

Abstract
Monitoring the levels of therapeutic antibodies in individual patients would allow patient-specific dose optimization, with the potential for major therapeutic and financial benefits. Our group recently developed a new platform of bioluminescent sensor proteins (LUMABS; LUMinescent AntiBody Sensor) that allow antibody detection directly in blood plasma. In this study, we targeted four clinically important therapeutic antibodies, the Her2-receptor targeting trastuzumab, the anti-CD20 antibodies rituximab and obinutuzumab, and the EGFR-blocking cetuximab. A strong correlation was found between the affinity of the antibody binding peptide and sensor performance. LUMABS sensors with physiologically relevant affinities and decent sensor responses were obtained for trastuzumab and cetuximab using mimotope and meditope peptides, respectively, with affinities in the 10–7 M range. The lower affinity of the CD20-derived cyclic peptide employed in the anti-CD20 LUMABS sensor (Kd = 10–5 M), translated in a LUMABS sensor with a strongly attenuated sensor response. The trastuzumab and cetuximab sensors were further characterized with respect to binding kinetics and their performance in undiluted blood plasma. For both antibodies, LUMABS-based detection directly in plasma compared well to the analytical performance of commercial ELISA kits. Besides identifying important design parameters for the development of new LUMABS sensors, this work demonstrates the potential of the LUMABS platform for point-of-care detection of therapeutic antibodies.
Therapeutic antibodies represent an important class of newly introduced drugs and have been particularly successful in cancer therapy and the treatment of inflammatory diseases. At the end of 2016, nearly 60 antibody drugs had been FDA approved and many more are currently in clinical trials.1 Because of their relatively long serum half-life,2,3 therapeutic antibodies are given via intravenous injection at a time interval of several weeks. At present, monoclonal antibodies are either administered using a fixed dose or by providing a body-size adjusted dose. However, interpatient variabilities in distribution as well as clearance have been shown for various therapeutic antibodies.4,5 Several studies have shown a strong correlation between clearance rates and treatment efficacy, suggesting that patient-specific therapeutic drug monitoring (TDM) would allow better efficacy by preventing both over- and under-dosing.6,7
Thus, far, TDM has been mainly explored in the area of autoimmune disorders,8−10 but it is relatively new to oncology.11,12 Patient-specific adjustment of the dosing regime in autoimmune disorders is possible by measuring antibody trough values just before a patient receives a new injection, but in oncology individual pharmacokinetic profiles are ideally determined during treatment following the first dose of antibody. For this to be practical and economically sustainable, a sensitive and easy to perform point-of-care assay for the detection of therapeutic antibodies is needed. The standard of practice in antibody detection is the enzyme-linked immuno-sorbent assay (ELISA), which is highly valued for its sensitivity and specificity. However, ELISA and other immunoassays require multiple washing and waiting steps, which makes it difficult to adapt them to the POC setting.13 While both miniaturization of ELISAs into POC testing devices14,15 and the use of alternate detection systems such as lateral flow tests,16 electrochemical sensors,17 surface plasmon resonance (SPR),18 and mass spectrometry19 are actively pursued, these assays are either semiquantitative or still require sophisticated equipment.
Our group recently developed a new platform of bioluminescent sensor proteins (LUMABS; LUMinescent AntiBody Sensor) that allow antibody detection directly in blood plasma using the camera of a smart phone as the sole piece of equipment.20,21 LUMABS consist of the blue-light emitting luciferase NanoLuc22 connected via a semiflexible linker23,24 to a green fluorescent acceptor protein mNeonGreen,25 which in the absence of an antibody are kept in close proximity by an interaction between an Src Homology 3 (SH3) domain and a proline rich peptide (PRP).26 Binding of an antibody to epitope sequences flanking the linker disrupts the interaction between these helper domains, resulting in a large decrease in BRET efficiency. The resulting change in color of the emitted light from green-blue to blue can be detected directly in blood plasma, even at low pM concentrations of antibody.20 The LUMABS sensors developed thus far targeted antibodies for which linear peptide epitopes with a relatively high affinity were available. Here we report the development of LUMABS proteins targeting four clinically important therapeutic antibodies, the Her2-binding trastuzumab, the anti-CD20 antibodies rituximab and obinutuzumab, and the EGFR-blocking cetuximab. Since no simple linear epitope sequences of sufficient affinity were available for these antibodies, we explored the use of disulfide-linked cyclic epitopes, mimotopes, and meditopes.27 In addition to providing clinically relevant LUMABS sensors for cetuximab and trastuzumab, our results provide further insight into the thermodynamics of the LUMABS–antibody interaction, in particular the relation between the monovalent antibody affinity and the dynamic range of the sensor response.
Experimental Section
NB: Details on molecular cloning, protein expression and purification, peptide synthesis, fluorescence polarization, and ELISA can be found in the Supporting Information.
General Reagents
Therapeutic antibodies trastuzumab (Herceptin, Roche), rituximab, (MabThera, Roche), obinutuzumab (Gazyva, Roche), and cetuximab (Erbitux, Merck) were obtained via the Catherina hospital pharmacy in Eindhoven, The Netherlands. Antibody concentrations expressed in units of molarity were obtained by dividing the concentrations given by the manufacturer in mg mL–1 by the molecular weight of 150 000 g mol–1. Pooled human blood plasma was obtained from Innovative Research Inc. NanoGlo luciferase substrate was from Promega.
Bioluminescence Spectroscopy
Spectra were recorded in PBS pH 7.4 with 0.1% (w/v) BSA at 100 pM sensor concentration and a NanoGlo substrate dilution of 1000-fold on a Varian Cary Eclipse spectrophotometer in bioluminescence scan mode with a 5 nm emission slit. Spectra were smoothed by averaging over a moving 5 data-point (5 nm) interval. Background at 600 nm was subtracted and spectra were normalized at 450 nm.
LUMABS Microtiter Plate Assays
Antibody titrations were performed using 100 pM LUMABS protein in PBS with 0.1% (w/v) BSA in a total volume of 50 μL in PerkinElmer flat white 384 well OptiPlate microtiter plates and incubated for 2 h at room temperature. Two microliters of 40× prediluted NanoGlo (final dilution 1000×) was then added to each well, and the plate was incubated at room temperature for another 30 min. Luminescence was then recorded on a Tecan Infinite F500 plate reader using an exposure time of 1000 ms. Emission was recorded in two channels, blue (400–450 nm) and green (500–550 nm). eq 1 was fit through the data to obtain apparent Kd values.
| 1 |
ER is the emission ratio at antibody concentration [Ab], ERmin is the emission ratio at sensor saturation. ERmax is the emission ratio in the absence of antibody. Kd.app is the apparent dissociation constant. Dynamic range (DR) was calculated as the total change in emission ratio divided by the lowest ratio:
| 2 |
Measurements in plasma were done by serially diluting cetuximab in PBS pH 7.4 and subsequently diluting each concentration 10× in pooled human plasma. LUMABS protein was added to a final concentration of 1 nM (final plasma content 85%). After 2 h at room temperature, NanoGlo was added to a final dilution factor of 1000×. After another 30 min incubation step, wells were read out as described above.
