

Abstract
An academic chemical screening approach was developed by using 2D protein-detected NMR, and a 352-chemical fragment library was screened against three different protein targets. The approach was optimized against two protein targets with known ligands: CXCL12 and BRD4. Principal component analysis reliably identified compounds that induced nonspecific NMR crosspeak broadening but did not unambiguously identify ligands with specific affinity (hits). For improved hit detection, a novel scoring metric—difference intensity analysis (DIA)—was devised that sums all positive and negative intensities from 2D difference spectra. Applying DIA quickly discriminated potential ligands from compounds inducing nonspecific NMR crosspeak broadening and other nonspecific effects. Subsequent NMR titrations validated chemotypes important for binding to CXCL12 and BRD4. A novel target, mitochondrial fission protein Fis1, was screened, and six hits were identified by using DIA. Screening these diverse protein targets identified quinones and catechols that induced nonspecific NMR cross-peak broadening, hampering NMR analyses, but are currently not computationally identified as pan-assay interference compounds. The results established a streamlined screening work-flow that can easily be scaled and adapted as part of a larger screening pipeline to identify fragment hits and assess relative binding affinities in the range of 0.3–1.6 mM. DIA could prove useful in library screening and other applications in which NMR chemical shift perturbations are measured.
Keywords: drug discovery, fragment-based screening, NMR spectroscopy, PAINS, principal component analysis
Can you tell the difference?
Difference intensity analysis of fragment-based screening by NMR 2D difference spectra enabled us to quickly discriminate potential chemical fragment hits from non-hits, compounds inducing nonspecific NMR crosspeak broadening, and other nonspecific effects. This quantitative NMR spectral analysis method could assist in library screening and other applications in which NMR chemical- shift perturbations are measured.
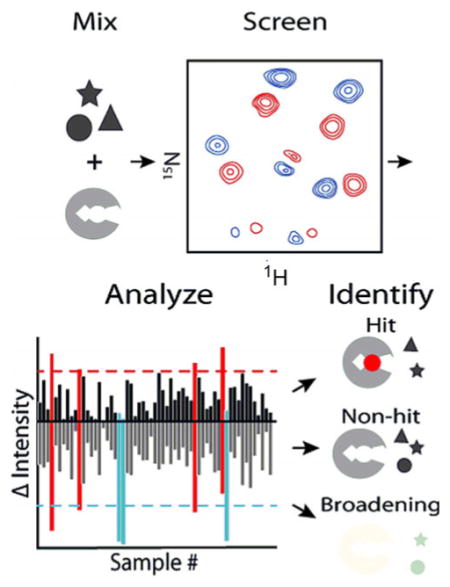
Introduction
Traditional drug discovery often involves screening a large chemical library (>50000 compounds) in high-throughput assays.[1] Obtaining such a library can be a limiting barrier, due to costs and resources associated with library construction and maintenance.[1, 2] An attractive way to address this barrier is fragment-based screening (FBS), which screens much smaller libraries (250–5000 compounds) of lower molecular weight compounds (<300 Da), termed fragments. The requirement for a low molecular weight fragment typically results in one chemotype present in each molecule and lower affinity compared to higher molecular weight drug-like compounds (<500 Da). However, fragments have reduced steric requirements compared to larger compounds, which allows efficient sampling of the available chemical space capable of binding a protein target. FBS has been used to discover two FDA-approved drugs, with more than thirty compounds currently in clinical trials.[3] Given the reduced library size, FBS is particularly well suited for academic drug discovery efforts, which typically have fewer available resources than industry. FBS has been increasingly used with NMR for hit-to-lead drug discovery efforts.[4] We sought to use 2D protein-detected NMR because of its numerous advantages over other screening techniques. Due to its high sensitivity and detection of protein residue chemical shifts, compounds with low affinity (high-millimolar range) can more readily be distinguished from false positives.[5] Further, protein-detected NMR can identify the binding location(s) of hits, as well as binding affinity by subsequent titration experiments. Here, we screened three distinct protein targets against a 352-fragment library by using 2D protein-detected NMR and identified fragments with binding constants in the range of 0.3–1.6 mM affinity.
We first validated our FBS approach against CXCL12, a chemokine implicated in cancer cell metastasis with known ligands.[6, 7] When CXCL12 binds its cognate receptor, CXCR4, chemotactic cell migration ensues, which is essential for metastatic cancer progression.[8] A novel therapeutic strategy is to disrupt the CXCL12:CXCR4 interaction. Previous in silico screening of CXCL12 identified fragment- and lead-like ligands with experimentally verified affinities in the micro- to millimolar range.[6, 7] Using CXCL12 as a testbed target protein, we questioned whether screening a 352-fragment library was sufficient to identify similar chemotypes or reveal new binding modalities.
We further validated our approach against the second bromodomain of BRD4, BRD4(BD2), an epigenetic regulatory protein of the bromodomain and extraterminal domain (BET) family (BRDT, BRD2, BRD3, and BRD4) implicated in the progression of various forms of cancer and inflammatory disease.[9] BET family proteins localize to the nucleus and specifically recognize and bind to acetylated lysine residues, influencing transcription by marking sites of acetylated chromatin throughout the genome.[10] Currently, small molecule inhibitors of BET bromodomains, which selectively displace BET proteins from acetylated histones, are in clinical trials for applications that include hematological malignancies, solid tumors, and inflammatory disorders.[9] However, these potential drugs are nonselective, as they bind all four BET family proteins with equal low nanomolar affinity.[11] As a result, more selective compounds are desired to elucidate the biological functions of individual BET proteins and to minimize side effects associated with current therapeutics targeting BET bromodomains.[12,13] We reasoned that an FBS approach might validate current molecules and possibly identify new chemotypes selective for BRD4.
The third—and novel—protein target we screened was Fis1, a mitochondrial outer membrane protein implicated in mitochondrial fission.[14,15] Excess mitochondrial fission is associated with several disease pathologies, including pulmonary arterial hypertension and diabetic cardiomyopathy.[16,17] Genetic inhibition of Fis1 prevents excess fission and improves mitochondrial function;[16,18,19] this suggests a novel pharmacological approach of inhibiting Fis1 to protect against cellular damage in various pathologies.
Here, we used an FBS by NMR approach, using 2D protein-detected heteronuclear experiments to identify fragment hits against CXCL12, BRD4(BD2), and Fis1. Limitations to principal component analysis (PCA) of 2D protein-detected NMR data were mitigated by a new metric: difference intensity analysis (DIA) of 2D difference spectra. DIA, in a protein-independent manner, readily identified potential hits from non-hits, including compounds inducing nonspecific NMR crosspeak broadening. From this, we identified unique chemical fragments against each target with binding affinities ranging from high micromolar to low millimolar values. The data suggest that a 352-compound library is a reasonable first step in building a fragment library, and FBS by NMR, aided by DIA, can serve as a powerful component of an academic FBS program.
