

Abstract
To evaluate the shared genetic etiology of type-2 diabetes (T2D) and coronary heart disease (CHD), we conducted a multi-ethnic study of genetic variation genome-wide for both diseases in up to 265,678 subjects for T2D and 260,365 subjects for CHD. We identify 16 previously unreported loci for T2D and one for CHD, including a novel T2D association at a missense variant in HLA-DRB5 (OR=1.29). We show that genetically mediated increase in T2D risk also confers higher CHD risk. Joint analysis of T2D loci demonstrated that 24% are associated with CHD, highlighting eight variants - two of which are coding - where T2D and CHD associations appear to co-localize, and a novel joint T2D/CHD association which also replicated for T2D. Variants associated with both outcomes implicate several novel pathways including cellular proliferation and cardiovascular development.
Introduction
The global epidemic of type 2 diabetes mellitus (T2D) is expected to worsen over the coming decades, and the number of people with T2D is projected to reach ~592 million by 20351. T2D is also a major vascular risk factor for coronary heart disease (CHD) which is the leading cause of death worldwide2. Patients with T2D are also at a twofold higher risk of mortality due to CHD compared to those that do not have T2D3, though the mechanisms that link T2D with increased risk of CHD remain inadequately understood. Recently, a coding variant in the gene encoding the glucagon-like peptide-1 receptor (GLP1R) was reported4 that was associated with lower fasting glucose, lower T2D susceptibility, and modestly with reduced risk for CHD, a result consistent with existing therapeutic perturbation of this gene. This type of result is intriguing, and motivates the search for additional loci with this type of genetic support: association with protective effects for both T2D and CHD in humans. Such targets would merit detailed molecular, functional, and therapeutic experimentation, but we need first to identify these candidate loci from existing and newly generated data sets.
Genome-wide association studies (GWAS) have advanced our understanding of the genetic architecture individually for each disease, yielding discovery of several dozen loci for T2D and CHD5,6. Previous work has also demonstrated a genetic correlation between both endpoints7,8, though no study has directly compared individual variants beyond established sites across the genome nor examined the pathways that are shared between the two outcomes. Regional association for multiple SNPs for both endpoints at a locus has been observed (e.g., CDKN2A/2B or APOE)5,6. These initial observations indicate that the genetic pathways that connect T2D and CHD may have a modest impact on disease risk, hence requiring large sample sizes to enable robust discovery.
We therefore assembled a discovery association set for T2D comprising of 73,337 T2D cases and 192,341 controls to first enable discovery of novel loci for T2D. Second, we used additional genetic data on 90,831 CHD cases and 169,534 controls to identify genetic pathways connected with both outcomes.
Results
Genome-wide association and replication testing for T2D
We used genetic data from 48,437 individuals (13,525 T2D cases and 34,912 controls) of South Asian (n = 28,139; 9,654 T2D cases and 18,485 controls) and European (n = 20,298; 3,871 T2D cases and 16,427 controls) descent. We utilized non-overlapping data for T2D from the DIAGRAM consortium5 and conducted combined discovery analysis on 198,258 participants (48,365 T2D cases and 149,893 controls). Characteristics of the participants and information on genotyping QC are summarized in Supplementary Tables 1-3 and Supplementary Figure 1. After removing known loci, we advanced 21 novel loci with suggestive association with T2D (P ≤ 5 × 10−6). We performed further testing of these SNPs in additional samples of up to 67,420 individuals (24,972 cases and 42,448 controls) of South Asian (n = 13,960; 4,587 T2D cases and 9,373 controls), European (n = 2,479; 387 T2D cases and 2,092 controls), and East-Asian descent (n = 50,981; 19,998 T2D cases and 30,983 controls). Our combined discovery and replication analyses included 265,678 participants (73,337 T2D cases and 192,341 controls) (Supplementary Figure 1a). In the combined analysis across both stages, 15 SNPs at previously unreported loci for T2D obtained genome-wide significance (fixed-effects meta-analysis P < 5 × 10−8, Table 1). A previous report found one of our variants (rs10507349) strongly associated with T2D, but we report genome-wide significance for this variant here for the first time9. Population-specific analyses (i.e., Europeans only or South Asians only) identified one additional locus where a sentinel variant obtained genome-wide significance in only European participants (Table 1, P Figure 1, Supplementary Figures 2 and 3, and Supplementary Tables 4 and 5). Aside from this case, there was little evidence of heterogeneity of effect between the ancestry groups in either our primary genetic analyses or across the two stages (Supplementary Figure 2). We replicated previously reported associations with T2D at 59 loci at genome-wide significance; a further 25 known loci were associated with T2D at < 0.05 (Figure 1 and Supplementary Table 4). We did not observe association at 3 loci (rs76895963, rs7330796, rs4523957) in our overall meta-analyses, owing to their previous discovery in subjects of East Asian ancestry (Supplementary Table 4). To nominate candidate genes and pathways, we obtained expression quantitative trait locus (eQTL) data from the MuTHER consortium and GTEx (v6, Supplementary Table 6)10,11. These data suggest a candidate gene at two loci (ITFG3 and PLEKHA1) where the lead eQTL association strongly tagged the T2D association (r2 = 1.0).
Table 1.
16 novel loci associated with T2D
| Lead variant | Closest gene | Chr | Pos (hg19) | EA | NEA | EAF | OR | 95% CI | P-value | I2 | Phet |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Loci associated with T2D in the combined analysis of Europeans, South Asians and East Asians at a P-value < 5×10−8 | |||||||||||
|
| |||||||||||
| rs2867125 | TMEM18 | 2 | 622,827 | C | T | 0.83 | 1.06 | 1.04 - 1.08 | 1.73 × 10−09 | 18 | 2.3 × 10−01 |
| rs11123406 | BCL2L11 | 2 | 111,950,541 | T | C | 0.36 | 1.04 | 1.03 - 1.06 | 9.10 × 10−09 | 2 | 4.4 × 10−01 |
| rs2706785 | TMEM155 | 4 | 122,660,250 | G | A | 0.05 | 1.13 | 1.08 - 1.17 | 2.74 × 10−08 | 0 | 9.0 × 10−01 |
| rs329122 | PHF15 | 5 | 133,864,599 | A | G | 0.43 | 1.04 | 1.03 - 1.06 | 2.90 × 10−09 | 0 | 5.1 × 10−01 |
| rs622217 | SLC22A3 | 6 | 160,766,770 | T | C | 0.52 | 1.05 | 1.03 - 1.07 | 5.81 × 10−09 | 0 | 7.0 × 10−01 |
| rs9648716 | BRAF | 7 | 140,612,163 | T | A | 0.15 | 1.06 | 1.04 - 1.09 | 2.82 × 10−09 | 0 | 4.8 × 10−01 |
| rs12681990 | KCNU1 | 8 | 36,859,186 | C | T | 0.15 | 1.05 | 1.04 - 1.07 | 2.28 × 10−09 | 0 | 6.4 × 10−01 |
| rs7111341 | INS | 11 | 2,213,166 | T | C | 0.26 | 1.07 | 1.05 - 1.09 | 2.13 × 10−11 | 8 | 3.7 × 10−01 |
| rs10507349 | RNF6 | 13 | 26,781,528 | G | A | 0.78 | 1.05 | 1.04 - 1.07 | 1.87 × 10−09 | 0 | 6.1 × 10−01 |
| rs576674 | KL | 13 | 33,554,302 | G | A | 0.16 | 1.07 | 1.05 - 1.10 | 1.07 × 10−12 | 4 | 4.0 × 10−01 |
| rs7985179 | MIR17HG | 13 | 91,940,169 | T | A | 0.72 | 1.07 | 1.05 - 1.10 | 3.72 × 10−09 | 0 | 6.2 × 10−01 |
| rs9940149 | ITFG3 | 16 | 300,641 | G | A | 0.83 | 1.05 | 1.04 - 1.07 | 1.70 × 10−09 | 0 | 9.2 × 10−01 |
| rs2050188 | HLA-DRB5* | 6 | 32,339,897 | T | C | 0.67 | 1.06 | 1.04 - 1.08 | 5.20 × 10−10 | 0 | 5.8 × 10−01 |
| rs2421016 | PLEKHA1 | 10 | 124,167,512 | C | T | 0.53 | 1.05 | 1.03 - 1.06 | 3.86 × 10−11 | 17 | 2.3 × 10−01 |
| rs2925979 | CMIP | 16 | 81,534,790 | T | C | 0.29 | 1.05 | 1.03 - 1.07 | 3.75 × 10−08 | 6 | 3.8 × 10−01 |
|
| |||||||||||
| Locus associated with T2D in Europeans at a P-value < 5×10−8 | |||||||||||
|
| |||||||||||
| rs7674212 | CISD2* | 4 | 103,988,899 | G | T | 0.58 | 1.07 | 1.04-1.09 | 6.85 × 10−09 | 0 | 7.2 × 10−01 |
Chr., chromosome; Position is under hg19; EA, effect allele; NEA, non-effect allele; EAF, risk allele frequency in Europeans – allele frequencies by ancestry are reported in Supplementary Table-2; OR, odds ratio; CI, confidence interval; I2, heterogeneity inconsistency index; Phet, P-value for heterogeneity across meta-analyzed datasets.