Results
Thermodynamic Considerations
The modular architecture of the LUMABS sensor allows one to change antibody specificity by simple exchange of the antibody-binding epitope sequences, without the need for extensive sensor optimization for each new antibody target. The two most important sensor properties, the overall affinity for the target antibody and the change in BRET ratio between the closed state and the antibody-bound state, are independent of the nature of the molecular interactions but are determined by the monovalent affinity of the interaction between antigen-binding domain and the epitope, the strength of the helper domain interaction and two effective concentration terms that describe the intramolecular interaction between the helper domains, and the binding of the sensor to the second antigen binding domain following initial complex formation. A thermodynamic scheme can be derived that allows one to obtain equations describing the overall dissociation constant of the antibody-sensor interaction and to model the change in emission ratio as a function of these parameters (Figure 1A, Figure S1).
| 3 |
Figure 1.
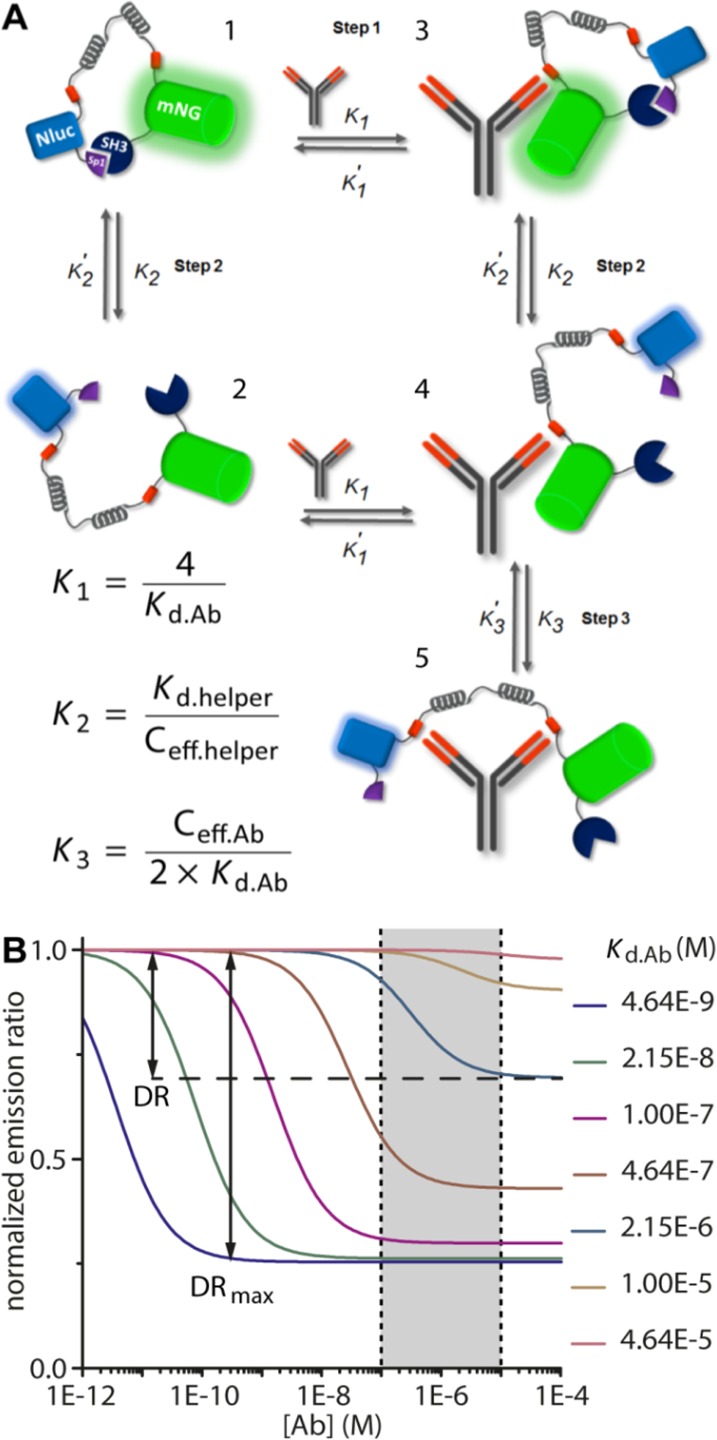
Thermodynamic model of LUMABS sensor mechanism. (A) Different states accessible to the sensor in the absence or presence of antibody. (B) Predicted response curves for sensors with epitopes of different Kd.Ab and a Ceff.helper/Ceff.Ab ratio of 10 (see Supporting Information for the exact parameters used). The gray area between dashed lines represents the global concentration range of therapeutic antibodies in patient serum. DR = dynamic range.
eq 3 shows that because of the bivalent interaction, the affinity of the antibody–sensor interaction depends very strongly on the monovalent antibody–epitope affinity, which in principle should increase the specificity of the sensor. The magnitude of this effect can be substantial, as we previously determined Kd values of 50–100 pM for the overall antibody–sensor interaction, using a monovalent peptide-antibody interaction of 40 nM.20 However, previous work also taught that when the monovalent interaction becomes too weak (Kd,Ab ≥ 10 μM), no ratiometric change is observed, even in the presence of very high antibody concentrations. The explanation for this observation is that even at high concentrations where the first antibody–sensor complex is formed (step 1), the intramolecular interaction between the second epitope and the second antigen binding site is not sufficient to compete (step 3) with the intramolecular interaction between the helper domains (step 2). Interestingly, therapeutic antibodies occur in patient serum at nanomolar to low micromolar concentrations. The thermodynamic model predicts that sensors with a Kd.app within this concentration range can be obtained using monovalent antibody-epitope interactions of moderate strength (Kd.Ab 0.1–10 μM), but the emission ratio change may be attenuated, because a certain percentage of sensors will remain in the closed state (Figure 1B) The first therapeutic antibody that we targeted, trastuzumab, demonstrates this fine balance between affinity and the sensor’s ratiometric response.