Results and Discussion
Validating approach against CXCL12
We established an academic-based approach to identify chemical fragments specific to protein targets implicated in various diseases, with the ultimate goal of discovering high-affinity chemical biology tools (Figure 1). We first optimized and validated our workflow by using the chemokine CXCL12. CXCL12 was chosen because we previously identified novel small molecules that bind to three distinct sites with affinities from 64–4000 μM.[6, 7] We screened 15N,13C-labeled CXCL12 against a commercially available library of 352 fragments in fragment mixtures with six compounds per NMR sample by collecting 1H,13C HSQC and 1H,15N SOFAST-HQMC spectra. Manual inspection of spectral overlays between fragment mixtures and DMSO control spectra of CXCL12 identified a subset of samples with clear chemical shift perturbations (CSP) from 1H,15N SOFAST-HMQC (Figure 2A), but not 1H,13C HSQC (Figure S1, Supporting Information), experiments. The lack of hit detection when using carbon-based methods was most likely due to the higher sensitivity of 15N chemical shifts (arising from chemical shift anisotropy) to ligand binding compared to 13C chemical shifts.[20] Carbon nuclei are less sensitive to through-space interactions, and an observable carbon chemical shift perturbation would require local conformational changes, which are not likely to be induced by a weak affinity fragment.[21–23]
Figure 1.

Schematic of fragment-based hit detection using 1H,15N SOFAST-HMQC or HSQC experiments. Six fragments (1 mM each) were mixed with 100 μM 15 N-labeled protein and screened by NMR. After data collection, spectral changes were quantified by a novel scoring metric: DIA. For DIA, 2D difference spectra were computed from DMSO control and each fragment mixture, which quantitatively discriminated hits, non-hits, and fragments that induced nonspecific NMR crosspeak broadening in a protein-independent manner. Individual fragments were identified by parsing experiments, and subsequent titrations allowed determination of binding affinities to develop initial structure–activity relationships.
Figure 2.

Fragment-based hit detection by NMR against 15N-CXCL12. A) Spectral overlay of fragment mixture 40. B) PCA for all 60 spectra using the NMRPipe module pcaNMR.[24, 25] The PCA score plot is annotated with 68 % (blue ellipse) and 95 % (red ellipse) CIs of CXCL12 1H,15N SOFAST-HMQC spectra. The percent explained variance is indicated on each axis for each PC. Samples that induced nonspecific NMR crosspeak broadening (gray), no change (black), or were identified as a hit (red) are indicated. C) DIA from 2D difference spectra showing potential hits (>+1 σ) and compounds that induced nonspecific NMR cross-peak broadening (<−1 σ). D) 1H,15N SOFAST-HMQC overlays of CXCL12 titrated with ZT0784 (inset), which was parsed from fragment mixture 40. E) Quantification of total chemical shift perturbations (1H/15N Δδ chemical shift) elicited by ZT0784 for individual amino acid residues of CXCL12. F) Fitted curves of indicated residues used to calculate binding affinity (Kd = 1.0 ± 0.1 mM) of ZT0784 for CXCL12. G) Mapping of substantially perturbed residues on the solution structure of CXCL12 (PDB ID: 2K05, monomer A). The in silico docked pose of ZT0784 is included.
Manual inspection of HSQCs is commonplace in identifying CSPs; however, it is cumbersome, subjective, and qualitative. PCA offers an attractive alternative.[24–26] Using automated scripts in R, Bruker TopSpin 3.5, or NMRPipe, we generated PCA score plots for the set of 60 1H,15N SOFAST-HMQC spectra of CXCL12. We found 81 % of the variance was contained in PC1 and PC2 and could not be attributed to baseplane noise (Figure S2). Eight NMR samples gave PC variances outside the 95% confidence interval (95 % CI; Figure 2 B). To determine the spectral basis of these variances, we manually inspected 2D spectral overlays of all datasets. Three of the eight NMR samples (31, 40, and 45) contained unambiguous CSPs. These three samples were also found in a cluster distinct from the remaining five samples (20–22, 42, and 48). By contrast, these other five samples (20–22, 42, and 48) displayed small, if any, changes in chemical shifts but had significantly attenuated crosspeak intensities. The intensity loss was not uniform and suggested that some fragments might have induced intermediate exchange, nonspecific protein modification (i.e., free amines), or aggregation of the protein sample.
The inability of PCA to unambiguously discriminate potential fragment hits from nonspecific interactions prompted us to explore alternative methods to quantify spectral changes and avoid subjective manual inspection of spectral overlays. We reasoned that 2D difference spectra computed from the DMSO control and each fragment mixture should quickly discriminate CSPs from intensity losses due to non-binding interactions.
For CXCL12 and the other proteins we tested to date, changes in 2D difference spectra were classified into three categories: little change, global intensity loss, or clear CSPs (Figure S3). An NMR sample with little to no change in chemical shift or intensities relative to the DMSO control resulted in few to no peaks observed in the 2D difference spectrum and was classified as a non-hit. In contrast, some NMR samples yielded spectra similar to the DMSO control but with negative intensities in the 2D difference spectrum, indicating a global loss of signal upon fragment addition, consistent with NMR crosspeak broadening, protein aggregation, or slow to intermediate exchange. 2D difference spectra that presented positive and negative intensities arose from clear CSPs and were classified as potential hits.
To quantify spectral differences, we consolidated each 2D difference spectrum into two values by summing all the positive crosspeak intensities into a single positive value and doing the same for all negative crosspeak intensities (Figure 2 C, Table S1). For 60 NMR samples, this analysis gave 60 positive and negative values. The uncertainty in these values was estimated from consecutive HSQC spectra on a control sample and found to be ≈6 %. CXCL12 samples that gave rise to significant positive and negative values of equal magnitude indicated a potential hit arising from multiple CSPs in 2D difference spectra. In contrast, samples that gave rise to a significant negative value indicated compound(s) that induced NMR crosspeak broadening, likely due to intermediate exchange or nonspecific interaction with/modification of the protein. To quantitatively discriminate potential hits from non-hits, we took the mean of the positive and negative intensities of all 60 samples, calculated the standard deviation, and set a threshold at one standard deviation from the mean. Any sample/spectrum with a positive intensity greater than this threshold signaled a potential hit for further consideration. Samples that gave rise to a negative intensity exceeding the negative threshold without a positive intensity higher than the positive threshold signaled a potential interfering compound that induced nonspecific NMR crosspeak broadening. We refer to this overall process as difference intensity analysis (DIA).