: candidate gene based on Exomechip lookup or Mendelian subform.
Figure 1.
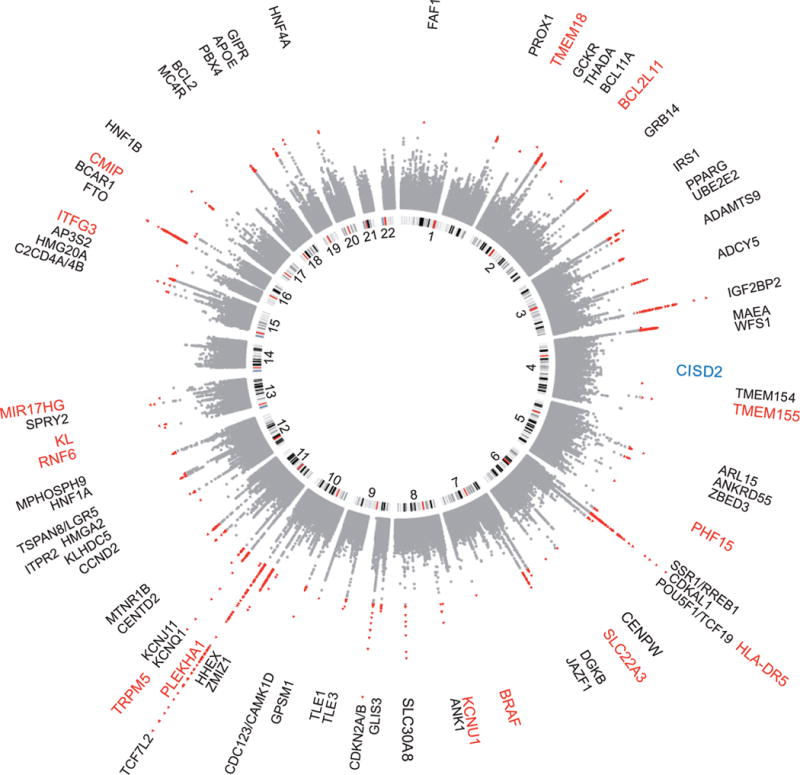
A circular Manhattan plot summarizing the association results for the T2D scan. Black: Previously established T2D loci, Red: Previously unreported T2D loci from trans-ethnic meta-analysis, Blue: Previously unreported T2D loci from EUR only meta-analysis.
Coding variants at new genetic loci
To identify coding variants that may influence protein structure at unreported T2D loci, we obtained data on up to 31,207 individuals (9,500 T2D cases, 21,707 controls) of South Asian (7,832 T2D cases, 16,703 controls) and European origin (1,668 T2D cases, 5,004 controls) (Methods) genotyped on the Exome-chip12. We investigated 505 variants captured by the exome-chip within ±250kbp flanking regions of the sentinel SNPs. We identified one missense variant in HLA-DRB5 (rs701884), that was associated with T2D risk at close to exome-wide levels of significance (fixed-effects meta-analysis P < 2 × 10−7) (Supplementary Table 7). The missense variant was found to have a relatively stronger impact on disease risk compared to the lead non-coding variant. At HLA-DRB5 the odds ratio (OR) for T2D for the missense variant (rs701884) was 1.29 (95% CI: 1.23 - 1.35; P = 4.8 × 10−7) compared to the T2D OR of 1.06 (95% CI: 1.04-1.08; P = 2.1 × 10−10) for the non-coding variant (rs2050188). Existing knowledge on gene function is summarized in Supplementary Table 8.
Variant association with traits and circulating biomarkers
To help understand the underlying biological mechanisms, we examined the association of genetic variation at newly discovered loci with a range of phenotypes and biomarkers (n = 70 traits) (Supplementary Table 9). We also used an association screen against a panel of 105 phenotypic traits measured in the PROMIS study in up to 17,542 participants (Supplementary Table 10)13. For these 17 loci, we conducted 4,275 variant-phenotype analyses using linear regression resulting in a Bonferroni-adjusted significance threshold of P = 1.5 × 10−5. Allelic variation that increased T2D risk was associated at TMEM18 with increased BMI (P = 4.39 × 10−52), BMI in childhood (P = 7.95 × 10−12), obesity (P = 2.50 × 10−25) and obesity in childhood (P = 2.85 × 10−20); at KL, with increased fasting glucose (P = 2.26 × 10−8); at PLEKHA1 with increased risk of neovascular disease (P = 2.71 × 10−94); at SLC22A1, with increased Lp(a) levels (P = 5.10 × 10−6); at CMIP, with decreased HDL-C (P = 1.32 × 10−19), increased triglycerides (P = 2.14 × 10−7) and decreased adiponectin (P = 1.87 × 10−18).
Genetic risk for T2D and CHD shared at established loci
We next examined the relationships of sentinel T2D SNPs with the risk of CHD at all T2D loci (Supplementary Table 11). For analyses in relation to CHD, we used data on up to 260,365 participants (90,831 CHD cases and 169,534 controls) (Methods). We found allelic variation at 17 T2D loci to be nominally associated with CHD risk at P < 0.01, which was more than expected (17 of 106 T2D SNPs, binomial test P = 5.9 × 10−13). In one case, we found that the T2D sentinel SNP rs7578326 (the IRS1 locus) was associated with both T2D and CHD at genome-wide levels of significance (Supplementary Table 11 and Supplementary Figure 4). To the best of our knowledge, this is the first report of genetic variation at IRS1 associated with CHD beyond a reasonable doubt (Supplementary Note)6. In what follows, we investigate the relationship between these two endpoints in more detail.
Genetically elevated T2D risk overall increases CHD risk
First, we examined if elevated T2D risk conferred a higher risk of CHD using the framework of Mendelian Randomization (MR)14-16 and examined if all genetic T2D risk pathways impact CHD susceptibility in a similar way. We calculated genetic risk scores comprising collections of SNPs associated with T2D and potentially a range of cardiometabolic traits (Methods). These analyses underscore three key findings. First, a genotype risk score based on variants exclusively associated with increased risk of T2D (Methods, Supplementary Table 12 and 13) was significantly associated with increased CHD risk (OR = 1.26, Wald-test P = 3.3 × 10−8), supporting a causal role for T2D in CHD etiology in a directionally consistent manner (Supplementary Table 14). Second, T2D risk scores that involved variants based on their association with established risk factors for CHD (blood pressure, BMI, lipids and anthropometric traits, Supplementary Table 13) revealed significant differences in their estimated effects in relation to CHD (OR = 1.07 to 1.43, Cochran’s Heterogeneity Test P = 1.4 × 10−5), indicating that the genetic mechanisms and underlying pathways that increase risk of T2D do not uniformly impact CHD risk in the same manner (Supplementary Table 14). Finally, in contrast to these scores, variants associated with T2D and glucose or insulin-related traits, but not other traits, were not associated with CHD (OR = 1.07, Wald-test P = 0.06) (Supplementary Table 14); though could be due to reduced power of this instrument relative to others, as has been observed previously15. These analyses indicate that pathways segregating genetic susceptibility for T2D may not have an equivalent impact on CHD risk.