Sensor Development
Four clinically important therapeutic antibodies were chosen as suitable targets for LUMABS sensor development: the Her2-receptor targeting trastuzumab, the EGFR-blocking cetuximab, and the anti-CD20 antibodies rituximab and obinutuzumab. Each of these antibodies has been implicated as useful targets for TDM, and for each target, antibody-binding peptides have been reported with affinities in the high nM to low μM range. Trastuzumab is used in the treatment of breast cancers overexpressing the cell surface receptor Her2. Population pharmacokinetics indicate that 10% of patients display fast clearance of trastuzumab, resulting in drug levels below the minimally effective concentration.28 Cetuximab has been clinically approved to treat colorectal carcinoma,29 nonsmall cell lung carcinoma,30 and squamous cell head and neck carcinoma.31 Several studies indicate that a high clearance rate of cetuximab correlates with poor clinical outcome.6,7 CD20 is a cell surface receptor expressed on B-cells and is targeted by the anti-CD20 antibodies rituximab and obinutuzumab in the treatment of various B-cell malignancies as well as autoimmune disorders. Intersubject variability in pharmacokinetic rates of clearance are typically large, and higher serum levels have been shown to correlate with greater tumor shrinkage,32,33 thus underscoring the potential benefit of therapeutic drug monitoring.
Because trastuzumab recognizes a discontinuous conformational epitope, no linear epitopes were available that would bind with sufficient affinity to be used in a LUMABS sensor.34 Trastuzumab binding mimotope peptides have been obtained from phage display screening,35 but their affinities for trastuzumab were not reported. We therefore synthesized the fluorescently labeled QLGPYELWELSH mimotope peptide and used fluorescence polarization titration experiments to determine the affinity of the trastuzumab-mimotope interaction, yielding a Kd of 294 ± 10 nM (Figure S2, Table 1). Since the model predicted that this affinity is sufficient to support antibody-induced conformational switching in a LUMABS sensor, a synthetic DNA fragment containing this mimotope sequence at each end of the semiflexible linker was cloned into a LUMABS expression plasmid. The resulting TRAS-LUMABS-1 sensor protein was expressed in E. coli and successfully purified in good yield. The bioluminescence emission spectrum of TRAS-LUMABS-1 showed efficient BRET in the absence of antibody, with the intensity of the mNeonGreen emission peak at 517 nm slightly higher than the NanoLuc peak at 460 nm (Figure 2A). Addition of saturating amounts of trastuzumab resulted in a clear decrease in BRET, corresponding to a 1.5-fold change in emission ratio. A titration experiment in which the emission ratio was monitored as a function of trastuzumab concentration revealed a Kd.app of 303 ± 15 nM (Figure 2B, Table 1). TRAS-LUMABS variants with mimotope sequences that bound trastuzumab with affinities of 1.2 and 1.6 μM were still responsive, but showed smaller changes in emission ratio (Supplementary Figures S2–S4, Table 1). The latter is expected based on the thermodynamic model and shows that these weaker binding mimotope sequences do not allow the sensor to switch completely to the open state. While all sensors were responsive in the clinically relevant concentration regime between 10–7 and 10–5 M,28,36,37 TRAS-LUMABS-1 clearly showed the largest change in emission ratio, making this sensor the preferred choice to quantify trastuzumab levels in patient samples.
Table 1. Affinities and Dynamic Range of LUMABS Sensors.
| target antibody | sensor name | Ab binding elementa | monovalent Kd.Abb | sensor Kd.app | sensor DR |
|---|---|---|---|---|---|
| trastuzumab | TRAS-LUMABS-1 | QLGPYELWELSH | 0.29 ± 0.01 μM | 303 ± 15 nM | 59 ± 4% |
| TRAS-LUMABS-2 | LWGPYEWWELHH | 1.6 ± 0.3 μM | 0.6 ± 0.2 μM | 20 ± 1% | |
| TRAS-LUMABS-3 | LWGPYEWWEFHH | 1.2 ± 0.2 μM | 0.16 ± 0.05 μM | 31 ± 2% | |
| obinutuzumab | CD20-LUMABS-1 | YNCEPANPSEKNSPSTQYCYSI | 7 μMd | 0.54 ± 0.13 nM | 23 ± 1% |
| CD20-LUMABS-2 | YNCAPATPSEKNSPSTQYCYSI | N.D. | 0.23 ± 0.06 nM | 26 ± 1% | |
| rituximab | CD20-LUMABS-1 | YNCEPANPSEKNSPSTQYCYSI | 28 μMd | 3.9 ± 0.7 μM | 22 ± 1% |
| CD20-LUMABS-2 | YNCAPATPSEKNSPSTQYCYSI | N.D. | N.B. | N.B. | |
| cetuximab | CTX-LUMABS-1 | CQFDLSTRRLKC | 0.27 ± 0.01 μMc | 376 ± 18 nM | 47.3 ± 0.6% |
| CTX-LUMABS-2 | CVFDLGTRRLRC | 61 ± 2 nMc | 55 ± 3 nM | 60.0 ± 0.8% | |
| CTX-LUMABS-3 | AVFDLGTRRLRA | N.D. | N.B. | N.B. |
Epitope, mimotope, or meditope as described in the text. For exact sequences of the proteins used in this study, refer to the Supporting Information.
Per binding site.
See ref43
See ref (30). DR dynamic range. N.D. not determined. N.B. no binding.
Figure 2.

Characterization of LUMABS assays for the detection of trastuzumab and cetuximab. (A,C,E) Luminescence emission spectra of 100 pM TRAS-LUMABS-1 (A) CTX-LUMABS-1 (C) or CTX-LUMABS-2 (E) with or without the indicated amount of the target antibody and normalized at 460 nm. (B,D,F) Antibody titrations to 100 pM of TRAS-LUMABS-1 (B) CTX-LUMABS-1 (D) or CTX-LUMABS-2 (F). Data represent mean ± SD from triplicate measurements.