For CXCL12, DIA identified ten NMR samples that gave positive intensities above the +1 σ threshold (1, 23, 24, 31, 39, 40, 45, 50, 53, and 54) and thirteen samples that gave negative intensities below the −1 σ threshold (1, 20, 21, 22, 31, 32, 39, 40, 41, 42, 45, 48, and 50, Figure 2C). Six of these samples (1, 31, 39, 40, 45, and 50) exceeded both thresholds and gave rise to the largest positive intensities, which is expected from ligand binding, as a perturbation that shifts a crosspeak from the DMSO control will result in equal intensity positive and negative peaks in the 2D difference spectrum. We assigned all ten samples exceeding the +1σ threshold as potential hits. Baseplane noise was not shown to contribute significantly to DIA values (Figures S2 and S4). Manual inspection of spectral overlays for these ten samples verified that the seven samples with the highest positive intensity values (1, 24, 31, 39, 40, 45, and 50) derived from distinct CSPs. By contrast, the three samples with the lowest positive intensity values above the +1 σ threshold (23, 53, and 54) did not give rise to substantial CSPs and were classified as false positives. Samples with negative intensities below the −1 σ threshold but not near the +1 σ threshold (20, 21, 22, 41, 42, and 48) were deemed to arise from substantial nonspecific NMR crosspeak broadening, based on manual inspection of 2D overlays that showed uniform loss of crosspeak intensity (data not shown).
The finding that three of the ten samples with positive intensity values above the +1 σ threshold were false positives suggested that the stringency of this threshold might need to be raised. However, we inspected all remaining overlays and discovered one more sample with CSPs suggestive of binding (32). This sample was below our positive, but not our negative, threshold. Interestingly, in the PCA score plot (Figure 2 B), sample 32 clustered near the seven samples identified from DIA and was included, along with the seven samples with the highest positive intensity values, to give 48 total fragments for further analysis. Each fragment was individually tested at 1 mM with 50 μM 15N-CXCL12, and eight fragments were found to give significant CSPs, including one from sample 32. One fragment mixture (24) yielded no individual fragment-induced CSPs. We concluded that DIA is useful in quickly highlighting samples for further analysis and could benefit from being coupled with PCA to evaluate samples near the threshold.
Of the eight fragments, seven contained one or more carboxylate groups, a moiety known to enhance chemical fragment binding to CXCL12.[6, 7] To determine binding affinities, these eight fragments were titrated (0–1600 μM) against 50 μM 15N-CXCL12 and monitored by 1H,15N SOFAST-HMQC experiments (Figure 2D). Each fragment induced CSPs encompassing two of three known ligand binding pockets on CXCL12—the so-called sY12 and I4/I6 sites[7]—which include residues Val18, Val23, Lys24, His25, and Lys27 (Figure 2E, G). Therefore, it appeared a 352-compound library was not of sufficient size to detect all possible binding sites, as only two of the three sites were detected for CXCL12. Global nonlinear fitting of the titration data from these residues for one of the fragments (ZT0784) yielded an apparent Kd value of 1.0 ± 0.1 mM (Figure 2 F). Similar affinities were found for the other fragments (Table 1). Unbiased molecular docking studies with these fragments against a high-resolution structure of CXCL12 placed the fragments within the area identified by the NMR titration experiments. Curiously, the I4/I6 site occupied by ZT0784 (Figure 2 G) was an ancillary site with respect to binding the cognate receptor of CXCL12[7] and lent support to the idea that fragment-based screening can identify alternative or cryptic binding sites that might be missed when screening with libraries of larger (for example, drug-like or lead-like) compounds. In summary, DIA was useful in interpreting CXCL12 screening data and identified eight fragments (2 % hit rate) with affinities expected of a fragment-based approach.
Table 1.
Summary of hit identification for CXCL12, BRD4(BD2), and Fis1.
| Target | CXCL12 | BRD4(BD2) | Fis1 |
|---|---|---|---|
| Fragment mixture samples for fragment parsing | 8 | 8 | 10 |
| Potential hits isolated from single fragments screened | 8/48 | 8/48 | 6/60 |
| Potential hits titrated | 8 | 1 | 6 |
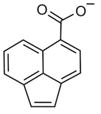 1.0 ± 0.1 mM [0.28] 1.0 ± 0.1 mM [0.28] |
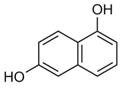 0.34 ± 0.2 mM [0.40] 0.34 ± 0.2 mM [0.40] |
||
1.0 ± 0.1 mM [0.35]
|
 1.6 ± 0.2 mM [0.36] |
0.85 ± 0.4 mM [0.36]
|
|
| Fragments for further study Kd [LE] |
 1.0 ± 0.2 mM [0.32] 1.0 ± 0.2 mM [0.32] |
 1.3 ± 0.4 mM [0.34] 1.3 ± 0.4 mM [0.34] |
|
1.2 ± 0.1 mM [0.37]
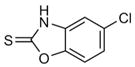
|
1.5 ± 0.3 mM [0.33]
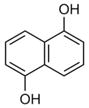
|
For each target screened, the number of fragment mixtures that led to individual isolated hits is shown. A subset of fragments that were assessed for binding by titration experiments are included, along with their binding affinities and ligand efficiencies (LE), which were calculated as LE=(1.4(−logKd))/N, where N =total number of non-hydrogen atoms.
Validation approach against BRD4(BD2)
We next evaluated whether our approach could identify fragments for an entirely different protein: a bromodomain involved in epigenetic gene regulation. We first evaluated FTMap analysis on the second bromodomain of BRD4 (BRD4(BD2)), which identified a strong hot spot (cluster strength [CS] =34) located within the acetylated lysine binding pocket (Figure 3 A). In addition, two neighboring hot spots (<8 Å away) were identified, thus suggesting ligands to BRD4(BD2) are discoverable, as expected.
Figure 3.

Fragment-based hit detection by NMR against 15N-BRD4(BD2). A) Potential ligand binding hot spots were identified by FTMap analysis on the crystal structure of BRD4(BD2) (PDB ID: 4Z93). Hot spot regions are ranked by the number of probes in each cluster (cluster strength; CS1–5). B) Spectral overlay of fragment mixture 52. C) PCA score plot with 68 % (blue ellipse) and 95 % (red ellipse) confidence intervals of BRD4(BD2) 1H,15N HSQC spectra. The percent explained variance is indicated on each axis for each PC. Fragment mixtures that induced nonspecific NMR crosspeak broadening (gray), no change (black), or were identified as a hit (red) are indicated. D) DIA from 2D difference spectra showing the potential hits (>+1 σ) and compounds that induced nonspecific NMR crosspeak broadening (<−1 σ). E) 1H,15N SOFAST-HMQC overlays of BRD4(BD2) titrated with ZT0769 (inset), which was parsed from fragment mixture 52. F) Quantification of total chemical shift perturbations (1H/15N Δδ chemical shift) elicited by ZT0769 for individual amino acid residues of BRD4(BD2). G) Fitted curves of indicated residues used to calculate binding affinity (Kd = 1.6 ± 0.2 mM) of ZT0769 for BRD4(BD2). H) Mapping of substantially perturbed residues on the crystal structure of BRD4(BD2) (PDB ID: 4Z93). The in silico docked pose of ZT0769 is included.