Genetic risk for T2D and CHD shared genome-wide
We next looked for enrichment in the consistency of the risk allele associated with both T2D and CHD across the genome. In our meta-analyses, of the 1,260 variants associated with T2D at a P < 5 × 10−8, we found that 76.1% of the T2D risk alleles were associated with higher risk of CHD as well, in comparison with an expectation of 50% under the null hypothesis (binomial test P = 2.6 × 10−33, Table 2). In contrast, variants associated with CHD at P < 5 × 10−8 were not enriched for directional consistency in allelic associations with T2D (48.2 vs 50% expected, binomial test P = 0.79). Among the loci nominally associated with T2D and CHD (P < 0.05 but excluding P < 5 × 10−8 associations above), 81.8% of the allelic variation associated with both the outcomes in a directionally consistent manner (binomial test P < 10−100). Furthermore, of the allelic variation that was not associated with both T2D and CHD at a P-value > 0.05, only 50.6% of the allelic variation (compared to 50% expected under the null hypothesis) was associated with the two outcomes in a directionally consistent manner (Table 2). To rule out any biases introduced due to allelic variations at a limited set of loci associated with both CHD and T2D, we conducted sensitivity analyses using genome-wide variants pruned for LD and found results consistent for an overall enrichment of loci associated with both T2D and CHD in a directionally consistent manner (Supplementary Table 15).
Table 2.
Enrichment in directional consistency for all SNPs in T2D and CHD association scan
| T2D and CHD in meta-analyses
|
|||||
|---|---|---|---|---|---|
| p-value cut point | # of SNPs in total | # of SNPs CHD/T2D consistent | % of SNPs CHD/T2D consistent | adjusted −log10(p-value)* | |
| T2D | CHD | ||||
| (0, 5×10−8] | – | 1,260 | 959 | 76.11% | 76.966 |
| – | (0, 5×10−8] | 595 | 287 | 48.24% | 0.062 |
| (0.5, 1] | (0.5, 1] | 1,874,138 | 948,292 | 50.60% | – |
| (5×10−8, 0.05] | (5×10−8, 0.05] | 36,242 | 29,634 | 81.77% | 3319.168 |
P-value values from the binomial sign test were reported. The probability used to estimate the p-values in the binomial sign test is the percentage highlighted in red.
Joint test reveals an additional locus for T2D and CHD
Motivated by the enrichment of directionally consistent associations of allelic variation between T2D and CHD SNPs, we performed a genome-wide association scan which modeled the joint distribution of association with both T2D and CHD (T2D-CHD, Methods), a test to help improve power for discovery of novel loci that are associated with both the outcomes (Supplementary Figure 5). After verifying that our test statistic was calibrated (Supplementary Figure 6), we used this approach to identify a set of loci that were associated with both T2D and CHD (both traits with fixed effects meta-analysis P < 10−3), and were overall associated at genome-wide levels of significance (bivariate P < 5 × 10−8, Supplementary Table 16). 19 loci met these criteria, which included many established loci for T2D or CHD.
We identified one association near CCDC92 (bivariate P = 2.7 × 10−9, Supplementary Figure 7a). The sentinel variant (rs825476) was associated with both T2D (fixed effects meta-analysis P = 2.2 × 10−6) and CHD (fixed effects meta-analysis P = 2.9 × 10−7) (Supplementary Figure 7b and 7c); rs825476-T at this locus increased risk for both the outcomes. To demonstrate conclusive association of rs825476 with T2D, we sought additional replication data from 8 additional cohorts, comprising 21,560 T2D cases and 42,814 controls. We observed marginal replication for this variant in those data alone (OR = 1.04, 95% CI: 1.01 - 1.07; fixed effects meta-analysis P = 5.5 × 10−3), and obtained genome-wide significance when combined with the previous data (OR = 1.04, 95% CI: CI: 1.03 - 1.06; fixed effects meta-analysis P = 4.3 × 10−8), Supplementary Figure 7b). Analyses conditioned on the lead SNP accounted for all the residual joint T2D-CHD association in the region (Methods), indicated that the underlying genetic associations for both endpoints co-localize to a shared genetic risk factor potentially tagged by the sentinel SNP (Supplementary Figure 8). rs825476-T allele also increased the expression of CCDC92 in the subcutaneous adipose tissue (Supplementary Table 6) in eQTL analyses conducted in the MuTHER consortium and GTEx [8,9], suggesting a possible candidate gene for the association.
We sought to reduce our list to a subset of loci that co-localized the T2D and CHD associations to a single underlying genetic risk variant by conducting formal co-localization analyses (Methods). 8 of these 19 met this criterion, and at 7 of those 8 loci, the risk allele for T2D also increased the risk for CHD (Table 3). This included loci with known associations with T2D (TCF7L2, HNF1A, and CTRB1/2) as well as previously unreported T2D loci reported here (MIR17HG and CCDC92), or known association with CHD (MRAS and ZC3HC1). Interestingly, this set of directionally consistent loci included coding variants in two transcription factors: the missense variant(s) I27L in HNF1A, and R326H in ZC3HC1. At the APOE locus, where the effect of association for T2D and CHD risk was opposite, localization was observed at rs4420638, but the tagging among the lead sentinel SNPs was incomplete, making it challenging to distinguish between multiple conditionally independent variant associations with both traits versus partial tagging of a single, common association. At the IRS1 locus, while we found rs7578326 to be associated with both T2D and CHD (P < 5 × 10−8), formal co-localization analyses failed to identify a single underlying genetic risk factor for the two outcomes at this locus.
Table 3.
Genome-wide significant loci by bivariate scan at sentinel SNPs that are associated with both T2D and CHD (P < 10−3) where leading associations co-localize
| T2D | CHD | BVN | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||
| Gene | Lead variant | Chr | Pos (hg19) | EA | NEA | OR | 95% CI | P-value | OR | 95% CI | P-value | P-value |
| Established Loci with T2D/CHD risk allele agreement and co-localization (r2 > 0.7 between T2D and CHD associations and COLOC Prob. > 0.5) | ||||||||||||
| TCF7L2 | rs7903146 | 10 | 114758349 | T | C | 1.35 | 1.33 - 1.38 | 1.3 × 10−219 | 1.04 | 1.02 - 1.05 | 2.9 × 10−5 | 2.6 × 10−212 |
| HNF1A (I27L) | rs1169288 | 12 | 12146650 | A | C | 1.06 | 1.04 - 1.08 | 9.3 × 10−10 | 1.04 | 1.03 - 1.06 | 3.9 × 10−7 | 2.0 × 10−12 |
| CTRB1/2 | rs7202877 | 16 | 75247245 | T | G | 1.06 | 1.03 - 1.08 | 4.0 × 10−6 | 1.06 | 1.04 - 1.09 | 2.9 × 10−6 | 1.0 × 10−8 |
| MRAS | rs2306374 | 3 | 138119952 | C | T | 1.05 | 1.02 - 1.07 | 6.5 × 10−4 | 1.06 | 1.04 - 1.08 | 2.3 × 10−8 | 9.8 × 10−9 |
| ZC3HC1 (R342H) | rs11556924 | 7 | 129663496 | C | T | 1.03 | 1.01 - 1.05 | 4.9 × 10−4 | 1.08 | 1.06 - 1.10 | 3.3 × 10−20 | 1.4 × 10−19 |
| Novel Loci with T2D/CHD risk allele agreement and co-localization (r2 > 0.7 between T2D and CHD associations and COLOC Prob. > 0.5) | ||||||||||||
| MIR17HG | rs7985179 | 13 | 91940169 | A | T | 1.07 | 1.05 - 1.10 | 3.7 × 10−9 | 1.05 | 1.02 - 1.08 | 6.4 × 10−4 | 1.5 × 10−9 |
| CCDC92 | rs825476 | 12 | 124568456 | T | C | 1.04 | 1.03 - 1.06 | 2.2 × 10−6 | 1.03 | 1.02 - 1.05 | 3.0 × 10−7 | 2.7 × 10−9 |
| Opposite Risk Allele for T2D/CHD with co-localization (r2 > 0.7 between T2D and CHD associations and COLOC Prob > 0.5) | ||||||||||||
| APOE | rs4420638 | 19 | 45422946 | A | G | 1.08 | 1.05 - 1.11 | 8.8 × 10−8 | 0.89 | 0.85 - 0.93 | 1.8 × 10−6 | 2.6 × 10−13 |
Chr., chromosome; Position is under hg19; EA, effect allele; NEA, non-effect allele; OR, odds ratio; CI, confidence interval; BVN, the bivariate normal distribution of T2D and CHD statistics.