Because cetuximab binds a conformational epitope on EGFR, linear epitope sequences are also not available for cetuximab.38 Cyclic mimotope sequences have been reported for cetuximab, but these cyclic peptides are relatively long (20 aa), and their interaction strength has not been well characterized.39 Therefore, we here tested whether we could instead use a so-called meditope peptide. Meditopes are peptides that specifically recognize an antibody by binding in a cavity at the interface between the constant and variable domains (Figure S5). Donaldson et al. reported the crystal structure of a cetuximab Fab fragment complexed with a 10 aa disulfide cyclized meditope peptide (CQFDLSTRRLKC, hereafter called meditope 1) that binds with a Kd of 260 nM.27 In a recent study using deep mutational scanning, we identified three mutations that together increased the affinity of the meditope peptide for cetuximab further to 60 nM (CVFDLGTRRLRC, meditope 2).40 Two LUMABS variants were cloned and expressed that contained either meditope 1 or meditope 2. To assess the importance of peptide cyclization, we also included a third variant, (CTX-LUMABS-3) that contains a linear version of meditope 2 in which the cysteines were replaced by alanines (AVFDLGTRRLRA, meditope 3). In the absence of cetuximab, the bioluminescence emission spectra of all three sensor variants were the same and similar to those observed for the other LUMABS sensors in the absence of antibody. Addition of cetuximab induced a robust decrease in emission ratio of 47 ± 0.6% and 60 ± 0.8% for CTX-LUMABS-1 and -2, respectively (Figure 2C,E), while no change in emission ratio was observed for CTX-LUMABS-3 up to 1 μM cetuximab (Supplementary Figure S6). These results are consistent with previous observations that disulfide-cyclization is required for high affinity binding to cetuximab41 and thus imply the correct formation of these disulfide bonds in CTX-LUMABS-1 and -2. Correct disulfide bond formation was also confirmed by ESI-MS, which showed a 4 Da increase in molecular weight for both CTX-LUMABS-1 and -2 following treatment with dithiothreitol (Supplementary Figure S7). Cetuximab titrations revealed apparent Kd values of 380 ± 20 nM and 55 ± 3 nM for CTX-LUMABS-1 and -2, respectively (Figure 2D,F, Table 1). While the 7-fold difference in cetuximab affinity is somewhat smaller than anticipated based on the thermodynamic model, these results still demonstrate the ability to tune the affinity of the LUMABS sensors in a predictable way. Importantly, the responsive concentration regime of these two sensors together covers the range of cetuximab concentrations that have been reported in patient serum (25 nM–2.3 μM.)6,7,42,43 The CTX-LUMABS sensors do not target the antigen binding domains of cetuximab, but rather, they bind a cavity inside the cetuximab Fab fragment. Although structural analysis suggests that the chimeric nature of cetuximab renders this binding site significantly different from the pocket present in other antibodies, we nonetheless tested the response of CTX-LUMABS-1 and CTX-LUMABS-2 against two humanized therapeutic antibodies, (trastuzumab and obinutuzumab) and two chimeric ones, rituximab and infliximab (Supplementary Figure S8). Neither sensor showed any response to micromolar concentrations of any of these four antibodies. We also tested binding to a mix of IgG from human serum and found no unspecific binding (Supplementary Figure S9).
The anti-CD20 antibodies obinutuzumab and rituximab recognize different, but overlapping epitopes within the second extracellular loop of CD20 (sequence 165-YNCEPANPSEKNSPSTQYCYSI-186).44−46 Previous binding studies reported affinities of 7 and 28 μM for binding of this disulfide-cyclized peptide to obinutuzumab and rituximab, respectively.46 Although these affinities were likely to be too low, we nonetheless constructed two LUMABS proteins, one incorporating the original cyclic CD20 epitope sequence (CD20-LUMABS-1), and one containing the same cyclic peptide with two mutations (E168A and N171T) that were reported to enhance the affinity for obinutuzumab but completely abrogated rituximab binding (CD20-LUMABS-2).46 Titration experiments with rituximab showed either a small response and weak binding (CD20-LUMABS-1), or no response at all (CD20-LUMABS-2) (Supplementary Figure S10). Addition of obinutuzumab induced a modest 20% change in emission ratio for both sensors, with Kd.app-values of 0.5 ± 0.1 μM and 0.23 ± 0.06 μM for CD20-LUMABS-1 and -2, respectively (Table 1 and Supplementary Figure S11). While the latter results show that relatively large disulfide-cyclized peptide epitopes can be successfully incorporated in LUMABS sensors, it is clear that epitopes with higher affinity are required to obtain CD20-LUMABS sensors with a more robust change in emission ratio. Development of a higher affinity sensor was attempted by incorporating the entire extracellular loop as an epitope, because the affinity of both antibodies to cells expressing full-length CD20 is in the nanomolar range.46 Unfortunately, this construct suffered from proteolytic degradation during expression.
TRAS-LUMABS and CTX-LUMABS Allow Direct Antibody Quantification in Blood Plasma
Because LUMABS sensors with physiologically relevant affinities and a sufficient change in emission ratio were obtained for both trastuzumab (TRAS-LUMBAS-1) and cetuximab (CTX-LUMABS-1 and -2), we next assessed the potential of these LUMABS sensors to allow direct antibody quantification in blood plasma. An important consideration for use in a point-of-care setting is whether the response of the sensor is fast enough to allow accurate quantification within a reasonable time frame. The kinetics of the sensors’ responses were determined for different antibody concentrations (Figure 3). Addition of trastuzumab to TRAS-LUMABS-1 induced a rapid response, showing essentially complete equilibration within minutes for physiologically relevant antibody concentrations. A similar rapid equilibration was observed for CTX-LUMABS-1, but the response of CTX-LUMABS-2 was considerably slower than that of CTX-LUMABS-1. Because all sensors have the same helper interaction, these clear differences in kinetics show that the dissociation of the helper interaction cannot be rate-limiting. Most likely, at low antibody concentrations, the kinetics of equilibration are dominated by the dissociation rate of the mimotope/meditope peptide-antibody interaction, which is expected to be lower for the higher affinity interaction in CTX-LUMABS-2. Based on its faster kinetics, CTX-LUMABS-1 would be the preferred sensor for a point-of-care application.
Figure 3.