Prior to screening 15N-labeled BRD4(BD2), we first collected a 1H,15N SOFAST-HMQC spectrum that exhibited NMR crosspeak broadening of resonances that could obscure interpretation of screening data. By contrast, 1H,15N HSQC spectra gave rise to higher quality data with a twofold increase in screening data collection time (Figure 3 B). We screened the 352-fragment library by collecting 1H,15N HSQC data and generated PCA score plots for all 60 HSQC spectra. A plot of PC1–PC2 (Figure 3C) clustered much more tightly than the CXCL12 PCA plot (Figure 2 B), although ten NMR samples lay away from this cluster and outside the 95% CI (14, 16, 19, 20–22, 25, 32, 48, and 59). We suspected that many of these BRD4 outliers derived from samples with nonspecific NMR crosspeak broadening, as found with CXCL12. Indeed, DIA of the BRD4(BD2) 2D difference spectra revealed that all but two of these samples (25 and 32) gave negative intensity values exceeding the −1σ threshold indicative of nonspecific NMR crosspeak broadening (Figure 3 D, Table S2). Four other NMR samples with negative intensity values less than −1σ (9, 11, 40, and 47) were also found. Two of these samples (11 and 40) also had positive intensities greater than +1 σ, indicative of ligand binding, and were outliers above the 68 % CI in PCA. One additional BRD4(BD2) sample (34) had sufficient total positive intensity (>+1σ) to warrant further examination, although it was not highlighted in PCA (Figure 3 C, D).
Manual inspection of 2D spectral overlays confirmed four samples (11, 34, 40, and 47) were worthy of further consideration. Also, four more samples with positive intensities just below our threshold were assessed to determine the stringency of the +1 σ threshold (41, 52, 53, and 55). Although these samples did not show significant variance in PCA, we identified chemical fragments in individual parsing experiments that were indicative of ligand binding. We interpreted this finding to indicate that DIA is potentially more useful than PCA in identifying hits from protein-detected NMR data.
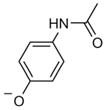
Perhaps most interesting was sample 52, which when parsed into individual fragments at 1 mM against 50 μM 15N-BRD4(BD2) identified acetaminophen as giving substantial CSPs (Figure 3 E). Acetaminophen has previously been shown to bind the first bromodomain of BRD4 with a Kd value of 290 μM.[27] Titrations of 0–1600 μM acetaminophen with BRD4(BD2), monitored by 1H,15N HSQC experiments, yielded data that were simultaneously fit to give an apparent Kd value of 1.6 ± 0.2 mM (Figure 3 F–H). The weaker affinity of acetaminophen for the second compared to the first bromodomain was consistent with previous observations that BRD4(BD2) generally harbors weaker ligand affinities compared to BRD4(BD1).[10] Individual compound parsing experiments also identified additional potential ligands for BRD4(BD2) spanning diverse chemotypes, for which Kd values were not determined.
Possible orientations for acetaminophen binding to the acetyl-lysine binding pocket of BRD4(BD2) were revealed from unbiased computational docking studies, which agreed with the experimentally identified binding pocket on BRD4(BD2) (Figure 3 H). These findings were consistent with previous observations that the relatively large and hydrophobic acetyl-lysine binding pockets of BET bromodomains are highly drug-gable compared to other bromodomains.[28] Indeed, many screening efforts have successfully identified small molecules for BET bromodomain acetyl-lysine binding pockets.[29–31] In summary, screening BRD4(BD2) against our chemical library identified acetaminophen, a compound known to interact with bromodomains, and demonstrated the feasibility and success of our approach in identifying compounds that interact with target proteins for further characterization.
FTMap analysis of Fis1
After validating our approach against CXCL12 and BRD4(BD2), we screened the novel protein target Fis1. To assess potential small molecule binding sites on Fis1, FTMap analysis was performed on crystallographic and NMR solution structures of the cytoplasmic domain of Fis1 (residues 1–125). FTMap analysis of a 2.0 Å crystallographic Fis1 structure identified five hot spots (xCS1–5) on the concave TPR-containing pocket of Fis1 (Figure 4 A, left). The strongest hot spot xCS1 (CS=17) was located within 8 Å of the xCS3, xCS4, and xCS5 hot spots, giving rise to a putative ligand binding site. As the conformations between crystallographic and solution structures of Fis1 differed, all 20 Fis1 conformations of the NMR ensemble were also analyzed by FTMap analysis (Figure S5). The same five hot spots were identified with one notable exception: a putative binding pocket (nCS1) on the convex face of Fis1 (Figure 4 A, right). Taken together, these results suggested that chemical fragments binding to the concave face of Fis1 could be discovered by using our screening approach.
Figure 4.

Fragment-based hit detection by NMR against Fis1. A) Comparison of computational solvent mapping analysis by FTMap on crystallographic (left, PDB ID: 1NZN) and solution (right, PDB ID: 1PC2) structures of hFis1. The Fis1 structures are positioned similarly. The C-terminal disordered region of the solution structure of Fis1 was removed for clarity. Hot spot regions are ranked by number of probes in each cluster (cluster strength; xCS =cluster strength for crystal structure; nCS =cluster strength for solution NMR structure). B) Spectral overlay of fragment mixture 4. C) PCA score plot with 68 % (blue ellipse) and 95 % (red ellipse) CIs of Fis1 1H,15N SOFAST-HMQC spectra. The percent explained variance is indicated on each axis for each PC. Samples that induced non-specific NMR crosspeak broadening (gray), no change (black), or were identified as a hit (red) are indicated. D) DIA from 2D difference spectra showing potential hits (>+1 σ) and compounds that induced nonspecific NMR crosspeak broadening (<−1 σ). E) 1H,15N SOFAST-HMQC overlays of Fis1 titrated with ZT0096 (inset), which was parsed from fragment mixture 4. F) Quantification of total chemical shift perturbations (1H/15N Δδ chemical shift) elicited by ZT0096 for individual amino acid residues of Fis1. G) Nonlinearly fitted curves of indicated residues used to calculate binding affinity (Kd = 1.5 ± 0.3 mM) of ZT0096 for Fis1. H) Mapping of substantially perturbed residues on the crystal structure of Fis1 (PDB ID: 1NZN). The in silico docked pose of ZT0096 is included.