Next, we used biomarker data to help understand the mechanisms linking T2D and CHD at two novel loci discovered through bivariate scan, MIR17HG, and CCDC92. The region around CCDC92 segregates numerous cardiometabolic trait associations, including T2D (rs1727313)9, HDL-C (rs4759375, rs838880), triglycerides (rs4765127)17, and waist-hip ratio adjusted for body mass index (rs4765219)18. However, variants in these previous reports were not strongly linked to our sentinel SNP (r2 < 0.02 in all cases). The risk variant for T2D-CHD at the CCDC92 locus also decreased HDL-C levels (fixed effects meta-analysis P = 2.2 × 10−9) in analyses by the Global Lipids Genetics Consortium (GLGC)17; this variant was in partial linkage with a variant (rs10773049, r2 = 0.6 and 0.3 in Europe and South Asia, respectively) previously known for association with body mass index. MIR17HG appeared to harbor only modest associations with HDL (fixed effects meta-analysis P = 1.3 × 10−4), fasting insulin levels (P = 6.4 × 10−4) and HOMA-IR (P = 7.9 × 10−4).
We also examined association at APOE where the T2D risk allele was associated with decreased CHD risk. The T2D risk allele was also found associated with increased HDL-C (fixed effects meta-analysis P = 1.72 × 10−21), decreased LDL-C (P = 1.51 × 10−178), decreased total cholesterol (P = 1.14 × 10−149), decreased triglycerides (P = 1.55 × 10−14), reduced LDL particle size (P = 3.80 × 10−11), increased waist-hip ratio (P = 1.80 × 10−6, BMI-adjusted), and neovascular disease (P = 2.78 × 10−8).
Joint T2D-CHD associations highlight novel pathways
We next aimed to identify a subset of highly connected loci that indicate unidentified pathways that jointly related to T2D-CHD. To achieve this, we used results from our bivariate T2D-CHD association scan and pruned SNPs for LD to obtain a set of unlinked regions across the genome (r2 < 0.05). From this list, we selected 299 LD independent SNPs that were found associated with T2D-CHD in our bivariate scan (P < 0.001, Supplementary Table 17 and 18) and sought to prioritize candidate genes implicated in the association using the text-mining approach, GRAIL19. 79 out of 299 regions were found to have prioritized specific genes in associated intervals (GRAIL P < 0.05), significantly more overall than expected (26.4%, binomial test P < 10−34). Next, protein-protein interaction connectivity analysis among these 79 genes20 demonstrates more direct and indirect connections than expected (permuted P < 10−4, Methods), thus motivating us to focus on this subset for further analysis. Several plausible candidates from this list emerge, including the hepatic glucose transporter SLC2A2, the Adipocyte fatty-acid-binding protein aP2, Lipin-1, PGC-1β, and the Free fatty acid receptor 1 FFAR1, among others (Supplementary Table 18).
We next performed ontology analysis on the set of 79 genes that emerged from T2D-CHD bivariate scan for connectivity21. To compare our findings, we also conducted similar ontological analysis on loci identified for T2D or CHD in previous GWAS for each of these traits. As expected, ontological analysis of established T2D loci alone indicated robust enrichment of diabetes, hyperglycemia, and insulin resistance disease annotations (enrichment test P < 10−55), as well as enrichment for pathways related to insulin secretion and transport, glucose homeostasis, and pancreas development (all P < 10−9). Also as expected, ontological analysis of CHD loci alone demonstrated robust enrichment of disease annotations related to coronary disease, myocardial infarction, and arteriosclerosis (P < 10−36), as well as enrichment for pathways related to lipid homeostasis and cholesterol transport (P < 10−8). As expected, the analysis of the 79 T2D-CHD associated gene intervals identified loci that were also modestly enriched for disease ontologies related to vascular resistance (P < 10−12), T2D, cardiovascular disease, fatty liver, obesity, gestational hypertension, and Pre-eclampsia (all P < 10−5), as well as cancer (P < 10−9). But in contrast to the pathways described above, we also observed enrichment for additional pathways related to cardiovascular system development, cell signaling, signal transduction, regulation of phosphorylation, and transmembrane receptor protein kinase signaling among the categories (adjusted P < 10−7) (Supplementary Figure 9).
Discussion
We report the discovery of 17 novel loci for T2D using discovery and replication studies in 265,678 participants. Using exome-chip data, we were able to identify a coding variant which was more strongly associated with T2D risk than the corresponding common variant. Using additional data on 260,365 participants, we report a novel locus for CHD, and identify genetic loci that are shared between T2D and CHD of which a subset co-localized to the same genetic variant (e.g., CCDC92, MIR17HG, HNF1A, ZC3HC1, APOE, Table 3). Finally, using a bivariate scan for T2D-CHD, genetic association data pointed to new pathways that are implicated in the etiology of both the disease outcomes.
Many of the loci discovered in the current meta-analyses suggest novel T2D biology or confirm pathways previously implicated in T2D. For instance, MIR17HG, KL, and BCL2L11 have been shown involved in cell-survival, apoptosis, and cellular aging, respectively22-24. Genetic variants near KL have also been shown associated with fasting glucose levels as well25. TMEM18 is involved in cellular migration; HLA-DR5 and CMIP play crucial roles in immune mediated responses and have been implicated in various immunological disorders26,27. Genetic variation at the HLA locus (rs9272346) has been previously implicated in type-1 diabetes (T1D); however, we did not find any evidence of association of rs9272346 with T2D in our meta-analyses. Additionally, rs9272346 was not in LD with the T2D sentinel SNP (rs2050188) at this locus (r2 = 0.06 in EUR and r2 = 0.01 in SA). However, rs7111341 has been previously reported as a risk factor for T1D28. We found that rs7111341-T is associated with increased risk for T2D, but decreased risk for T1D, a similar pattern of association to a previously established association (rs7202877, nearby CTRB1).
The contrasting associations of APOE with T2D and CHD were puzzling. APOE encodes apolipoprotein-E found in the chylomicron and intermediate-density lipoprotein (IDLs). Genetic variation at the APOE locus is associated with major lipids and CHD6,17. Here, the T2D risk variant was associated with decreased CHD risk and LDL-C, and reduced LDL particle size. These observations are consistent with recent studies indicating that reduction in LDL-C levels, a major CHD risk factor, may confer a higher risk of T2D. Evidence from a meta-analysis of randomized controlled trials has shown that reduction of LDL-C by statin treatment, compared to placebo, led to a higher, but a very small absolute, risk of T2D29. Moreover, genetic variants associated with reduced expression of HMG-CoA reductase, the target of statins, and reduced LDL-C levels have been shown associated with increased risk of T2D30. Also, two MR studies concluded that genetically mediated decreases in LDL-C associated with a higher risk of T2D31,32. Furthermore, it has been shown that genetic variants in the PCSK9 gene that lower LDL-C levels associated with a higher risk of T2D, fasting glucose concentration, body weight, and waist-to-hip ratio33. In contrast to the findings from our overall meta-analyses, these results suggest that LDL-C may represent one of a small subset of discrete pathways that display opposing associations for the two outcomes. These findings underscore how human genetics can help focus future investigations on T2D therapeutics that have either neutral or beneficial effects on vascular outcomes.