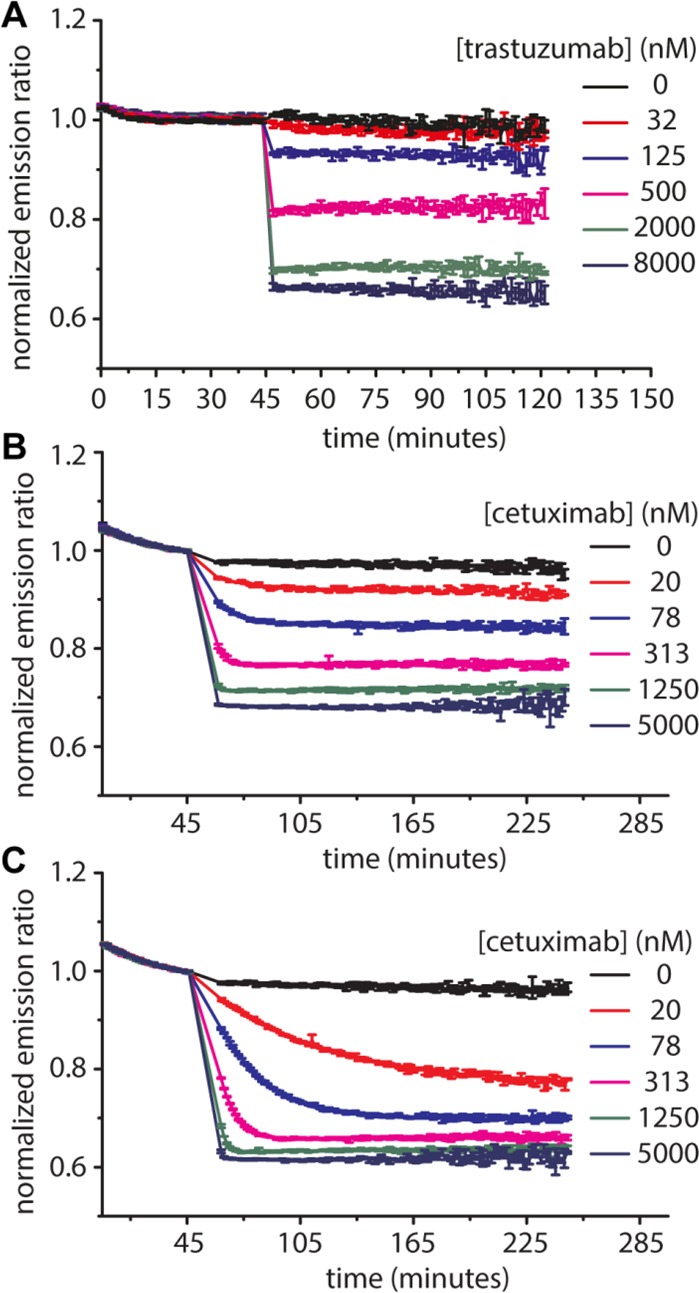
Kinetics of TRAS-LUMABS and CTX-LUMABS sensors. Green/blue luminescence emission ratio was monitored at 100 pM of TRAS-LUMABS-1 (A), CTX-LUMABS-1 (B) or CTX-LUMABS-2 (C) in 50 μL of luminescence buffer and a NanoGlo substrate dilution of 1000×. Error bars represent mean ± SD of triplicate measurements. Data were normalized to the average ratio of the last 10 data before antibody addition.
Because the LUMABS sensors are intended to be used directly in patient plasma, we next assessed their performance in pooled human plasma spiked with different concentrations of trastuzumab and cetuximab. Because some of the emitted light is reabsorbed in plasma, the absolute intensity ratios were different between buffer and plasma, but the relative response was comparable (41.0 ± 1.2%, 47.2 ± 0.6% and 57.6 ± 1.9% for TRAS-LUMABS-1, CTX-LUMABS-1 and -2, respectively). The apparent affinity of TRAS-LUMABS-1 was found to be 10-fold lower in blood plasma (Kd.app = 3.61 ± 0.21 μM), whereas the apparent affinities of CTX-LUMABS-1 and -2 were found to be slightly increased, yielding Kd.app values of 100 ± 5 nM and 34 ± 4 nM for CTX-LUMABS-1 and -2, respectively (Figure 4A,B). These results show that the performance of both sensors in plasma was comparable to that in buffer, although a calibration curve should be obtained in plasma since the Kd values and absolute emission ratios cannot be directly compared between buffer and plasma.
Figure 4.

Performance of TRAS-LUMABS and CTX-LUMABS assays in blood plasma. (A,B) Calibration of TRAS-LUMABS-1 (A) and CTX-LUMABS-1 and -2 (B) in 90% plasma. Error bars represent mean ± SD from three measurements of the same calibration sample. (C,D) Correlation of antibody concentrations measured by ELISA and by TRAS-LUMABS-1 (C) or CTX-LUMABS-1 and -2 (D). Concentrations were calculated from each measurement using the calibration curve. Error bars represent mean ± SD of these concentrations.
Finally, we compared the analytical performance of the TRAS-LUMABS and CTX-LUMABS sensors with commercially available ELISAs that are currently used to quantify trastuzumab and cetuximab concentrations in clinical samples. Test samples were prepared in human blood plasma at four different trastuzumab concentrations, representing typical peak measurements (2.3 μM), two levels corresponding to slow- and fast-clearance patients (1.38 μM and 500 nM), and a low trough level (138 nM). The trastuzumab concentrations in the test samples were determined by ELISA after appropriate dilution, resulting in an average deviation from the spiked concentrations of 22%. The deviation was highest for the 138 nM sample (38%). In parallel, test samples were directly measured by TRAS-LUMABS-1 without dilution by comparing the measured emission ratios with the sigmoidal fit for the calibration curve obtained in Figure 4A. The concentrations obtained deviated from the spiked concentrations by 8% on average, with the highest deviation of 13% for the 500 nM sample. A good correlation was observed between the ELISA and Tras-LUMABS-1, R2 = 0.9959, p < 0.003 (Figure 4C). Similarly, test samples were simulated by spiking pooled human blood plasma with four different cetuximab concentrations, representing typical peak measurements (2.3 μM), trough concentrations from patients with relatively slow clearance (500 nM), trough levels from fast-clearance patients (200 nM) and the very low trough levels (12 nM) found when patients must forego a dose.42,43,47,48 Determination of the cetuximab concentrations in the test samples using the commercial ELISA was again done after proper dilution, yielding an average deviation from the true concentration of 14% (Supplementary Figure S13). No measurement deviated by more than 20% from the spiked concentration. In parallel, cetuximab concentrations were determined directly in the spiked plasma samples with CTX-LUMABS-1 and -2 in a microtiter well plate assay by comparing the measured emission ratios with the sigmoidal fit for the calibration curve obtained in Figure 4B. On average, the concentrations obtained using CTX-LUMABS-1 deviated from the spiked concentrations by 9% (Figure 4D). The deviation was highest for the 2.3 μM sample (24%), which is probably due to the fact that the sigmoidal curve levels off at the high concentration end. For CTX-LUMABS-2, the average deviation from the spiked concentrations was 21%. This higher deviation is due to the insensitivity of CTX-LUMABS-2 in the high concentration regime, as reflected by the high deviations observed for the 500 nM sample (deviation 24%) and particularly the 2.3 μM sample (49%). As a result, a very good correlation was observed between the ELISA and CTX-LUMABS-1, R2 = 0.9998, p < 0.001 and a slightly lower correlation between the ELISA and CTX-LUMABS-2, R2 = 0.985 p < 0.05 (Figure 4D).