Identification of fragment hits against Fis1
Fis1 was screened against the 352-fragment library by 1H,15N SOFAST-HMQC experiments in a similar manner to CXCL12 and BRD4(BD2) (Figure 4B). 1H,13C HSQC experiments were also collected on 15N,13C-Fis1 and, like CXCL12, 13C-based protein detection methods were unable to discriminate hits from non-hits (Figure S6). PCA of the resulting 1H,15N SOFAST-HMQC spectra identified four NMR samples (9, 21, 22, and 48) outside the 95% CI (Figure 4C). Samples 21–22 and 48 gave the most negative total crosspeak intensity, exceeded the −1σ threshold (Figure 4 D), and caused nonspecific NMR crosspeak broadening. DIA revealed that only four NMR samples (4, 11, 40, and 47) had sufficient positive intensity (>+1 σ) to warrant further examination (Figure 4D, Table S3), which was confirmed by manual inspection of 2D overlays. To assess whether samples below the +1σ threshold might give rise to CSPs, we included five additional NMR samples for further analysis: two samples just below our threshold (31 and 52), and three randomly selected (01, 03, and 59). The 95% CI PCA outlier 9 was also included, to give a total of ten NMR samples for parsing.
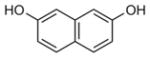
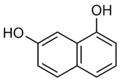
To determine which individual fragment hits were contained within these ten NMR samples, 60 fragments plus a DMSO control were individually screened at 1 mM with 50 μM 15N-Fis1 by 1H,15N SOFAST-HMQC experiments. Six chemical fragments elicited CSPs indicative of binding, which originated from fragment mixture samples near or at the +1 σ threshold (4, 11, 32, 40, and 47). By contrast, the other samples examined showed no substantial spectral changes upon addition of individual ligands, supporting the use of the +1 σ threshold. Each of the six fragments were titrated (0–1600 μM) against 50 μM 15N-Fis1 and monitored by 1H,15N SOFAST-HMQC experiments (Figure 4 E). The resulting data were fit to determine binding affinities that ranged from 0.34 to 1.50 mM (Table 1). An example is shown for Fragment ZT0096 (isolated from sample 4), in which global fitting CSPs of individual residues resulted in a Kd value of 1.5 ± 0.3 mM (Figure 4F–H). The CSPs from each fragment highlighted the same putative binding site on the concave pocket of Fis1. Curiously, four of the fragments were dihydroxynaphthalene isomers with similar affinities, regardless of the position of the hydroxy groups. Three other naphthalene-containing compounds (1,8-diaminonaphthalene, 3-amino-2-naphthol, and naphthalene-2-sulfonamide) were present in our library, none of which bound appreciably.
To further inform fragment binding location, we performed unbiased fragment docking with a high-resolution structure of Fis1. In agreement with CSP mapping, all six fragments docked to the concave pocket of Fis1 in a similar manner, which was also predicted by FTMap (Figure 4 A, H). We concluded that screening a 352-compound library and analyzing the resulting spectra by PCA and 2D difference data by DIA can rapidly identify distinct chemical moieties with affinities expected for fragments with an average molecular weight of 150 Da.
Identification of common interfering compounds inducing nonspecific NMR crosspeak broadening
Across all targets screened, we identified 18 possible compounds that induced nonspecific NMR crosspeak broadening contained in fragment mixtures 21, 22, and 48. These samples were easily discernible in DIA plots (largest negative intensity, Figures 2 C, 3D, and 4D) and clustered away from the bulk of other spectra in the PCA plots (Figures 2 B, 3C, and 4C). To identify which of the 18 fragments induced nonspecific NMR crosspeak broadening, we conducted 1H,15N SOFAST-HMQC experiments on 50 μM 15N-Fis1 and 100 μM 15N-CXCL12 and 1H,15N HSQC experiments on 100 μM 15N-BRD4(BD2) with 1 mM each of individual compounds. Six fragments consisting of quinones, catechols, and ninhydrin induced nonspecific NMR crosspeak broadening of Fis1 (Table S4) and were not deemed colloidal aggregators, as addition of 0.1% (v/v) Triton X-100 had no effect on spectral quality (data not shown). To determine if these compounds induced aggregation of Fis1, dynamic light scattering (DLS) measurements were collected on Fis1 in the absence and presence of each of the six interfering compounds. Size distribution analysis demonstrated that all six interfering compounds induced a large change in Fis1 particle size, from a radius of 2.6 nm in the absence of compound to 80–130 nm in the presence of interfering compounds (Figure S7). To evaluate whether the increase in Fis1 particle size was due to a direct chemical modification, mass spectrometry experiments were conducted on Fis1 with and without each of the six interfering compounds. Only one interfering compound (2-methylbenzene-1,4-diol) caused a mass shift (119.7 Da) from the observed molecular weight of apo Fis1 (14 487.2 Da, Figure S8); this suggested direct chemical modification of Fis1. The diol of 2-methylbenzene-1,4-diol could be oxidized to a quinone, which could then react with nucleophiles on the protein.[32,33] If the remaining hydroxy group was again oxidized to the quinone, then the expected mass shift would be 120 Da, which explains the measured mass shift of Fis1.
None of the six interfering compounds against Fis1 were identified by the recently developed software Aggregation Advisor.[34] However, one fragment, 4-aminophenol, was highlighted by Aggregation Advisor, although it did not induce nonspecific NMR crosspeak broadening, as seen in fragment mixtures 21, 22, or 48 (Table S5). Only ninhydrin was found to induce nonspecific NMR crosspeak broadening across all targets screened, which was most likely due to ninhydrin-induced protein aggregation, as was seen for Fis1 by DLS measurements. Another possibility was that ninhydrin reacted with free amines on CXCL12 and BRD4BD2 targets. Given its widespread use in chemical detection of free amines, incorporation of ninhydrin in a commercial fragment library is surprising. Notably, ninhydrin was not identified in a computational search filter for PAINS compounds.
Conclusion
In summary, we presented an accessible workflow to perform fragment-based screening by 2D protein-detected NMR to identify small molecules with affinities ranging from high micromolar to low millimolar against three distinct protein targets. To readily discriminate hits from non-hits, we developed a rapid and quantitative method for consolidating NMR spectra, which we termed difference intensity analysis (DIA). Through DIA, we reduced the complex spectral comparison of a control sample with each fragment mixture to two values reflecting the overall summation of positive and negative intensities in 2D difference spectra. We demonstrated the utility of DIA by robustly differentiating chemical fragments that bound with measurable affinities from those that did not.
The strength of DIA is that it is objectively quantitative and does not rely on subjective manual inspection of spectral overlays, which is advantageous when screening larger chemical libraries. DIA detects chemical shift perturbations independent of the NMR chemical shift timescale and, in principle, is sensitive to slow, intermediate, and fast exchange. For the proteins screened in this study, only fast exchange was detected, as expected for fragments with weak affinity. Our initial threshold for detection of −1 σ worked well but might require adjustment, depending on the protein screened and screening capabilities. For example, applying −1 σ threshold to CXCL12 identified seven fragment mixtures, containing 42 fragments, which, upon individual fragment screening and titration experiments, identified seven hits. Lowering our threshold to +0.75 σ identified five additional NMR samples, but only one of 30 fragments gave detectable CSPs, supporting the choice of a +1 σ threshold. An alternative to a −1σ threshold is to calculate the chi-square value for crosspeaks in the difference spectra, which would take into account the probability that the difference peak could arise from random noise alone. Another approach, although restricting the total possible hits, would be to take the top 5–10 highest positive intensity samples from the DIA plot and follow up on fragments contained within those fragment mixtures. However, this runs the risk of missing important chemotypes for further studies.
DIA performed better than PCA in our hands, with an average false positive rate for DIA of 5.7% compared to 29.9 % for PCA alone (Figure S9). PCA alone did not readily identify fragments that induced substantial CSPs. For each protein, the first principal component represented −80–90 % of the total variance and appeared to reflect changes in peak intensities. By contrast, the second principal component represented <3% of total variance and appears to reflect CSPs. Others have made similar conclusions from ligand binding studies,[24] which supports focusing on the variance in the second principal component to identify potential hits. One way to accomplish this is to identify samples from DIA with large negative and small positive values, remove fragment mixtures that induce nonspecific NMR crosspeak broadening from the analysis, and repeat PCA. This approach removed large variances due to intensity differences and better highlighted potential hits from non-hits for all three screened proteins (Figure S10). Another method we evaluated was to analyze the 2D difference spectra by PCA, which did not directly eliminate the crosspeak intensity issue but did highlight the largest variances in these datasets (Figure S11). From our analyses, DIA in conjunction with PCA after removal of compounds that induced nonspecific NMR cross-peak broadening was most useful. Collectively, DIA could prove useful in library screening and other applications in which NMR chemical shift perturbations are measured.
Experimental Section
Protein expression, purification, and stability
Recombinant human 15N-CXCL12 was purchased from Protein Foundry (Milwaukee, WI, USA). The cytosolic domain of human Fis1 (residues 1–125) was subcloned into the pQE30 expression system as an N-terminal His6-SUMO fusion protein and purified by Ni-affinity chromatography as previously described.[35] The recombinant N-terminally His6-tagged BRD4(BD2) bromodomain construct (Addgene 38943; residues 333–460) was purified from BL21(DE3) Escherichia coli by using nickel affinity chromatography. Both Fis1 and BRD4(BD2) were expressed in minimal medium (1 L) supplemented with 15N ammonium chloride (3.0 g) as the only source of nitrogen (see the Supporting Information for a full description of recombinant protein expression and purification for Fis1 and BRD4(BD2)). The stability of each protein was evaluated by 1H,15N HSQC experiments at 0, 3, and 14 days at 25°C. Inspection of spectral overlays showed negligible changes in either chemical shift or intensity.
FTMap analysis
The potential ligand binding sites of each protein target were assessed by using computational solvent mapping with FTMap.[36] The structures of Fis1 (PDB ID: 1NZN and 1PC2) and BRD4(BD2) (PDB ID: 4Z93) were submitted to the FTMap server with default settings (http://ftmap.bu.edu).[36] Results were visually inspected, and figures were rendered by using PyMol.[37]
Library selection
The Zenobia Therapeutics Library 1 (Zenobia Therapeutics, San Diego, CA) contains 352 fragments, all in accordance with the “rule of three”,[38] with the average molecular weight of fragments being 154 Da, ranging from 94–286 Da. Fragments were received in 96-well plates and solubilized to 200 mM in [D6]DMSO. All fragments were soluble to 200 mM in DMSO and 1 mM in H2O. For the Zenobia library, each compound (10 μL; 352 each in 200 mM [D6]DMSO) was mixed into 59 wells of a 96-well v-bottomed plate, with each well containing six chemical fragments at 33.3 mM each (Figure S12). A sixtieth well contained DMSO only. This step was performed manually resulting in a mother matrix plate with 60 wells containing a total of 60 μL of fragments (or DMSO) in [D6]DMSO. Then, 7.2 μL of each fragment mixture (or DMSO) from the mother matrix plate was transferred to single-use daughter plate 96-well v-bottomed plates. Mother and daughter plates were sealed and stored at −80°C. Chemicals for subsequent NMR titration experiments were purchased from Enamine, AK Scientific, or Sigma–Aldrich. For individual titration experiments, compounds were initially solubilized to 200 mM in [D6]DMSO and stored at −80 °C until further use.
Identification of PAINS and aggregators in fragment library
No pan-assay interference compounds (PAINS) were identified by the PAINS-Remover filter using default parameters (www.cbligand.org/PAINS).[39] Chemicals known to cause to aggregation were identified by using Aggregation Advisor (http://advisor.bkslab.org/), which performs a computational substructure search for compounds previously reported to cause aggregation.[34] Results are scored based on chemical similarity to known aggregation-inducing compounds by using the Tanimoto coefficient (Tc).[34] Based on a threshold of Tc > 0.85,[34] the 352-compound Zenobia library contains ten aggregation-prone fragments (Table S5).
Fragment screening and semi-automated preparation of NMR samples
For NMR-based screening, 15N-labeled protein (−15 mL, 100 μM) in appropriate buffer with 10% (v/v) D2O was required. In each well, 15N-labeled protein (232.8 μL, 100 μM) was added to fragment mixture (7.2 μL) to give final concentrations of 3 % (v/v) [D6]DMSO, 1 mM of each chemical fragment, and 97 μM protein. Subsequent steps used a PAL liquid handling robot (LEAP Technologies), configured in-house for solution mixing and loading of 3 mm diameter NMR sample tubes. To avoid cross-contamination, the syringe was washed with acetonitrile and water between samples. Each NMR sample was mixed and transferred from the 96-well v-bottomed plate to 3 mm NMR tubes. Upon completion of sample preparation, NMR tubes were capped, and any signs of precipitation/aggregation were noted. Total time spent for sample preparation for NMR screening was −4 h, or 4 min/sample.
Primary screening identified a subset of NMR samples containing chemical fragment(s) with affinity to the protein target. To identify these, each of the six fragments from a given sample (1.2 μL) were aliquoted individually into six wells of a 96-well v-bottomed plate. Then, 15N-labeled protein (238.8 μL, 50 or 100 μM) was added, and the solution was gently mixed and transferred into 3 mm NMR tubes by using the PAL liquid-handling robot to give final concentrations of 0.5% (v/v) [D6]DMSO, 1 mM chemical fragment, and 49.75 or 99.5 μM protein. A protein concentration of 50 or 100 μM conserved 15N-labeled protein while maintaining sufficient signal/noise to discriminate non-binders from binders.
To determine binding affinities of chemical fragments against protein targets, titration experiments by NMR were performed as previously described.[7] Briefly, the PAL robot was used to set up one DMSO control (2 %, v/v) and six titration points per fragment at 25, 50, 150, 400, 800, and 1600 μM with 15N-labeled protein (50 μM).
NMR spectroscopy
Automated NMR data collection was performed on 15N-labeled protein samples (50 or 100 μM) by using 1H,15N SOFAST-HMQC experiments for CXCL12 and Fis1 or 1H,15N HSQC experiments for BRD4(BD2). Data was collected at 25°C on a Bruker Avance II 600 MHz spectrometer equipped with a triple resonance z-axis gradient cryoprobe and SampleJet autosampler, which allowed automatic tuning, matching, and shimming for each sample. 1H,15N SOFAST-HMQC experiments consisted of 16 scans with 1024 and 210 complex points in the 1H and 15N dimensions, respectively. The total time for screening one target by 1H,15N SOFAST-HMQC was −16 h (−16 min/sample), whereas screening by an equivalent 1H,15N HSQC experiment increased NMR time ≈ twofold. NMR samples of 15N-CXCL12 (50 μM) contained sodium phosphate (50 mM) pH 6.0, sodium azide (0.02%, w/v), and D2O (10%, v/v); 15N-Fis1 (50 μM, residues 1–125) contained sodium phosphate (50 mM) pH 7.0, NaCl (175 mM), DTT (2 mM), and D2O (10%, v/v); and 15N-BRD4(BD2) (50 μM) contained sodium phosphate (100 mM) pH 6.5, NaCl (50 mM), and DTT (5 mM). Resonance assignments for CXCL12[7] and BRD4(BD2)[40] were obtained from published values. Backbone amide resonance assignments for Fis1 (residues 1–125) were obtained from the following experiments: 15N HSQC, HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, HN(CO)CACB, C(CO)NH. All NMR data were processed by using the NMRPipe.[25] XEASY was used for resonance assignment.[41] Spectra were processed by using automated python scripts in NMRPipe, and chemical shifts were measured by using TitrView and CARA software.[42] Total 1H/15N chemical shift perturbations were calculated by using Equation (1). Equilibrium dissociation constants (Kd values) were determined by using nonlinear fitting to a ligand depletion function [Eq. (2)] with the protein concentration held constant at 50 μM. For each fragment, residues (δmax > 0.2 ppm) were fitted individually, and the resulting Kd values were averaged to determine reported affinities and standard errors of the mean. Spectral overlays were generated by using XEASY software and Adobe Illustrator:
| (1) |
where Δδ chemical shift =total chemical shift change, and ΔδH and ΔδNH represent amide proton and nitrogen chemical shifts, respectively.
| (2) |
where Δ=adjusted chemical shift change, δmax = maximum chemical shift perturbation, Kd = binding dissociation constant, p = [protein], and l =[ligand].
Principal component analysis
2D NMR spectra of 512 and 256 complex points in proton and nitrogen dimensions, respectively, were used as input data for PCA performed by using the pcaNMR module in NMRPipe.[25] The spectral regions 6–10 ppm 1H and 105–135 ppm 15N (100–130 ppm 15N for Fis1) were included for PCA. After completion of PCA, score plots were generated with CIs by using automated scripts in R.
Difference intensity analysis (DIA)
By using the addNMR module of NMRPipe, 2D difference spectra were generated from hetero-nuclear data from each fragment mixture compared to the DMSO control. Total crosspeak intensities were calculated from 2D difference spectra in NMRPipe by using in-house Python and R scripts. Only crosspeak intensities ±200000 were included. The resulting 2D difference spectra for each fragment mixture were condensed into a single positive (and negative) value representing the total sum of all positive (and negative) crosspeak intensities. Accordingly, each NMR sample gave two values. NMR samples containing chemical fragments that gave rise to total (positive and negative) crosspeak intensity value(s) greater than one standard deviation from the mean across all 60 samples were considered as either potential binders (+1σ) or compounds that induced nonspecific NMR crosspeak broadening (−1 σ). We refer to this process as DIA. All scripts used in data processing and analysis are available upon request.
Computational fragment docking
Structures of Fis1, CXCL12, and BRD4(BD2) (PDB IDs: 1NZN, 2K05, and 4Z93, respectively) were imported into the Schrödinger Maestro suite (v. 10.7; Schrödinger, LLC, New York, NY) by using the Protein Preparation wizard and subjected to restrained minimization by using the OPLS3 force field.[43] Ligand structures were prepared for docking by using the LigPrep application in Maestro (v. 3.9; Schrödinger, New York, NY), and ligand ionization states at pH 7.0±2.0 were generated by using Epik (v. 3.7; Schrödinger, New York, NY). Glide (v. 7.2; Schrödinger, New York, NY)[43] was used to generate a receptor grid surrounding the centroid of each target protein. Finally, flexible ligand docking was performed by using the Glide docking protocol in standard precision mode.
Supplementary Material
Acknowledgments
We thank Drs. Katherine M. Collins and Andres Ramos for providing initial scripts for PCA in NMRPipe. We thank the Medical College of Wisconsin (MCW) Office of Research and Research Computing Center for help with Schrödinger and computational server resources. We thank Aye Myat Myat Thinn for assistance in collecting DLS measurements and Dr. Jieqing Zhu and the Blood Research Institute Protein Core for time on their DLS instrument. This project was supported by the following sources: the Advancing a Healthier Wisconsin Endowment, National Center for Advancing Translational Sciences, National Institutes of Health (NIH; grant no. UL1 TR001436), NIH grants R01 GM067180 (R.B.H.), R01 HL128240 (R.B.H.), R01 GM097381 (B.F.V.), R01 AI058072 (B.F.V.), R01 AI120655 (B.F.V.), S10 OD020000 (B.F.V.), American Heart Association Scientist Development grant 15SDG25830057 (B.C.S.), the Medical College of Wisconsin Research Affairs Committee (BCS), and Institutional Research grants 14-247-29-IRG and 86-004-26-IRG from the American Cancer Society (B.C.S.). M.D.O. is a member of the Medical Scientist Training Program at MCW, which is partially supported by a training grant from NIGMS T32 GM080202.