The collection of 79 regions identified through our joint T2D-CHD bivariate scan involves targets of existing drugs. This includes icosapent, a polyunsaturated fatty acid found in fish oil, which is FFAR1 and PPARG agonist and a COX-1/COX-2 inhibitor34. The ANCHOR trial showed that icosapent ethyl, marketed as the drug Vascepa, has efficacy in lowering triglycerides in patients with high TG levels35 as well as non-HDL-C and HDL-C36. A second plausible candidate gene is the adipocyte fatty-acid-binding protein (FABP4, also known as aP2). Mouse models deficient in aP2 display protection against atherosclerosis and anti-diabetic phenotypes37-39. Moreover, small-molecule inhibition of aP2 has been shown to reduce atherosclerosis, glucose and insulin levels, and triglycerides in a mouse model40, inhibition of this pathway through a monoclonal antibody also appear efficacious in mouse models41.
Careful evaluation of the pathways or biological process where T2D, CHD, and related traits overlap could help to highlight new avenues for therapeutic targeting. First, using gene discovery and biomarker studies, we have identified new pathways, outside of the established, glucose and cholesterol homeostatic networks, which could be investigated in more detail. Second, we have found that some genetic variants associated with T2D singly or in aggregate are enriched for associations with CHD. With one exception (e.g., pathways involving LDL-C), genetic pathways that increase T2D risk tend to overall increase CHD risk. Hence, existing or future therapeutic programs designed for the prevention of T2D could be better guided by evidence from genetic studies, either to prioritize targets that have either neutral or directionally consistent effects on vascular outcomes, or to review the disease risk profile of existing targets that may be under development. Overall, identification of genetic loci associated with both T2D and CHD risk in a directionally consistent manner could provide therapeutic opportunities to lower the risk of both outcomes.
Online Methods
Study subjects
In the discovery phase, we meta-analyzed data from eight different studies; four studies (PROMIS, RACE, BRAVE and EPIDREAM) include participants of South Asian origin living in Pakistan, Bangladesh and Canada whereas four studies (FINRISK, MedStar, MDC and PennCATH) include subjects of European origin (Supplementary Table 1 and Supplementary Note). GWAS/Metabochip data and information on T2D risk was available on 48,437 individuals (13,525 T2D cases and 34,912 controls) from these eight studies. We further used published data from the DIAGRAM consortium and conducted combined discovery analysis on 198,258 participants (48,365 T2D cases and 149,893 controls). Characteristics of the participants, information on genotyping arrays and imputation are summarized in Supplementary Tables 1-3. Replication studies were completed in participants enrolled in the LOLIPOP, SINDI, SDS, MSSE, TAICHI and BBJ studies (Supplementary Table-1), collectively composed of 67,420 individuals (24,972 cases and 42,448 controls) who were of South Asian (n = 13,960; 4,587 T2D cases and 9,373 controls), European (n = 2,479; 387 T2D cases and 2,092 controls), and East-Asian descent (n = 50,981; 19,998 T2D cases and 30,983 controls). Hence, our combined discovery and replication analyses included 265,678 participants (73,337 T2D cases and 192,341 controls). All studies were approved by the relevant institutional review boards and all participants provided written informed consent. Further details of the contributing cohorts and characteristics of the participants are provided in the Supplementary Note and Supplementary Table 1.
Institutional Review Board and Informed consent
All participating studies were approved by the relevant local institutional review boards. All participants enrolled in each of the participating studies provided informed consent.
Genotyping and quality control in the discovery stage
All studies used a high density genotyping array (GWAS / Metabochip) (Supplementary Table 2 and Supplementary Note). Quality control procedures were performed for each individual study. Details on study specific QC are provided in Supplementary Table 2. Each study individually assessed and controlled for any population stratification using principal component analysis.
Imputation
In all studies, genomic locations of all variants were first harmonized using the NCBI Build 37/UCSC hg19 coordinates. Only studies that contributed GWAS data underwent imputation. Imputation of genotypes across the genome was computed using the 1000 Genomes Project (phase 1 integrated release 3, March 2012)42. Imputed SNPs were removed if they had: (i) a minor allele frequency of < 0.01; (ii) info score of < 0.90; (iii) average maximum posterior call < 0.90. Supplementary Table 2 provides further details on the imputation protocol used by each of the participating studies.
Statistical analysis in the discovery stage
To test for an association between each SNP and risk of T2D, a logistic regression model was computed with adjustment for age, sex, and the first study-specific principal components using SNPTEST43. The SNP was modeled under an additive genetic model and imputation uncertainty was accounted for under an allele dosage approach. Inflation of association statistics was assessed within each study by the genomic control method (Supplementary Table 3 and Supplementary Figure 1). Variants that were retained in at least two studies were meta-analyzed using the METAL program44. We used a weighted inverse normal fixed-effects method, weighting by sample size; heterogeneity was assessed by the Cochran’s Q statistic and the I2 heterogeneity index. Pairwise linkage disequilibrium between SNPs was assessed and visualized using the 1000 Genomes European reference panel42. Regional association plots were visualized using the LocusZoom software. After removing regions harboring known loci, the top associated SNP and one or more SNPs based on linkage disequilibrium with the lead variant found in association with any of the above phenotypes (P < 5×10−6) were selected for the replication studies.
Analyses in the replication stage
Studies that participated in the replication stage had conducted genotyping on GWAS or Metabochip arrays. The association of SNPs with T2D was calculated separately using trend test, with heterogeneity between studies assessed using the Cochran’s Q statistic. The weighted inverse normal fixed effects meta-analysis was then computed to combine the results across all replication studies and with the discovery stage. For the combined analysis of discovery and replication data, genome-wide significance was inferred at P < 5 × 10−8.
Expression QTL and functional prioritization
To determine whether the identified risk variants influenced expression of any nearby genes, we accessed a variety of sources, including: (i) GTEx cis-eQTL data in all available tissues, including liver, brain, endothelial cells, and whole blood10, and (ii) cis-eQTL data for adipose, lymphoblastoid cell lines, and skin from the MuTHER consortium11.
Exome-chip analysis
To assess if there are coding variants associated with T2D in the proximity of the newly-discovered sentinel T2D SNPs, we performed an Exomechip-based meta-analysis in four studies (PROMIS, BRAVE, CIHDS-CGPS, and PROSPER) in the ±500 kbp region of the sentinel T2D SNPs. For all the studies, genotyping and QC were done centrally at the University of Cambridge, UK. In each study, samples with extreme intensity values, and outlying plates or arrays were removed prior to genotype calling. Genotype calling was initially performed with optiCall. Samples with call rate (CR) less than (mean CR - 3 standard deviations) were removed prior to post-processing optiCall calls with zCall. Scanner specific Z-values (calculated using 1,000 samples with the highest optiCall CR) were adopted as they gave the best global concordance within each batch. Rare variants (optiCall, Minor Allele Frequency (MAF) < 0.05) were then post processed with zCall using the scanner specific Z-values. Within each genotyping batch, variants were removed if variant CR < 0.97; HWE P < 1×10−6 for common variants or HWE P < 1×10−15 for rare variants (MAF < 0.05). Variants within each genotyping batch were aligned to human genome reference sequence plus strand and the standardized files were used for sample QC. Samples were excluded from each batch/study if sample heterozygosity > ±3 standard deviations from the mean heterogeneity or sample call rate >3 standard deviations from the mean call rate. Variants were further selected based on stringent QC thresholds (CR < 0.99; Hardy-Weinberg Equilibrium (HWE) P < 1×10−4) MAF > 0.05 and LD pruned (r2 <0.2) for PCA and kinship calculations. Duplicates within each collection (kinship coefficient>0.45) and ancestral outliers identified by PCA were removed. Samples and variants that failed QC were removed from individual batches. Where studies were analyzed in multiple batches, the batches were combined and any SNVs out of HWE across the study were removed.