Discussion and Conclusions
In this work, we successfully demonstrated the development of dual color bioluminescent sensor proteins that allow homogeneous detection of two well-known therapeutic antibodies directly in solution. LUMABS sensors with physiologically relevant affinities and a robust response were obtained for trastuzumab and cetuximab, whereas a relatively weak antibody-epitope interaction yielded suboptimal LUMABS variants for the anti-CD20 antibodies obinutuzumab and rituximab. The strength of the monovalent antibody-peptide interaction strongly correlated with the relative change in bioluminescent emission ratio observed upon antibody binding, which can be understood using a thermodynamic model that describes the binding of the sensor to the antibody in three different steps. Further characterization of the trastuzumab and cetuximab LUMABS sensors in buffer showed excellent antibody selectivity and sufficiently rapid equilibration. Importantly, because the signal is ratiometric and based on bioluminescence, both LUMABS sensors could be used directly in blood plasma, where their analytical performance compared well to that obtained using a classical ELISA.
The thermodynamic model introduced in this work can be used to guide the development of LUMABS for other therapeutic antibodies. Our results showed a good correlation between the affinity of the antibodies for the fluorescently labeled peptides determined in fluorescence anisotropy titration experiment and the properties of the sensors in which these peptides were genetically encoded. With decreasing monovalent affinity (higher Kd.Ab) the apparent sensor Kd shifted toward higher concentrations, whereas the difference in emission ratio was attenuated. Of course, the model predictions are not absolute as antibody binding may also be affected by interactions with surrounding residues, such as the label in the fluorescent peptides or interactions with flanking amino acids in the sensor. Nonetheless, our work shows that, as a rule of thumb, the antigen–antibody interaction should preferably have a Kd of 1 μM or lower.
The most obvious strategy to improve the dynamic response of LUMABS sensors is to increase the affinity of the antibody epitope, mimotope, or meditope interaction. The example of the anti-CD20 antibodies shows that this can be challenging as many therapeutic antibodies recognize complex discontinuous epitopes whose structural features may not be well preserved in peptide analogues. The successful introduction of disulfide constrained peptides, suggests that it may be worth to explore other structurally constrained epitopes such small protein domains or bicyclic peptides, which afford high affinity and selectivity and can be screened using phage display.49 An alternative strategy to improve the dynamic response of LUMABS sensors may be to adjust the ratio of Ceff.helper/Ceff.Ab. On the basis of earlier work, we estimated the effective concentration for formation of the enzyme–inhibitor complex (Ceff.EI, equivalent to Ceff.helper in this work) to be 10-fold higher than the effective concentration for formation of the intramolecular antibody-peptide interaction (Ceff.Ab).50 However, Ceff.Ab may also depend on the orientation of antibody–peptide interaction and differences in conformational preferences between different antibody classes, as determined by the flexibility of the hinge region.51,52 Modeling the relationship between the dynamic range and Kd.app for different values of Ceff.helper/Ceff.Ab shows that the concentration where the dynamic range starts to decrease from its maximum is higher for lower Ceff.helper/Ceff.Ab (Supplementary Figure S1). One way to decrease this ratio would be to make the linker stiffer, which should results in an increase in Ceff.Ab.
The trastuzumab and cetuximab LUMABS sensors developed in this work are attractive candidates for the development of a low cost point-of-care assay for TDM applications. Because bioluminescence does not require external excitation, LUMABS detection does not suffer from background signals or light scattering, making detection in complex matrices relatively straightforward. The bright and stable luminescence produced by NanoLuc allows sensitive detection both in heterogeneous and homogeneous assay formats, as demonstrated by several recent applications including the use of antibody-NanoLuc fusions in the heterogeneous detection of antidrug antibodies and the high-throughput screening of antibody libraries.53,54 Moreover, the two-color, ratiometric detection renders LUMABS-based detection independent of sensor concentration and much less sensitive to matrix effects that affect the absolute signal intensity such as temperature, pH, substrate concentration, and product inhibition. Nonetheless, in the next step, it will be important to demonstrate that these principle advantages also hold-up when measuring antibody concentrations in patient samples. Even though we have previously shown that the LUMABS signal is readily detected using as a standard smart phone camera, reabsorption of light by components in blood plasma can affect the absolute ratio. Because individual patient samples can vary considerably in absorbance, we are currently exploring to combine the LUMABS sensors with paper-based detection devices, where the short path length that the light travels through the sample will effectively suppress light reabsorption.55
Acknowledgments
This work was supported by an ERC starting grant (ERC-2011-Stg 280255) and ERC proof of concept grants (ERC-2013-PoC 632274 and ERC-2016-PoC 755471).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b00041.