Footnotes
Conflicts of Interest
F.C.P. and B.F.V. have significant financial interests in Protein Foundry, LLC. The other authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of all authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting information and the ORCID identification numbers for the authors of this article can be found under https://doi.org/10.1002/cbic.201700386.
References
- 1.Scannell JW, Blanckley A, Boldon H, Warrington B. Nat Rev Drug Discovery. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 2.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Nat Rev Drug Discovery. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- 4.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 5.Harner MJ, Frank AO, Fesik SW. J Biomol NMR. 2013;56:65–75. doi: 10.1007/s10858-013-9740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veldkamp CT, Ziarek JJ, Peterson FC, Chen Y, Volkman BF. J Am Chem Soc. 2010;132:7242–7243. doi: 10.1021/ja1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith EW, Nevins AM, Qiao Z, Liu Y, Getschman AE, Vankayala SL, Kemp MT, Peterson FC, Li R, Volkman BF, Chen Y. J Med Chem. 2016;59:4342–4351. doi: 10.1021/acs.jmedchem.5b02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleist AB, Getschman AE, Ziarek JJ, Nevins AM, Gauthier P-A, Chevigné A, Szpakowska M, Volkman BF. Biochem Pharmacol. 2016;114:53–68. doi: 10.1016/j.bcp.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waring MJ, Chen H, Rabow AA, Walker G, Bobby R, Boiko S, Bradbury RH, Callis R, Clark E, Dale I, et al. Nat Chem Biol. 2016;12:1097–1104. doi: 10.1038/nchembio.2210. [DOI] [PubMed] [Google Scholar]
- 10.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras A-C, Arrowsmith CH, Knapp S. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theodoulou NH, Tomkinson NCO, Prinjha RK, Humphreys PG. Curr Opin Chem Biol. 2016;33:58–66. doi: 10.1016/j.cbpa.2016.05.028. [DOI] [PubMed] [Google Scholar]
- 12.Korb E, Herre M, Zucker Scharff I, Darnell RB, Allis CD. Nat Neurosci. 2015;18:1464–1473. doi: 10.1038/nn.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee DU, Katavolos P, Palanisamy G, Katewa A, Sioson C, Corpuz J, Pang J, DeMent K, Choo E, Ghilardi N, Diaz D, Danilenko DM. Toxicol Appl Pharmacol. 2016;300:47–54. doi: 10.1016/j.taap.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 14.James DI, Parone PA, Mattenberger Y, Martinou J-C. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 15.Dohm JA, Lee SJ, Hardwick JM, Hill RB, Gittis AG. Proteins Struct, Funct, Bioinf. 2004;54:153–156. doi: 10.1002/prot.10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess M-A, Levit A, Kim B, Hartman M-L, Joseph L, Shirihai JA, Vita OS. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archer SL. N Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 18.Disatnik M-H, Ferreira JCB, Cruz Campos J, Sampaio Gomes K, Dourado PMM, Qi X, Mochly-Rosen D. J Am Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K, Long B, Jiao J-Q, Wang J-X, Liu J-P, Li Q, Li P-F. Nat Commun. 2012;3:781. doi: 10.1038/ncomms1770. [DOI] [PubMed] [Google Scholar]
- 20.Kleckner IR, Foster MP. Biochim Biophys Acta Proteins Proteomics. 2011;1814:942–968. doi: 10.1016/j.bbapap.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williamson MP, Asakura T. J Magn Reson Ser B. 1993;101:63–71. [Google Scholar]
- 22.Iwadate M, Asakura T, Williamson MP. J Biomol NMR. 1999;13:199–211. doi: 10.1023/a:1008376710086. [DOI] [PubMed] [Google Scholar]
- 23.Williamson MP. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Collins KM, Oregioni A, Robertson LE, Kelly G, Ramos A. Nucleic Acids Res. 2015;43:e41. doi: 10.1093/nar/gku1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 26.Trbovic N, Dancea F, Langer T, Günther U. J Magn Reson. 2005;173:280–287. doi: 10.1016/j.jmr.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 27.Mishra NK, Urick AK, Ember SWJ, Schönbrunn E, Pomerantz WC. ACS Chem Biol. 2014;9:2755–2760. doi: 10.1021/cb5007344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vidler LR, Brown N, Knapp S, Hoelder S. J Med Chem. 2012;55:7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fish PV, Filippakopoulos P, Bish G, Brennan PE, Bunnage ME, Cook AS, Federov O, Gerstenberger BS, Jones H, Knapp S, Marsden B, Nocka K, Owen DR, Philpott M, Picaud S, Primiano MJ, Ralph MJ, Sciammetta N, Trzupek JD. J Med Chem. 2012;55:9831–9837. doi: 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L, Cao D, Chen T, Wang Y, Miao Z, Xu Y, Chen W, Wang X, Li Y, Du Z, Xiong B, Li J, Xu C, Zhang N, He J, Shen J. J Med Chem. 2013;56:3833–3851. doi: 10.1021/jm301793a. [DOI] [PubMed] [Google Scholar]
- 31.Taylor AM, Vaswani RG, Gehling VS, Hewitt MC, Leblanc Y, Audia JE, Bellon S, Cummings RT, Côté A, Harmange J-C, et al. ACS Med Chem Lett. 2016;7:145–150. doi: 10.1021/ml500411h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mason HS. Nature. 1955;175:863–864. [Google Scholar]
- 33.Morrison M, Steele W, Danner DJ. Arch Biochem Biophys. 1969;134:515–523. doi: 10.1016/0003-9861(69)90313-0. [DOI] [PubMed] [Google Scholar]
- 34.Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, Shoichet BK. J Med Chem. 2015;58:7076–7087. doi: 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koppenol-Raab M, Harwig MC, Posey AE, Egner JM, MacKenzie KR, Hill RB. J Biol Chem. 2016;291:20329–20344. doi: 10.1074/jbc.M116.724005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D, Vajda S. Nat Protoc. 2015;10:733–755. doi: 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015. [Google Scholar]
- 38.Congreve M, Carr R, Murray C, Jhoti H. Drug Discovery Today. 2003;8:876–877. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 39.Baell JB, Holloway GA. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 40.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, et al. Cancer Cell. 2014;25:210–225. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 42.Keller R. The Computer Aided Resonance Assignment Tutorial. 2004. [Google Scholar]
- 43.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.