotic rat glomeruli. It is conceivable that the promoted local RAS action participated in SD dysfunction, and that ARB treatment ameliorated SD injury by blocking the positive feedback loop of the activated local Ang II action.
Materials and methods
Animals and cultured podocytes
All in vivo experiments were performed using specific pathogen-free female Wistar rats weighing 100–110 g at the age of 7–8 weeks. All rats used in this study were purchased from Charles River Japan (Atsugi, Japan), and fed with a normal salt diet. All animal experiments conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Procedures for the present study were approved by the Animal Committee according to the guidelines for animal experimentation of Niigata University. All the animal experiments were conducted in compliance with the protocol which was reviewed by the Institutional Animal Care and Use Committee and approved by the President of Niigata University (Permit Number: #26, Niigata Univ. Res.378-2). Cultivation of conditionally immortalized mouse podocytes kindly donated by Dr. P. Mundel (Albert Einstein College of Medicine, Bronx, NY) was conducted as reported previously.19
Experimental protocols
Experiment 1: Analyses of the expression of RAS components in nephrotic syndrome models
ANA nephropathy
Two sets of experiments were done. In each set nine rats were used. Six rats were intravenously injected with murine monoclonal antibody against rat nephrin (ANA: mAb 5-1-6),20,21 and three rats each were killed under pentobarbital anesthesia at 1 h and 5 days after the injection. Another three rats were injected with vehicle (PBS) and sacrificed at 1 h. After blood sampling, the kidneys were removed, weighed, cut into portions and used in immunofluorescence (IF) studies. The remaining kidney materials from three rats in each group were pooled and used for preparation of glomeruli, and the glomerular materials were used for reverse transcription polymerase chain reaction (RT-PCR) and for Western blot analysis.
PAN nephropathy
The same as for the study on ANA nephropathy, two sets of experiments were performed for PAN nephropathy. In each set nine rats were used. Six rats were intravenously injected with PAN (100 mg/kg b.w.), and three rats each were killed under pentobarbital anesthesia at 1 h and 10 days after the injection. Another three rats were injected with vehicle (PBS) and sacrificed at 1 h. The samples were prepared as in the ANA nephropathy experiment.
Experiment 2: Analysis of the effect of ARB on the expression of AGT and nephrin
A total of 12 rats were injected with PAN, and six rats were orally administered with irbesartan (15 mg/kg b.w. per day) from 3 days before the injection with PAN, and the remaining six rats were treated with distilled water. Irbesartan was kindly gifted from Dainippon Sumitomo Pharma Co., Ltd, Osaka, Japan. The treatment of 15 mg/kg b.w. per day of irbesartan was confirmed not to affect systemic blood pressure18 and to ameliorate proteinuria (day 8: 237.6 ± 97.0 vs. 359.0 ± 63.3 mg/day, p < 0.05). All rats were sacrificed on day 10. Kidney materials were used for RT-PCR and IF analyses. The other three rats injected with PBS instead of PAN were treated with ARB as described above and sacrificed 10 days after PBS injection.
RT-PCR
Real-time RT-PCR analysis and semi-quantitative RT-PCR with glomerular RNA was performed according to the method described previously.17,18 The data are shown as ratios relative to control findings and expressed as means ± SD of two samples. The primer sequences are summarized in Table 1.
Table 1.
PCR primers used in this study.
| Probe | Sense primer, 5’ to 3’ | Anti-sense primer, 5’ to 3’ | Size (bp) |
|---|---|---|---|
| AGT | CTG TGC CCA TTC AGG CCA AG | TCT TCC ACC CTG TCA CAG CC | 581 |
| Renin* | ACT ACA GCA GGG AGT CCC AC | AGA GCC AGT ATG CAC AGG TC | 412 |
| ACE | CAG TGG CGC TGG AGG GTC TTT G | TAC TCC GGC CAG CCC AGT GTC TC | 460 |
| ACE2* | GAC CAA AAA GTG GTG GGA GAT G | ATG GGC TCC AGT CAG TGC TC | 419 |
| AT1R | GGA AAC AGC TTG GTG GTG ATT G | CCA ATG GGG AGT GTT GAG TTC | 450 |
| AT2R | CCG GCA GAT AAG CAT TTG GAA G | GAA TGC CAA CAC AAC AGC AGC | 680 |
| Neprilysin | TGC GAG TGA TGT CAG GTC AC | GTG TCT GTA CAG CGG AAT GG | 151 |
| Nephrin* | ATC TGT GGA ATG TGA CCC GCG C | CTG GGG GGC AAA TCT GAC AAC AAG | 404 |
| Nephrin* | CTG ACT GGG CTG AAG CCT TCT | AAG AGC ACA GGC AGC AGG GG | 203 |
| GAPDH* | CTC TAC CCA CGG CAA GTT CAA | GGA TGA CCT TGC CCA CAG C | 515 |
| Renin# | GCC TCA GCA AGA CTG ATT CC | CCT GGC TAC AGC TCA CAA CA | 207 |
| ACE2# | GAC CAA AAA GTG GTG GGA GAT G | ATG GGC TCC ATT CAG TGT TCC | 419 |
| Nephrin# | AGC TGT GGA ATG TAA CCC GAG C | TGG GGG GCA AAT CGG ACG ACA AG | 404 |
| GAPDH# | CTC CAC TCA CGG CAA ATT CAA | GGA TGA CCT TGC CCA CAG C | 516 |
Primer for rat; #Primer for mouse; others, for rat and mouse.