Build and strand for each study was checked using checkVCF against build37 plus strand. The reference and alternate alleles were aligned with the reference. Study specific analyses were conducted using RAREMETALWORKER45,46 incorporating the kinship matrix and adjusting for age and sex. In each study, variants with a MAC < 10 were removed before meta-analysis. Meta-analysis was done in METAL. In the meta-analysis, the sample-size weighted approach was used to estimate the P-values and an inverse-variance weighted approach was used to calculate the pooled effect estimates and corresponding standard errors. Study specific information is provided in Supplementary Table 19.
Phenome/Biomarker scan analyses
We downloaded online-available GWAS data from 12 consortia for 70 traits (Supplementary Table 9) and harmonized the genome position to build 37/hg19. We then performed a lookup for the 17 newly discovered T2D SNPs using these harmonized data sets. We also performed a phenotypic scan for the same 17 SNPs across 105 biomarkers measured in the PROMIS participants using a linear regression model adjusted for the first principal components (Supplementary Table 10). We used a Bonferroni-adjusted P-value cut point of 1.7 × 10−5 (= 0.05 / 175 traits / 17 SNPs) to declare statistical significance.
Coronary heart disease (CHD) meta-analysis
We assembled 56,354 samples of European, East Asian and South Asian ancestries genotyped on the CardioMetabochip to identify genetic determinants of CHD. These results were combined with those reported by CARDIoGRAMplusC4D to yield analyses comprising of 260,365 subjects (90,831 CHD cases) for CHD. The Cambridge MI studies comprised of 16,093 CHD cases and 16,616 unaffecteds from the EPIC-CVD study, a case-cohort study recruited across 10 European countries, the Copenhagen City Heart Study (CCHS), the Copenhagen Ischemic Heart Disease Study (CIHDS) and the Copenhagen General Population Study (CGPS) all recruited within Copenhagen, Denmark. The Cambridge MI SAS studies comprised up to 7,654 CHD cases and 7,014 controls from the Pakistan Risk of Myocardial Infarction Study (PROMIS) a case-control study that recruited samples from 9 sites in Pakistan, and the Bangladesh Risk of Acute Vascular Events (BRAVE) study based in Dhaka, Bangladesh. The EA studies comprised 4,129 CHD cases and 6,369 controls recruited from 7 studies across Taiwan that collectively comprise the TAIwan metaboCHIp (TAICHI) Consortium. Samples from EPIC-CVD, CCHS, CIHDS, CGPS, BRAVE and PROMIS were all genotyped on a customized version of the Illumina CardioMetabochip (manufactured by Illumina, San Diego, USA) referred to as the Metabochip+, in two Illumina-certified laboratories located in Cambridge, UK, and Copenhagen, Denmark. TAICHI samples were genotyped using the latest version of the CardioMetabochip. For each study, samples were removed if they had a call rate < 0.97, average heterozygosity >±3 standard deviations away from the overall mean heterozygosity or their genotypic sex did not match their reported sex. One of each pair of duplicate samples and first-degree relatives (assessed with a kinship co-efficient > 0.2) were removed. Cardio-metabochip data were also obtained from the Women Health Initiative Study and the ARIC study; the two studies underwent same QC as described for the TAICHI study. Across all studies, SNP exclusions were based on minor allele frequency (MAF) < 0.01, P < 1×10−6 for Hardy-Weinberg Equilibrium or call rate (CR) less than 0.97. CARDIoGRAMplusC4D Consortium data were obtained online (see URLs). Only non-overlapping samples were used for meta-analyses. Fixed effects inverse variance weighted meta-analysis was used to combine the effects across studies in METAL44.
Genetic Risk Score Analysis
We utilized a two-sample MR method47 to estimate effects for a multi-SNP genetic instrument by using summary statistics. This method has been previously validated to infer causal effects (odds ratio) and associated standard error48. Briefly, association data for both T2D and CHD were obtained using data from two separate genome-wide meta-analyses. For T2D, we used the data from the current meta-analyses; whereas we used data from the most recent CHD meta-analyses as described in the Supplementary Note. Using sentinel SNPs for all established T2D associations, we identified a set of variants (n=16) exclusively associated with T2D, by screening against GWAS catalog of publicly available data49 for anthropometric traits (i.e., body mass index (BMI), waist-hip ratio (WHR), waist circumference (WC), WHR adjusted for BMI, WC adjusted for BMI, and hip), glucose/insulin (fasting glucose, 2 hour glucose, fasting insulin, and proinsulin levels), blood lipids (high, low-density lipoprotein cholesterol and triglycerides), and blood pressure (systolic and diastolic). We next attempted to group the remaining pleiotropic T2D SNPs into different categories based on their observed associations for various cardiometabolic intermediate traits (P < 0.01). These groupings included: (i) variants associated with glucose/insulin traits only (n=13), (ii) variants associated with TG/HDL-C and waist circumference / WHR but not glucose/insulin, blood pressure, LDL-C, or BMI (n=6), (iii) TG/HDL-C and obesity/anthropometric traits, but not glucose/insulin, blood pressure, or LDL-C (n=6), (iv) TG/HDL-C, blood pressure, and BMI, but not glucose/insulin or LDL-C (n=8), and (v) TG/HDL-C, blood pressure, BMI, and glucose/insulin but not blood pressure or LDL-C (n=24, Supplemental Table 12). Established T2D SNPs that did not fall into any of these categories were excluded. Heterogeneity in odds ratios was assessed via Cochran’s Q test for heterogeneity.
T2D and CHD enrichment analysis
We used a binomial distribution with a baseline enrichment probability Pb to derive the density for the test statistic E ~ Binomial(n, Pb), where n is the number of SNPs in a variant set. E is the number of SNPs with a directionally consistent effect on T2D and CHD (the allele that increases the risk for T2D also increases the risk for CHD). Using the SNPs that are not associated with T2D or CHD (P-values ≥ 0.05 for T2D and CHD), we calculated the percentage of SNPs with a directionally consistent effect in T2D and CHD and used it as an estimate for Pb. We then performed the enrichment analysis in two variant sets: (i) the variant set with all variants available and (ii) the variant set with LD-clumped variants. The results are shown in Supplementary Table 15. In the LD-clumping procedure, the SNPs with more significant T2D P-values were retained as seeds and the other SNPs that were in LD (r2 > 0.1 in based on data from the 1000 Genomes Project (Phase 3, v5 variant set) with the seed SNPs were removed42.
Estimating the T2D-CHD bivariate normal density
To establish the T2D-CHD bivariate normal density, we used all variants that we identified in our analyses on T2D and CHD; we further pruned them for LD using the 1000 Genomes project (Phase 3, v5 variant set) to r2 ≤ 0.1 using PLINK50,51. The reference and alternate alleles of the variants survived LD-pruning were retrieved from the same 1000 Genomes VCF file used for pruning, and the variants’ effects on CHD and T2D were aligned to their reference alleles. The statistics used to estimate the bivariate normal density were produced by using the following formula:
| (1) |
where Φ−1 is the inverse-cumulative distribution function of standard normal distribution, PCHD and PT2D are the P-values of CHD and T2D respectively, and βCHD and βT2D are the effect sizes of the reference allele on CHD and T2D respectively. Since a successful estimation of the bivariate distribution depends on both positive and negative Z-scores, we used the signs of the corresponding effect estimates (β/|β|) to determine the signs of ZCHD and ZT2D. The distribution of ZCHD and ZT2D are shown in Supplementary Figure 5. Parameters for the bivariate normal density were estimated by using the mvn.ub() function in the R package miscF. The estimated bivariate normal density has the following parameter values:
| (2) |
Two-degree-of-freedom test under the bivariate normal density
Assuming that Z is distributed as a Bivariate Normal, e.g., then:
| (3) |
where N2(μ, Σ) denotes a bivariate normal distribution with a vector of means μ and variance/covariance matrix Σ, and is the chi-square distribution with 2 degrees-of-freedom. Using Y as the test statistic, we performed a two-degree-of-freedom test on Z = (ZCHD, ZT2D), and our null hypothesis is that a SNP is not associated with any of thw two traits. Supplementary Figure 5 depicts the rejection region of the two-degree-of-freedom test.