Supplementary experimental procedures, a detailed description of the therapeutic model, supplementary Figures S1–S13, and the sequences of proteins used in this study (PDF)
Author Present Address
† M.v.R: Merus N.V., Yalelaan 62, 3584 CM Utrecht, The Netherlands
Author Present Address
†† S.K.J.L.: Janssen Vaccines and Prevention B.V., Archimedesweg 4–6, 2301 CA Leiden, The Netherlands
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Rodgers K. R.; Chou R. C. Biotechnol. Adv. 2016, 34, 1149–1158. 10.1016/j.biotechadv.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Batra S. K.; Jain M.; Wittel U. A.; Chauhan S. C.; Colcher D. Curr. Opin. Biotechnol. 2002, 13, 603–608. 10.1016/S0958-1669(02)00352-X. [DOI] [PubMed] [Google Scholar]
- Roskos L. K.; Davis C. G.; Schwab G. M. Drug Dev. Res. 2004, 61, 108–120. 10.1002/ddr.10346. [DOI] [Google Scholar]
- Ma P.; Yang B.-B.; Wang Y.-M.; Peterson M.; Narayanan A.; Sutjandra L.; Rodriguez R.; Chow A. J. Clin. Pharmacol. 2009, 49, 1142–1156. 10.1177/0091270009344989. [DOI] [PubMed] [Google Scholar]
- Gill K. L.; Machavaram K. K.; Rose R. H.; Chetty M. Clin. Pharmacokinet. 2016, 55, 789–805. 10.1007/s40262-015-0361-4. [DOI] [PubMed] [Google Scholar]
- Azzopardi N.; Lecomte T.; Ternant D.; Boisdron-Celle M.; Piller F.; Morel A.; Gouilleux-Gruart V.; Vignault-Desvignes C.; Watier H.; Gamelin E.; Paintaud G. Clin. Cancer Res. 2011, 17, 6329–6337. 10.1158/1078-0432.CCR-11-1081. [DOI] [PubMed] [Google Scholar]
- Pointreau Y.; Azzopardi N.; Ternant D.; Calais G.; Paintaud G. Ther. Drug Monit. 2017, 38, 567–572. 10.1097/FTD.0000000000000321. [DOI] [PubMed] [Google Scholar]
- Bendtzen K. Front. Immunol. 2015, 6, 109. 10.3389/fimmu.2015.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Ferreira F.; Afonso J.; Pinto-Lopes P.; Magro F. Inflamm. Bowel Dis. 2016, 22, 2289–2301. 10.1097/MIB.0000000000000855. [DOI] [PubMed] [Google Scholar]
- Schmitz E. M. H.; Van De Kerkhof D.; Hamann D.; Van Dongen J. L. J.; Kuijper P. H. M.; Brunsveld L.; Scharnhorst V.; Broeren M. A. C. Clin. Chem. Lab. Med. 2016, 54, 1211–1219. 10.1515/cclm-2015-0987. [DOI] [PubMed] [Google Scholar]
- Widmer N.; Bardin C.; Chatelut E.; Paci A.; Beijnen J.; Levêque D.; Veal G.; Astier A. Eur. J. Cancer 2014, 50, 2020–2036. 10.1016/j.ejca.2014.04.015. [DOI] [PubMed] [Google Scholar]
- Oude Munnink T.; Henstra M. J.; Segerink L. I.; Movig K. L. L.; Brummelhuis-Visser P. Clin. Pharmacol. Ther. 2016, 99, 419–431. 10.1002/cpt.211. [DOI] [PubMed] [Google Scholar]
- Gubala V.; Harris L. F.; Ricco A. J.; Tan M. X.; Williams D. E. Anal. Chem. 2012, 84, 487–515. 10.1021/ac2030199. [DOI] [PubMed] [Google Scholar]
- Ludwig S. K. J.; Tokarski C.; Lang S. N.; van Ginkel L. A.; Zhu H.; Ozcan A.; Nielen M. W. F. PLoS One 2015, 10, e0134360. 10.1371/journal.pone.0134360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadimisetty K.; Malla S.; Sardesai N. P.; Joshi A. A.; Faria R. C.; Lee N. H.; Rusling J. F. Anal. Chem. 2015, 87, 4472–4478. 10.1021/acs.analchem.5b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Mehta S.; Erickson D. Anal. Chem. 2016, 88, 8359–8363. 10.1021/acs.analchem.6b01828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R. J.; Kallewaard H. M.; Hsieh W.; Patterson A. S.; Kasehagen J. B.; Cash K. J.; Uzawa T.; Soh H. T.; Plaxco K. W. Anal. Chem. 2012, 84, 1098–1103. 10.1021/ac202757c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Van Stappen T.; Spasic D.; Delport F.; Vermeire S.; Gils A.; Lammertyn J. Biosens. Bioelectron. 2016, 79, 173–179. 10.1016/j.bios.2015.11.087. [DOI] [PubMed] [Google Scholar]
- Russo R.; Rega C.; Caporale A.; Tonon G.; Scaramuzza S.; Selis F.; Ruvo M.; Chambery A. Rapid Commun. Mass Spectrom. 2017, 31, 1184–1192. 10.1002/rcm.7898. [DOI] [PubMed] [Google Scholar]
- Arts R.; den Hartog I.; Zijlema S. E.; Thijssen V.; van der Beelen S. H. E.; Merkx M. Anal. Chem. 2016, 88, 4525–4532. 10.1021/acs.analchem.6b00534. [DOI] [PubMed] [Google Scholar]
- Arts R.; Ludwig S. K. J.; van Gerven B. C. B.; Estirado E. M.; Milroy L.-G.; Merkx M. ACS Sens. 2017, 2, 1730–1736. 10.1021/acssensors.7b00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; MacHleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. ACS Chem. Biol. 2012, 7, 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amet N.; Lee H.-F.; Shen W.-C. Pharm. Res. 2009, 26, 523–528. 10.1007/s11095-008-9767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golynskiy M. V.; Rurup W. F.; Merkx M. ChemBioChem 2010, 11, 2264–2267. 10.1002/cbic.201000143. [DOI] [PubMed] [Google Scholar]
- Shaner N. C.; Lambert G. G.; Chammas A.; Ni Y.; Cranfill P. J.; Baird M. A.; Sell B. R.; Allen J. R.; Day R. N.; Israelsson M.; Davidson M. W.; Wang J. Nat. Methods 2013, 10, 407–409. 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünberg R.; Burnier J. V.; Ferrar T.; Beltran-Sastre V.; Stricher F.; Van Der Sloot A. M.; Garcia-Olivas R.; Mallabiabarrena A.; Sanjuan X.; Zimmermann T.; Serrano L. Nat. Methods 2013, 10, 1021–1027. 10.1038/nmeth.2625. [DOI] [PubMed] [Google Scholar]
- Donaldson J. M.; Zer C.; Avery K. N.; Bzymek K. P.; Horne D. A.; Williams J. C. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17456–17461. 10.1073/pnas.1307309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno R.; Washington C. B.; Lu J.-F.; Lieberman G.; Banken L.; Klein P. Cancer Chemother. Pharmacol. 2005, 56, 361–369. 10.1007/s00280-005-1026-z. [DOI] [PubMed] [Google Scholar]
- Geng F.; Wang Z.; Yin H.; Yu J.; Cao B. Cancer Biother.Radiopharm. 2017, 32 (5), 149–160. 10.1089/cbr.2017.2210. [DOI] [PubMed] [Google Scholar]