Immunofluorescence
IF studies were performed according to the method previously reported.21 Antibodies used in this study are as follows: rabbit anti-AGT antibody was purchased from Abcam (Cambridge, MA, USA), and mouse anti-nephrin antibody (ANA: mAb 5-1-6) was prepared as previously described.20,21 The following antibodies were used as the markers for glomerular cells: mouse anti-RECA1 antibody (Serotec, Oxford, JE, UK), mouse anti-synaptopodin antibody (PROGEN, Heidelberg, Germany), mouse anti-Thy1.1 (OX7).22 FITC-conjugated anti-rabbit IgG, FITC-conjugated anti-mouse IgG and RITC-conjugated anti-mouse IgG were purchased from DakoCytomation (Glostrup, Denmark).
Western blot analysis
Western blot analysis was performed according to the method described previously.23 Strips of the membrane were exposed to rabbit anti-AGT antibody or rabbit anti-β-actin antibody. Anti-β-actin antibody was purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA). The strips were then washed and incubated with alkaline phosphatase-conjugated anti-rabbit immunoglobulins (Zymed Laboratory Inc. San Francisco, CA, USA).
Results
mRNA expression of RAS components was detected in glomeruli and cultured podocytes
mRNA expression of AGT, renin, ACE, ACE2, neprilysin, AT1R and AT2R was detected in isolated rat glomeruli and in differentiated cultured podocytes by RT-PCR. The findings are shown in Figure 1.
Figure 1.
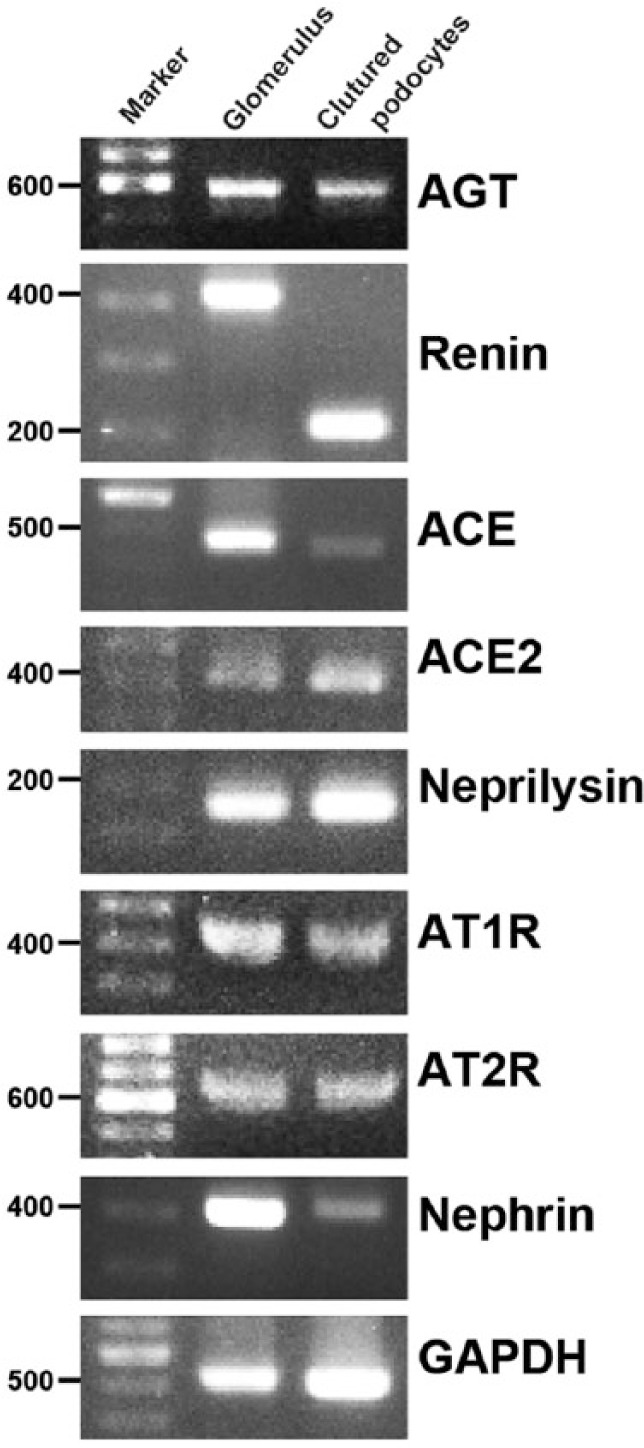
mRNA expression of RAS components.
Representative agarose-gel electrophoretic patterns of RT-PCR. mRNA expression of AGT, renin, ACE, ACE2, neprilysin, AT1R and AT2R was detected in isolated rat glomeruli and in differentiated cultured podocytes.
Glomerular mRNA expression of RAS components was altered in nephrotic rats
Glomerular mRNA expression of AGT was clearly increased more than 50 times in both nephrotic models. The expression levels were already increased just after disease induction (1 h) in both models. The expression levels of Ang II receptors were also increased. The expression of AT1R was clearly doubled when proteinuria peaked in both models, although the increase was not evident at the early phase (1 h). Expression of AT2R increased by around 10 times at 1 h after disease induction in both models. On the other hand, expression levels of the enzymes RAS, renin, ACE, ACE1 and neprilysin were decreased. The features of the changes in expression of RAS components were very similar in the two models. The findings are shown in Figure 2.
Figure 2.

Glomerular mRNA expression of RAS components in nephrotic rats.
Glomerular mRNA expression of AGT is evidently increased in both models. Expression levels of AT1R and AT2R were increased. Expression of renin, ACE, ACE1 and neprilysin was decreased. The data are shown as ratios relative to control findings and expressed as means ± SD of two samples.
Glomerular expression of AGT was increased at the protein level in nephrotic models
Bands around 50 kd were detected in normal rat glomerular lysate with anti-AGT antibody, and the intensity of the bands was increased in the glomerular lysates of rats in the nephrotic models. Representative Western blot findings and semi-quantitative data are shown in Figure 3(a). The immunostaining of AGT was observed in a discontinuous patchy pattern in normal rat glomeruli; the intensity of the AGT staining increased and its staining pattern changed to a continuous pattern along the capillary loop in the nephrotic models (Figure 3(b)). Dual-labeling IF analysis showed that the AGT staining on day 10 of PAN nephropathy was clearly different from endothelial cell marker RECA-1 and mesangial cell marker Thy1, and major portions of AGT were co-stained with a podocyte marker synaptopodin (Figure 3(c)). Positive AGT staining was detected at the apical surface of tubular cells in proteinuric states (Figure 3(b)). Positive AGT staining was observed in normal rat liver section. No clear increase in AGT staining was detected in the nephrotic models used in this study (data not shown).
Figure 3.

Glomerular expression of AGT and nephrin in nephrotic rats.
(a) Western blot analysis. AGT-specific bands around 50 kd were detected in normal rat glomerular lysate, and the intensity of the bands was increased in the glomerular lysates of rats in the nephrotic models. Semi-quantitative data of the band density are shown. (b) IF findings of AGT and nephrin. AGT staining was increased and its staining pattern changed to a continuous linear-like pattern along the capillary loop. Positive AGT staining was detected at the apical surface of tubular cells in proteinuric states. (c) Dual-labeling IF analysis: AGT staining on day 10 of PAN nephropathy was clearly different from the endothelial cell marker RECA-1 and the mesangial cell marker Thy1, and major portions of AGT were co-stained with a podocyte marker synaptopodin.
ARB treatment suppressed the increased AGT expression in PAN nephropathy
In experiment 1, we showed that AGT was clearly upregulated, and renin, ACE and nephrin were downregulated in nephrotic glomeruli. Next, we analyzed the effect of ARB treatment on the expression of RAS components and nephrin. The increase of mRNA expression of AGT and the reduction of renin, ACE and nephrin at 1 h of PAN nephropathy were suppressed by the ARB treatment (Figure 4(a)). The increase in the immunostaining of AGT on day 10 of PAN nephropathy was suppressed by ARB treatment (Figure 4(b)).
Figure 4.

ARB treatment suppressed the increased AGT expression in PAN nephropathy.
(a) Representative agarose-gel electrophoretic patterns of RT-PCR and semi-quantitative data for the three independent analyses. ARB treatment suppressed the increase of mRNA expression of AGT and the reduction of renin, ACE and nephrin expression at 1 h of PAN nephropathy. (b) IF findings of AGT and nephrin. Increase in the immunostaining of AGT on day 10 of PAN nephropathy was suppressed by ARB treatment.
Discussion
It is understood that Ang II contracts efferent arterioles of glomeruli more effectively than afferent arterioles, and that the consequent glomerular hypertension damages the glomeruli and enhances proteinuria. Thus, it has been accepted that RAS inhibitors have a protective role for kidney glomeruli and ameliorate proteinuria by reducing glomerular hypertension. However, some recent reports have shown that RAS inhibitors reduce proteinuria independently to intra-glomerular pressure.10,11 It is also reported that Ang II acts directly on glomerular podocytes.17,24 From these observations, it is now accepted that the action of Ang II directly damages the barrier function of podocytes, and RAS inhibitors ameliorate proteinuria by protecting podocytes. However, the pathogenic mechanism of how Ang II acts on podocytes is still unclear. Durvasula et al. showed that cultured podocytes actively synthesize AGT if they are stimulated by stretch or a high-glucose condition.25,26 The reports suggested that the local RAS participates in the development of podocyte injury. On the other hand, Matsusaka et al. reported that Ang II action-dependent nephropathy could be induced in podocyte-specific AGT knockout mice, indicating that the systemic RAS plays a major role in inducing podocyte damage.27 Thus the role of the local RAS in podocyte injury is still controversial.
In this study we first analyzed the expression of RAS components in isolated glomeruli and in cultured podocytes. mRNA expression of all major components of the RAS was detected (Figure 1). Expression of AGT in normal rat glomeruli was also observed with immunohistochemical analysis, although the staining was not very intense. It is conceivable that these observations suggest that the local RAS might have a physiological role in maintaining glomerular function.
Next, to elucidate the role of the local RAS in the development of podocyte injury, we analyzed the expression of RAS components in nephrotic models. We adopted two nephrotic models, ANA nephropathy and PAN nephropathy. ANA nephropathy is caused by a one-shot injection of the antibody against nephrin, a key molecule of the SD. Proteinuria in ANA nephropathy is induced by the rearrangement of nephrin and other functional molecules of the SD without participation of any inflammatory factors or complement.21,23,28,29 PAN nephropathy is widely used as an experimental model of human minimal change nephrotic syndrome. PAN has toxicity specifically to podocytes. Although the precise pathogenic mechanism of PAN nephropathy has not yet been elucidated, it is accepted that the disarrangement of the SD molecules is involved in the development of proteinuria in this nephropathy.21,23,30 We found that glomerular mRNA expression of AGT is dramatically increased in both models from just after disease induction, and that the increase of AGT expression was maintained when proteinuria peaked (Figure 2). The increased expression of AGT in glomeruli was also detected in the immunohistochemical analysis and Western blot study with glomerular lysates (Figure 3(a, b)). Dual-labeling IF study with glomerular cell markers showed AGT was mainly expressed in podocytes in glomeruli (Figure 3(c)).
In this study we also showed that the mRNA expression of AT1R was clearly increased in the nephrotic stage in both models. By contrast, the expression of renin and ACE was decreased (Figure 2). These enzymes might be decreased to suppress the effect of the dramatic increase of AGT production. Although the meaning of the changes in the expression of the RAS molecules in glomeruli is not well explained, these observations suggest that local RAS activity is basically regulated by the amount of the substrate, AGT, and that the increased production of AGT in podocytes contributes to podocyte injury in an autocrine manner.
It should be mentioned that the increase of the protein level of AGT was not remarkable if compared with the decrease of the level of mRNA. Although it is difficult to explain the precise reason for this discrepancy, it is plausible that some post-translational pathway suppressed the increase of the AGT protein. It is also plausible that some negative feedback pathway enhanced the degradation of AGT.
Another important finding we should mention is that ATR2 expression also increased in both nephrotic models. We have previously reported that AT2R-mediated Ang II action has a function opposite to AT1R-mediated Ang II action.17 It is thought that the increase of AT2R is a protective response to suppress AT1R-mediated Ang II action.
It is reported that the local tissue RAS is enhanced if the cells are stimulated,25,26,31 and that the activated local RAS participates in tissue injury in an autocrine or paracrine manner. A recent report implied that if the cells are stimulated with Ang II, the Ca++ channel was activated and the consequent increase of Ca++ influx enhanced the local RAS in a positive feedback loop.31 In this study we observed that glomerular mRNA expression of AGT increased remarkably in nephrotic models, and the increase of AGT was also observed by immunohistochemistry. We observed that the increase in AGT staining in liver is not clear in the nephrotic models. These observations suggest that the local RAS may be involved in the development of the nephrotic models. ARB is widely used for patients showing proteinuria, including in steroid-resistant nephropathy.32 In this study, we also observed that the ARB treatment suppressed the increase of the glomerular expression of AGT, and inhibited the decrease of the expression of SD molecules, and then ameliorated proteinuria in the nephrotic model (Figure 4). The observations suggest that the local RAS participated in SD dysfunction, and that ARB treatment protected the SD by blocking the positive feedback loop of the local action of Ang II.
This is the first report showing that the expression of AGT in podocytes was evidently increased in nephrotic states and the increased expression of glomerular AGT was suppressed by ARB treatment. It is conceivable that the promoted local RAS participates in the development of nephrotic syndrome.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Grant-Aids for Scientific Research (B: 23390224 to H. Kawachi) and Grant-Aid for Young Sciences (B: 24790839 and 16K197479 to Y. Fukusumi) from Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Holtkamp FA, de Zeeuw D, de Graeff PA, et al. Albuminuria and blood pressure, independent targets for cardioprotective therapy in patients with diabetes and nephropathy: A post hoc analysis of the combined RENAAL and IDNT trials. Eur Heart J 2011; 32: 1493–1499. [DOI] [PubMed] [Google Scholar]
- 2. Casas JP, Chua W, Loukogeorgakis S, et al. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: Systematic review and meta-analysis. Lancet 2005; 366: 2026–2033. [DOI] [PubMed] [Google Scholar]
- 3. Appel LJ, Wright JT, Jr., Greene T, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med 2010; 363: 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Contreras G, Greene T, Agodoa LY, et al. Blood pressure control, drug therapy, and kidney disease. Hypertension 2005; 46: 44–50. [DOI] [PubMed] [Google Scholar]
- 5. Fan YY, Baba R, Nagai Y, et al. Augmentation of intrarenal angiotensin II levels in uninephrectomized aldosterone/salt-treated hypertensive rats; renoprotective effects of an ultrahigh dose of olmesartan. Hypertens Res 2006; 29: 169–178. [DOI] [PubMed] [Google Scholar]
- 6. Aki K, Shimizu A, Masuda Y, et al. ANG II receptor blockade enhances anti-inflammatory macrophages in anti-glomerular basement membrane glomerulonephritis. Am J Physiol Renal Physiol 2010; 298: F870–F882. [DOI] [PubMed] [Google Scholar]
- 7. Urushihara M, Ohashi N, Miyata K, et al. Addition of angiotensin II type 1 receptor blocker to CCR2 antagonist markedly attenuates crescentic glomerulonephritis. Hypertension 2011; 57: 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Remuzzi A, Gagliardini E, Sangalli F, et al. ACE inhibition reduces glomerulosclerosis and regenerates glomerular tissue in a model of progressive renal disease. Kidney Int 2006; 69: 1124–1130. [DOI] [PubMed] [Google Scholar]
- 9. Macconi D, Sangalli F, Bonomelli M, et al. Podocyte repopulation contributes to regression of glomerular injury induced by ACE inhibition. Am J Pathol 2009; 174: 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goodfriend TL, Elliott ME, Catt KJ. Angiotensin receptors and their antagonists. N Engl J Med 1996; 334: 1649–1654. [DOI] [PubMed] [Google Scholar]
- 11. Ruster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol 2011; 22: 1189–1199. [DOI] [PubMed] [Google Scholar]
- 12. Liebau MC, Lang D, Bohm J, et al. Functional expression of the renin-angiotensin system in human podocytes. Am J Physiol Renal Physiol 2006; 290: F710–F719. [DOI] [PubMed] [Google Scholar]
- 13. Tojo A, Kinugasa S, Fujita T, et al. A local renal renin-angiotensin system activation via renal uptake of prorenin and angiotensinogen in diabetic rats. Diabetes Metab Syndr Obesity 2016; 9: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tryggvason K, Pettersson E. Causes and consequences of proteinuria: The kidney filtration barrier and progressive renal failure. J Intern Med 2003; 254: 216–224. [DOI] [PubMed] [Google Scholar]
- 15. Kawachi H, Shimizu F. Molecular composition and function of the slit diaphragm: Nephrin, the molecule responsible for proteinuria. Clin Exp Nephrol 2000; 4: 161–172. [Google Scholar]
- 16. Pavenstadt H. Roles of the podocyte in glomerular function. Am J Physiol Renal Physiol 2000; 278: F173–F179. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki K, Han GD, Miyauchi N, et al. Angiotensin II type 1 and type 2 receptors play opposite roles in regulating the barrier function of kidney glomerular capillary wall. Am J Pathol 2007; 170: 1841–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahashi A, Fukusumi Y, Yamazaki M, et al. Angiotensin II type 1 receptor blockade ameliorates proteinuria in puromycin aminonucleoside nephropathy by inhibiting the reduction of NEPH1 and nephrin. J Nephrol 2014; 27: 627–634. [DOI] [PubMed] [Google Scholar]
- 19. Mundel P, Reiser J, Zuniga Mejia Borja A, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res 1997; 236: 248–258. [DOI] [PubMed] [Google Scholar]
- 20. Orikasa M, Matsui K, Oite T, et al. Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J Immunol 1988; 141: 807–814. [PubMed] [Google Scholar]
- 21. Kawachi H, Koike H, Kurihara H, et al. Cloning of rat nephrin: expression in developing glomeruli and in proteinuric states. Kidney Int 2000; 57: 1949–1961. [DOI] [PubMed] [Google Scholar]
- 22. Nakayama H, Oite T, Kawachi H, et al. Comparative nephritogenicity of two monoclonal antibodies that recognize different epitopes of rat Thy-1.1 molecule. Nephron 1998; 78: 453–463. [DOI] [PubMed] [Google Scholar]
- 23. Kawachi H, Koike H, Kurihara H, et al. Cloning of rat homologue of podocin: Expression in proteinuric states and in developing glomeruli. J Am S Nephrol 2003; 14: 46–56. [DOI] [PubMed] [Google Scholar]
- 24. Hidaka T, Suzuki Y, Yamashita M, et al. Amelioration of crescentic glomerulonephritis by RhoA kinase inhibitor, Fasudil, through podocyte protection and prevention of leukocyte migration. Am J Pathol 2008; 172: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol 2008; 294: F830–F839. [DOI] [PubMed] [Google Scholar]
- 26. Durvasula RV, Petermann AT, Hiromura K, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 2004; 65: 30–39. [DOI] [PubMed] [Google Scholar]
- 27. Matsusaka T, Asano T, Niimura F, et al. Angiotensin receptor blocker protection against podocyte-induced sclerosis is podocyte angiotensin II type 1 receptor-independent. Hypertension 2010; 55: 967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawachi H, Han GD, Miyauchi N, et al. Therapeutic targets in the podocyte: Findings in anti-slit diaphragm antibody-induced nephropathy. J Nephrol 2009; 22: 450–456. [PubMed] [Google Scholar]
- 29. Kawachi H, Kurihara H, Topham PS, et al. Slit diaphragm-reactive nephritogenic MAb 5–1–6 alters expression of ZO-1 in rat podocytes. Am J Physiol 1997; 273: F984–F993. [DOI] [PubMed] [Google Scholar]
- 30. Holthofer H, Ahola H, Solin ML, et al. Nephrin localizes at the podocyte filtration slit area and is characteristically spliced in the human kidney. Am J Pathol 1999; 155: 1681–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nitschke R, Henger A, Ricken S, et al. Angiotensin II increases the intracellular calcium activity in podocytes of the intact glomerulus. Kidney Int 2000; 57: 41–49. [DOI] [PubMed] [Google Scholar]
- 32. Charlesworth JA, Gracey DM, Pussell BA. Adult nephrotic syndrome: Non-specific strategies for treatment. Nephrology 2008; 13: 45–50. [DOI] [PubMed] [Google Scholar]