Conditional Analysis for CCDC92
We performed approximate conditional analysis for the CCDC92 locus using the software package GCTA52. We used the summary meta-analysis data for our primary T2D and CHD scans (prior to replication) as data input from each continental group (European, South Asian, East Asian). As reference input, we utilized population data from the 1000 Genomes Project (version 3) which matched the continental ancestry for the respective conditional analysis. We then conditioned on rs825476 – the lead SNP associated with CHD and T2D – for each continental group. We then combined summary results from each continental group via inverse-weighted fixed-effects meta-analysis. Locus zoom plots for these conditional, meta-analyzed association results are presented in Supplementary Figure 8.
Co-localization Analysis
To determine if the T2D and CHD association signals co-localized to the same genetic variant, we utilized the R package coloc. For each of the 19 loci that met our T2D/CHD association criteria, we obtained association data from all SNPs within 500kb around the sentinel bivariate associated SNP (Supplementary Table 16). From there, we used the coloc.abf() function to calculate the probability that both traits are associated and share a single causal variant (H4), using the P-values given from the overall inverse-variance fixed effects meta-analysis for T2D (without replication) and CHD, the overall case/control sample sizes for both scans, and the allele frequencies for the variant based on all 1000 Genomes data (version 3). We call variants co-localized if the H4 co-localization probability was greater than 0.5.
Selection of loci for connectivity and ontology analyses
For T2D, we used the previously reported loci5 (n = 87) and the loci discovered in this report (n = 17). For CHD, we used the previously reported loci described in the most recent report published by the CARDIoGRAMplusC4D consortium (n = 58). Prioritization of genes from this list of established loci for T2D and CHD (Supplementary Table 17) was based on evidence from monogenic association with disease52, coding mutations in nearby genes, functional evidence implicating genes, or based on the gene nearest to the sentinel SNP. For T2D-CHD associations arising from our bivariate scan, we first pruned the dataset for LD (r2 < 0.1). We further selected 299 LD independent SNPs that were found associated with T2D-CHD with a P < 0.001 in our bivariate scan and used them to identify underlying candidate genes using GRAIL19. For protein-protein interaction connectivity analysis, we used DAPPLE20 on the 79 loci that were found significant in GRAIL19. Empirical significance for excess connectivity in protein-protein interactions was assessed by 10,000 permutations.
Ontology analysis and drug target annotations
We used the online tool WebGestalt21 to perform ontology enrichment analysis. For analysis of the query loci, we nominated genes (n=79) that were prioritized from text mining (GRAIL P < 0.05). We also performed ontology analyses using separate gene lists for T2D (n=104) and CHD (n=58) loci separately. The hypergeometric distribution was used to assess significance, and adjustment for multiple testing was controlled using the Benjamini-Hochberg procedure53 implemented in WebGestalt21.
Data Availability Statement
Summary GWAS estimates for the T2D meta-analysis and bivariate summary data are publicly available at the following:
Supplementary Material
Acknowledgments
D. Saleheen has received support from NHLBI, NINDS, Pfizer, Regeneron Pharmaceuticals, Genenetech and Eli Lilly. Genotyping in PROMIS was funded by the Wellcome Trust, UK and Pfizer. Biomarker assays in PROMIS have been funded through grants awarded by the NIH (RC2HL101834 and RC1TW008485) and the Fogarty International (RC1TW008485). The RACE study has been funded by the National Institute of Neurological Disorders (R21NS064908), the Fogarty International (R21NS064908) and the Center for Non-Communicable Diseases, Karachi, Pakistan. B. Voight was supported by funding from the American Heart Association (13SDG14330006), the W.W. Smith Charitable Trust (H1201), and US National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Disorders (R01DK101478). J Danesh is a British Heart Foundation Professor, European Research Council Senior Investigator, and NIHR Senior Investigator. VS was supported by the Finnish Foundation for Cardiovascular Research. SR was supported by the Academy of Finland [251217 and 255847], Center of Excellence in Complex Disease Genetics, EU FP7 projects ENGAGE [201413] and BioSHaRE [261433], the Finnish Foundation for Cardiovascular Research, Biocentrum Helsinki, and the Sigrid Juselius Foundation. The Mount Sinai IPM Biobank Program is supported by the Andrea and Charles Bronfman Philanthropies. Dr. Anand is supported by grants from Canada Research Chair in Ethnic Diversity and CVD, and the Heart and Stroke Michael G DeGroote chair in Population Health, McMaster University. Data contributed by the Biobank Japan was partly supported by a grant from the Leading Project of Ministry of Education, Culture, Sports, Science and Technology-Japan. We thank participants and staff of the Copenhagen Ischemic Heart Disease Study and the Copenhagen General Population Study for their important contributions. CHD Exome+ Consortium was funded by the UK Medical Research Council (G0800270), British Heart Foundation (SP/09/002), UK National Institute for Health Research Cambridge Biomedical Research Centre, European Research Council (268834), European Commission Framework Programme 7 (HEALTH-F2-2012-279233), Merck and Pfizer. PROSPER has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n° HEALTH-F2-2009-223004.