- Silva A. P.; Coelho P. V.; Anazetti M.; Simioni P. U. Hum. Vaccines Immunother. 2017, 13, 843–853. 10.1080/21645515.2016.1249551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira J.; Tobias A.; O’Brien M. P.; Agulnik M. Drugs 2017, 77, 843–857. 10.1007/s40265-017-0734-0. [DOI] [PubMed] [Google Scholar]
- Berinstein N. L.; Grillo-López A. J.; White C. A.; Bence-Bruckler I.; Maloney D.; Czuczman M.; Green D.; Rosenberg J.; McLaughlin P.; Shen D. Ann. Oncol. 1998, 9, 995–1001. 10.1023/A:1008416911099. [DOI] [PubMed] [Google Scholar]
- Cartron G.; Hourcade-Potelleret F.; Morschhauser F.; Salles G.; Wenger M.; Truppel-Hartmann A.; Carlile D. J. Haematologica 2016, 101, 226–234. 10.3324/haematol.2015.133421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.-S.; Mason K.; Ramyar K. X.; Stanley A. M.; Gabelli S. B.; Denney D. W. Jr.; Leahy D. J. Nature 2003, 421, 756–760. 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- Jiang B.; Liu W.; Qu H.; Meng L.; Song S.; Ouyang T.; Shou C. J. Biol. Chem. 2005, 280, 4656–4662. 10.1074/jbc.M411047200. [DOI] [PubMed] [Google Scholar]
- Leyland-Jones B.; Colomer R.; Trudeau M. E.; Wardley A.; Latreille J.; Cameron D.; Cubedo R.; Al-Sakaff N.; Feyereislova A.; Catalani O.; Fukushima Y.; Brewster M.; Cortés J. J. Clin. Oncol. 2010, 28, 960–966. 10.1200/JCO.2009.23.1910. [DOI] [PubMed] [Google Scholar]
- Leyland-Jones B.; Gelmon K.; Ayoub J.-P.; Arnold A.; Verma S.; Dias R.; Ghahramani P. J. Clin. Oncol. 2003, 21, 3965–3971. 10.1200/JCO.2003.12.109. [DOI] [PubMed] [Google Scholar]
- Li S.; Schmitz K. R.; Jeffrey P. D.; Wiltzius J. J. W.; Kussie P.; Ferguson K. M. Cancer Cell 2005, 7, 301–311. 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Desnoyers L. R.; Vasiljeva O.; Richardson J. H.; Yang A.; Menendez E. E. M.; Liang T. W.; Wong C.; Bessette P. H.; Kamath K.; Moore S. J.; Sagert J. G.; Hostetter D. R.; Han F.; Gee J.; Flandez J.; Markham K.; Nguyen M.; Krimm M.; Wong K. R.; Liu S.; Daugherty P. S.; West J. W.; Lowman H. B. Sci. Transl. Med. 2013, 5, 207ra144. 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- van Rosmalen M.; Janssen B. M. G.; Hendrikse N. M.; van der Linden A. J.; Pieters P. A.; Wanders D.; de Greef T. F. A.; Merkx M. J. Biol. Chem. 2017, 292, 1477–1489. 10.1074/jbc.M116.764225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek K. P.; Ma Y.; Avery K. A.; Horne D. A.; Williams J. C. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 434–442. 10.1107/S2053230X16007202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fracasso P. M.; Burris H.; Arquette M. A.; Govindan R.; Gao F.; Wright L. P.; Goodner S. A.; Greco F. A.; Jones S. F.; Willcut N.; Chodkiewicz C.; Pathak A.; Springett G. M.; Simon G. R.; Sullivan D. M.; Marcelpoil R.; Mayfield S. D.; Mauro D.; Garrett C. R. Clin. Cancer Res. 2007, 13, 986–993. 10.1158/1078-0432.CCR-06-1542. [DOI] [PubMed] [Google Scholar]
- Robert F.; Ezekiel M. P.; Spencer S. A.; Meredith R. F.; Bonner J. A.; Khazaeli M. B.; Saleh M. N.; Carey D.; Lobuglio A. F.; Wheeler R. H.; Cooper M. R.; Waksal H. W. J. Clin. Oncol. 2001, 19, 3234–3243. 10.1200/JCO.2001.19.13.3234. [DOI] [PubMed] [Google Scholar]
- Teeling J. L.; Mackus W. J. M.; Wiegman L. J. J. M.; van den Brakel J. H. N.; Beers S. A.; French R. R.; van Meerten T.; Ebeling S.; Vink T.; Slootstra J. W.; Parren P. W. H. I.; Glennie M. J.; van de Winkel J. G. J. J. Immunol. 2006, 177, 362–371. 10.4049/jimmunol.177.1.362. [DOI] [PubMed] [Google Scholar]
- Klein C.; Lammens A.; Schäfer W.; Georges G.; Schwaiger M.; Mössner E.; Hopfner K.-P.; Umaña P.; Niederfellner G. mAbs 2013, 5, 22–33. 10.4161/mabs.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederfellner G.; Lammens A.; Mundigl O.; Georges G. J.; Schaefer W.; Schwaiger M.; Franke A.; Wiechmann K.; Jenewein S.; Slootstra J. W.; Timmerman P.; Brännström A.; Lindstrom F.; Mössner E.; Umana P.; Hopfner K.-P.; Klein C. Blood 2011, 118, 358–367. 10.1182/blood-2010-09-305847. [DOI] [PubMed] [Google Scholar]
- Baselga J.; Pfister D.; Cooper M. R.; Cohen R.; Burtness B.; Bos M.; D’Andrea G.; Seidman A.; Norton L.; Gunnett K.; Falcey J.; Anderson V.; Waksal H.; Mendelsohn J. J. Clin. Oncol. 2000, 18, 904–914. 10.1200/JCO.2000.18.4.904. [DOI] [PubMed] [Google Scholar]
- Wang C.; He X.; Zhou B.; Li J.; Li B.; Qian W.; Hou S.; Wang H.; Shi Y.; Guo Y. mAbs 2011, 3, 67–75. 10.4161/mabs.3.1.14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentero Rebollo I.; Heinis C. Methods 2013, 60, 46–54. 10.1016/j.ymeth.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Banala S.; Aper S. J. A.; Schalk W.; Merkx M. ACS Chem. Biol. 2013, 8, 2127–2132. 10.1021/cb400406x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphire E. O.; Parren P. W. H. I.; Pantophlet R.; Zwick M. B.; Morris G. M.; Rudd P. M.; Dwek R. A.; Stanfield R. L.; Burton D. R.; Wilson I. A. Science 2001, 293, 1155. 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- Rayner L. E.; Hui G. K.; Gor J.; Heenan R. K.; Dalby P. A.; Perkins S. J. J. Biol. Chem. 2015, 290, 8420–8438. 10.1074/jbc.M114.631002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N.; Flemming R.; Godat B.; Urh M. J. Immunol. Methods 2017, 450, 17–26. 10.1016/j.jim.2017.07.006. [DOI] [PubMed] [Google Scholar]
- Boute N.; Lowe P.; Berger S.; Malissard M.; Robert A.; Tesar M. Front. Pharmacol. 2016, 7, 27. 10.3389/fphar.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss R.; Schena A.; Reymond L.; Patiny L.; Werner D.; Tinberg C. E.; Baker D.; Johnsson K. Nat. Chem. Biol. 2014, 10, 598–603. 10.1038/nchembio.1554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.