Footnotes
URLs
CARDIoGRAMC4D Consortium: http://www.cardiogramplusc4d.org/, DIAGRAM Consortium: http://diagram-consortium.org/, COLOC Tool: https://github.com/chr1swallace/coloc, Bivariate scan analysis code: https://github.com/WWinstonZ/bivariate_scan/. WebGestalt: http://www.webgestalt.org/option.php, GRAIL: http://software.broadinstitute.org/mpg/grail/, DAPPLE: http://archive.broadinstitute.org/mpg/dapple/dapple.php, SNPTEST: https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html, LocusZoom: http://locuszoom.org/, METAL: http://genome.sph.umich.edu/wiki/METAL, GTEx: https://gtexportal.org/home/, optiCall: https://opticall.bitbucket.io/, zCall: https://github.com/jigold/zCall, RAREMETALWORKER: http://genome.sph.umich.edu/wiki/RAREMETALWORKER, 1000 Genomes: http://www.internationalgenome.org/, PLINK: https://www.cog-genomics.org/plink2, GCTA: http://cnsgenomics.com/software/gcta/, Summary Data availability: http://www.med.upenn.edu/ccebfiles//t2d_meta_cleaned.zip
Author Contributions
A.R., B.F.V., B.G.N., D.J.R., D.S., D.S.A., J.I.R., M.R., O.M., P.F., R.C., R.J.F.L., S.Anand, S.E., S.M., S.Ripatti, T-D.W., W.H-H.S, and W.Zhao conceived of and designed the experiments. A.I., A.M., A.R., A.S.B., B.F.V., B.G.N., B.R.S., C.A.H, D.J.R., D.K.S., D.S., D.S.A., E.P.B., E.S.T., E.T., F.M., F.u.R.M., G.P., I-T.L, I.H.Q., J-J.L., J.C.C., J.I.R., J.M.M.H., J.S.K., J.W.J., J.Z.K, K-W.L, K.D.T., K.M., K.T., M.B., N.M., M.O-M., N.A., N.H.M., N.K.M., N.Q., N.Sattar, O.M., P.C., P.F., P.S., R-H.C., R.C., R.F-S., R.J.F.L., S.Abbas, S.Anand, S.E., S.J., S.M., S.N.H.R., S.Ralhan, S.Ripatti, S.Z.R., T-D.W., T-u.S., T.K., T.L.A., T.S., T.Y., U.M., W.H-H.S, W.I., W.Zhang, W.Zhao, X.G, Y-D.I.C., Y-J.H., Y.L., Y.Y.T., and Z.Y. performed the experiments. A.R., A.S.B., B.F.V., C.A.H, D.S., E.D.A., E.P.B., E.T., I-T.L, J-J.L., J-M.J.J., J.C.C., J.D., J.M.M.H., J.S.K., J.W.J., J.Z.K, K-W.L, K.D.T., M.B., M.I., M.O-M., M.R., N.K.M., N.Sattar, N.Shah, O.M., P.C., P.F., P.R.K., P.S., R-H.C., R.C., R.F-S., R.J.F.L., R.S., R.Y., S.Anand, S.Asma, S.D., S.F.N., S.M., S.Ralhan, T-D.W., N.M., T.L.A., T.Q., T.S., T.Y., U.M., V.S., W-J.L., W.H-H.S, W.Zhang, W.Zhao, X.G, Y-D.I.C., Y-J.H., Y.L., and Y.Y.T. performed statistical analyses. A.I., A.R., A.S., A.S.B., A.T-H., B.F.V., B.R.S., C-C.H., C.A.H, D.K.S., D.S., E.D.A., E.P.B., E.S.T., E.T., F.M., G.P., I-T.L, J-J.L., J-M.J.J., J.C.C., J.D., J.I.R., J.S.K., J.Z.K, K-W.L, K.D.T., K.T., M.I., M.O-M., M.R., N.H.M., N.Q., N.Sattar, N.Shah, O.M., P.C., P.R.K., P.S., R-H.C., R.F-S., R.J.F.L., R.S., R.Y., S.Abbas, S.Asma, S.D., S.J., S.Ralhan, T.K., T.L.A., T.Q., T.S., T.Y., U.M., V.S., W-J.L., W.Zhao, X.G, Y-D.I.C., Y-J.H., Y.L., Y.Y.T., and Z.Y. analyzed the data. A.M., A.S., A.T-H., B.F.V., B.G.N., D.J.R., D.S., F.u.R.M., G.P., I.H.Q., J-M.J.J., J.D., J.W.J., N.M., K.M., M.B., M.R., N.A., P.R.K., R.S., S.E., S.N.H.R., S.Ripatti, S.Z.R., T-u.S., V.S., W.I., and W.Zhao contributed-reagents, materials, and/or analysis tools. A.R., A.S.B., B.F.V., D.J.R., D.K.S., D.S., E.T., G.P., J-J.L., J.D., J.I.R., J.M.M.H., M.R., N.Sattar, V.S., W.Zhao wrote the paper. D.S. and B.F.V. lead the writing group. W.Zhao, A.R., and E.T., B.F.V. and D.S. were equal contributors. B.F.V. and D.S. jointly supervised all aspects of the work.
References
- 1.Guariguata L, et al. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–49. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Xu J, Murphy SL, Kochanek KD, Bastian BA. Deaths: Final Data for 2013. Natl Vital Stat Rep. 2016;64:1–119. [PubMed] [Google Scholar]
- 3.Emerging Risk Factors, C. et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011;364:829–41. doi: 10.1056/NEJMoa1008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott RA, et al. A genomic approach to therapeutic target validation identifies a glucose-lowering GLP1R variant protective for coronary heart disease. Sci Transl Med. 2016;8:341ra76. doi: 10.1126/scitranslmed.aad3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikpay M, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bulik-Sullivan B, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen H, et al. Genetic variants primarily associated with type 2 diabetes are related to coronary artery disease risk. Atherosclerosis. 2015;241:419–26. doi: 10.1016/j.atherosclerosis.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Replication DIG, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–44. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundberg E, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huyghe JR, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet. 2013;45:197–201. doi: 10.1038/ng.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saleheen D, et al. The Pakistan Risk of Myocardial Infarction Study: a resource for the study of genetic, lifestyle and other determinants of myocardial infarction in South Asia. Eur J Epidemiol. 2009;24:329–38. doi: 10.1007/s10654-009-9334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 15.Ross S, et al. Mendelian randomization analysis supports the causal role of dysglycaemia and diabetes in the risk of coronary artery disease. Eur Heart J. 2015;36:1454–62. doi: 10.1093/eurheartj/ehv083. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad OS, et al. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat Commun. 2015;6:7060. doi: 10.1038/ncomms8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Global Lipids Genetics, C. et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shungin D, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–96. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raychaudhuri S, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossin EJ, et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Pontual L, et al. Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat Genet. 2011;43:1026–30. doi: 10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuro-o M, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor L, et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–95. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki M, et al. Plasma FGF21 concentrations, adipose fibroblast growth factor receptor-1 and beta-klotho expression decrease with fasting in northern elephant seals. Gen Comp Endocrinol. 2015;216:86–9. doi: 10.1016/j.ygcen.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimbert P, et al. Truncation of C-mip (Tc-mip), a new proximal signaling protein, induces c-maf Th2 transcription factor and cytoskeleton reorganization. J Exp Med. 2003;198:797–807. doi: 10.1084/jem.20030566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madsen LS, et al. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet. 1999;23:343–7. doi: 10.1038/15525. [DOI] [PubMed] [Google Scholar]
- 28.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–7. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sattar N, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- 30.Swerdlow DI, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385:351–61. doi: 10.1016/S0140-6736(14)61183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White J, et al. Association of Lipid Fractions With Risks for Coronary Artery Disease and Diabetes. JAMA Cardiol. 2016;1:692–9. doi: 10.1001/jamacardio.2016.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fall T, et al. Using Genetic Variants to Assess the Relationship Between Circulating Lipids and Type 2 Diabetes. Diabetes. 2015;64:2676–84. doi: 10.2337/db14-1710. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt AF, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105. doi: 10.1016/S2213-8587(16)30396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–7. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ballantyne CM, et al. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study) Am J Cardiol. 2012;110:984–92. doi: 10.1016/j.amjcard.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 36.Ballantyne CM, et al. Effects of icosapent ethyl on lipoprotein particle concentration and size in statin-treated patients with persistent high triglycerides (the ANCHOR Study) J Clin Lipidol. 2015;9:377–83. doi: 10.1016/j.jacl.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Boord JB, et al. Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:1686–91. doi: 10.1161/01.atv.0000033090.81345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotamisligil GS, et al. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–9. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 39.Makowski L, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furuhashi M, et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–65. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burak MF, et al. Development of a therapeutic monoclonal antibody that targets secreted fatty acid-binding protein aP2 to treat type 2 diabetes. Sci Transl Med. 2015;7:319ra205. doi: 10.1126/scitranslmed.aac6336. [DOI] [PubMed] [Google Scholar]
References Online-Methods
- 42.Genomes Project, C. et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng S, Liu D, Zhan X, Wing MK, Abecasis GR. RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics. 2014;30:2828–9. doi: 10.1093/bioinformatics/btu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu DJ, et al. Meta-analysis of gene-level tests for rare variant association. Nat Genet. 2014;46:200–4. doi: 10.1038/ng.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dastani Z, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8:e1002607. doi: 10.1371/journal.pgen.1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans DM, Davey Smith G. Mendelian Randomization: New Applications in the Coming Age of Hypothesis-Free Causality. Annu Rev Genomics Hum Genet. 2015;16:327–50. doi: 10.1146/annurev-genom-090314-050016. [DOI] [PubMed] [Google Scholar]
- 49.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–75. S1–3. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008;8:186–200. doi: 10.1016/j.cmet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary GWAS estimates for the T2D meta-analysis and bivariate summary data are publicly available at the following: