

Abstract
Poliovirus is a prototype member of the Enterovirus genus of the Picornaviridae family of small positive strand RNA viruses, which include important human and animal pathogens. Quantitative assessment of viral replication is very important for investigation of the virus biology and the development of anti-viral strategies. The poliovirus genome structure allows replacement of structural genes with a reporter protein, such as a luciferase or a fluorescent protein, whose signals can be detected and quantified in vivo, thus permitting observation of replication kinetics in live cells. This paper presents protocols for poliovirus replicon RNA production, purification, packaging and transfection, as well as techniques for monitoring Renilla luciferase replication signal in living cells.
Keywords: picornaviruses, RNA replication, replicon assay, replicon packaging, Renilla luciferase
INTRODUCTION
Poliovirus is a representative member of the Picornaviridae family of small (+)RNA viruses, which have non-enveloped icosahedral capsid and infect vertebrate animals. Picornaviruses include important human and animal pathogens associated with broad spectrum of diseases, from hepatitis induced by hepatitis A virus to the common cold, attributed to numerous rhinoviruses. Research history of poliovirus covers more than a century, which makes it one of the best-developed systems to study molecular mechanisms of viral replication and virus-host interaction. One of the important tools in the arsenal of poliovirus research is the replicon system allowing monitoring virus replication by the signal of a reporter protein expressed from polio RNA. The poliovirus genome RNA of ~7500 nt is translated by cellular ribosomes to produce a single polyprotein. This polyprotein undergoes co- and post-translational cleavages by three virus-encoded proteases, 2A, 3C and 3CD, generating about a dozen mature peptides and intermediate cleavage products, enabling replication of the viral RNA and assembly of progeny virions (Fig. 1). The very N-terminal part of the poliovirus polyprotein contains the capsid protein precursor P1, which is cleaved off the nascent polypeptide chain by the protease 2A in cis during the translation process. The capsid proteins do not participate in RNA replication, nor does their coding region contains any cis-acting RNA elements, thus this region of the genome can be deleted, or substituted with a heterogeneous sequence, without affecting the replication capacity of such an RNA.
Figure 1.
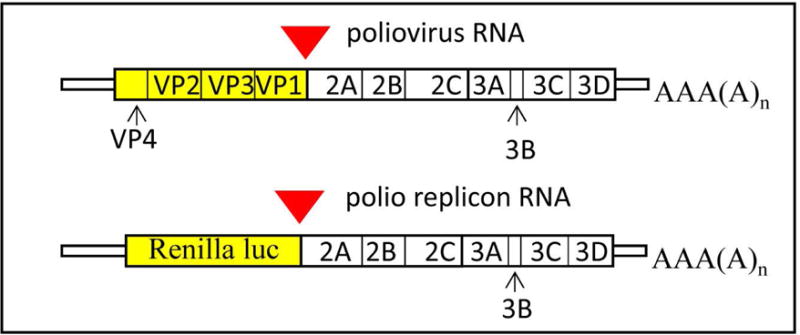
Scheme of poliovirus genome and replicon RNA. Red triangle indicates VP1-2A cleavage site preserved in the replicon construct.
The first polio replicons with deletions in the capsid region were described by Kaplan and Racaniello (Kaplan & Racaniello, 1988); replicons containing reporter proteins, such as chloramphenicol acetyl transferase, firefly luciferase or green fluorescent protein (GFP) were developed later by different research groups (Jackson, Cobbs, Peduzzi, Novak, & Morrow, 2001; Percy, Barclay, Sullivan, & Almond, 1992; Porter et al., 1998). However, these replicon systems are not well adapted for replication kinetic studies because the enzymatic assays required lysis of the cells, and GFP needs maturation time comparable to the duration of the whole poliovirus infectious cycle before it can be detected by fluorescence. To develop a replication system that can be monitored in live cells over extended period of time we replaced capsid-coding region of poliovirus with Renilla luciferase sequence. Renilla luciferase was chosen because of the availability of cell-permeable substrates that can be simply added to the incubation medium. For replicon construction we first modified the plasmid pXpA coding for the cDNA of poliovirus type I Mahoney under control of T7 RNA polymerase promotor (Herold & Andino, 2000) so that it contains two unique restriction sites SalI and HpaI flanking the protein-coding sequence of poliovirus genome (Belov, Fogg, & Ehrenfeld, 2005). Renilla luciferase gene optimized for expression in human cells from the plasmid phRL-CMV (Promega) was amplified by PCR and introduced between SalI and XhoI sites, generating plasmid pXpA-RenR (Belov et al., 2007). The Renilla sequence is placed in frame with the poliovirus polyprotein sequence, without a stop codon, so that the luciferase C-terminus is fused to the last 5 amino-acids of poliovirus VP1 sequence, preserving VP1-2A cleavage site (Fig. 1). Similar design can be easily adopted for development of replicons of poliovirus, or other related viruses, coding for novel reporter proteins should those become available.
Here we describe protocols for polio replicon RNA generation, purification, encapsidation and transfection as well as for the in vivo measurement of Renilla luciferase signal.
STRATEGIC PLANNING
The protocols described here require proficiency in general molecular biology and cell culture techniques.
BASIC PROTOCOL 1: Generation of poliovirus replicon RNA using T7 RNA polymerase transcription
The success of replicon RNA transfection and reproducibility of the results strongly depend on the quality of replicon RNA. RNA is a very labile molecule, and even one break will render replicon RNA non-functional, therefore, researchers working with long RNAs should pay maximum attention to maintaining an RNAase-free working environment and using the highest purity reagents available. The pXpA-RenR plasmid coding for Renilla luciferase polio replicon was constructed using the backbone of the pXpA plasmid which contains poliovirus cDNA flanked by a hammerhead ribozyme on the 5′ end to ensure generation of the authentic 5′ RNA end, and a poly-A sequence at the 3′ end (Herold & Andino, 2000). It has a T7 RNA polymerase promotor but does not have a T7 RNA polymerase terminator, so the plasmid needs to be linearized before transcription. The EcoRI site located just downstream of the poly-A sequence is the most convenient site for linearization of this plasmid (Fig. 2).
Figure 2.
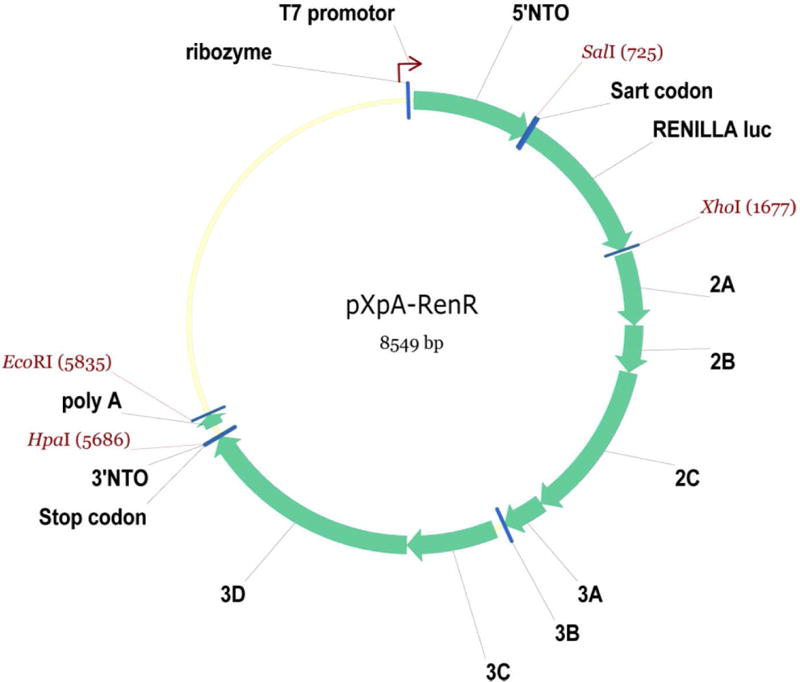
Scheme of the pXpA-RenR plasmid. Unique restriction sites used for plasmid construction as well as EcorI site used for plasmid linearization are shown. The map is generated by Vector NTI software (Invitrogen).
CAUTION: Phenol can cause severe burns; it should be handled and discarded according to appropriate regulations as a hazardous chemical. General good laboratory techniques should always be executed when working with chemicals and human cell cultures.
Materials
3 μg of plasmid pXpA-RenR in water (plasmid prepared using any standard plasmid purification method can be used. (The plasmid is available upon request from the authors.)
EcoRI and 10× buffer (20,000 units/ml, New England BioLabs)
Molecular biology grade water
Agarose (electrophoresis grade, low EEO)
Ethidium bromide solution (10mg/ml; 1000×)
10×TBE buffer (we recommend using a certified DNAse- RNAse-free buffer from a reputable manufacturer, but it can be prepared in house (see recipe)
*DNA phenol. 25:24:1 (v/v/v) saturated phenol/chloroform/isoamyl alcohol, pH 7.9; for DNA extraction (Thermo Fisher, AM9730)
*RNA phenol. 25:24:1 (v/v/v) saturated phenol/chloroform/isoamyl alcohol, pH 4.5; for RNA extraction (Thermo Fisher, AM9720)
Chloroform (ultra pure, Sigma Aldrich)
5M sodium acetate (it is highly recommended to use 5M sodium acetate solution provided in
T7 MEGA script transcription kit, see below)
Ethanol (pure, molecular biology grade)
DEPC-treated high purity water
80% (v/v) ethanol/water solution
10% SDS solution in water
T7 MEGA script transcription kit (Thermo Fisher, AM1333)
RNaseZap wipes (Thermo Fisher, AM9786)
Croma Spin 100 DEPC-water columns (Takara Clontech, cat#636089)
37◦C incubator
Vortex shaker
NanoDrop spectrophotometer
Minifuge for Eppendorf tubes
Speedvac, or equivalent vacuum drier
Tabletop centrifuge with a bucket rotor with adaptors suitable for 15 ml tubes
1.5 ml Eppendorf tubes
15ml tubes with screw cap
Electrophoresis apparatus for agarose electrophoresis
*It is highly recommended upon receiving the phenol mix to aliquot it in sterile plastic tubes (10 ml/each) and store them at −20 °C, keeping a working tube at 4 °C for no longer than a month. If the phenol mix starts acquiring reddish color, it should be discarded immediately.
Protocol steps
-
Linearize plasmid.
- Assemble restriction reaction: in an Eppendorf tube combine the plasmid prep, molecular biology grade water and 10X restriction buffer. The mix should contain 3μg of pXpA-RenR plasmid in 30–50 μl final volume.
- Add 1μl of EcoRI enzyme (20units)
- Incubate restriction reaction for 2 h at 37 °C.
- During the incubation step prepare 0.8% agarose gel with 1xTBE buffer for analysis of restricton product
- Prepare two samples for electrophoresis using available DNA loading dye. One sample should contain 1 μl of the restriction reaction, the other should have the equivalent amount of the uncut plasmid.
- Perform electrophoresis in 0.8% agarose gel in TBE buffer, the linearized plasmid should run as one band with mobility different from that of the uncut plasmid.
It is possible to linearize more plasmid and use it as a stock for several transcription reactions if necessary, increasing the volume of water in the step f) of section 3 (transcription), accordingly. The volume of restriction reaction should not be less than 30μl in order to avoid star activity of the restriction enzyme.
- Perform phenol extraction
- Add molecular grade DEPC-treated water to the restriction reaction to bring the volume to 200μl.
-
Add 200μl of DNA phenol, vortex the tube vigorously for ~1 min.Make sure that fine phenol/water suspension is formed (the content of the tube should have milk-white appearance), rather than just swirling the content (in this case the material in the tube will be semi-transparent).
- Centrifuge tube for 1 min at max speed in a minifuge to separate the top (aqueous) and bottom (organic) phases should be clearly visible in the tube after centrifugation.
- Transfer the top (aqueous) phase into a new Eppendorf tube, repeat steps b) and c).
- Transfer the top (aqueous) phase into a new Eppendorf tube. Add 200μl of chloroform to the water phase, vortex vigorously for 30 sec, centrifuge like in step c) and transfer top (water) phase in a new Eppendorf tube.
-
Add 20μl of 5M ammonium acetate and 800 μl of ethanol, mix well and place overnight at −20 °C.If necessary, incubation at −20 °C could be as short as 2h, however it is usually convenient to start transcription the next day.
- Perform transcription
- IMPORTANT: Prepare working station by carefully wiping table surface, pipettes, minifuge, as well as your gloves with RNAseZAP wipes. This is a critical step for generating high quality RNA. Be careful not to accidentally introduce the RNAseZAP solution into working tubes.
- Pellet the linearized plasmid DNA by centrifugation at max speed in a minifuge for 15 min, carefully remove and discard the supernatant making sure not to disturb the pellet.
- Add 800μl of 80% ethanol to the tube, close the tube and carefully rotate the tube so that the ethanol washes traces of precipitation solution from the walls.
-
Centrifuge for 10 min at max speed in a minifuge, carefully remove supernatant and dry the pellet in a Speedvac for 5 min (do not use heated drying regime).The following steps are performed according to the MEGA script T7 kit manual, using chemicals provided in the kit:
-
Thaw water, 10x reaction buffer and four nucleotide solutions, make sure the buffer is dissolved completely, it may require intensive vortexing.IMPORTANT: Do not keep the thawed solutions on ice. Reaction should be assembled at room temperature.
- Dissolve DNA pellet in 8μl of H2O by vortexing and spinning down the tube several times
- Add 2μl of each of nucleotide solutions followed by 2μl of 10x reaction buffer, spin down, vortex and spin down the tube again to mix the components.
-
Add 2μl of T7 enzyme mix, gently vortex and spin down the tube, place in an incubator at 37 °C.It is important to either place the tube containing the transcription reaction in an air incubator, or in a heating block with a heated lid so that the tube is heated evenly. In a usual open thermal block the lid is exposed to cooler air leading to water vapor condensation, which can significantly change concentration of components in a small volume reaction.
- After 2.5 h of incubation start preparing 1% agarose gel with 1% TBE and 10μg/ml ethidium bromide. Make sure to wipe the electrophoresis tank, comb and gel plate with RNAseZAP wipes and rinse with distilled water.
- At 3 h of incubation prepare samples for electrophoresis using 1μl of transcription reaction, 4μl of DEPC-treated water and 1μl of loading dye (provided in the MEGA script transcription kit).
-
Run electrophoresis using 1xTBE buffer supplemented with 0.005% SDS (add 250μl of 10%SDS to 500ml of 1×TBE buffer).The transcription sample should appear as a bright high molecular weight band and a fainter low molecular weight band (Fig. 3, lane 1). The low molecular weight band is due to a specific sequence in the 5′ end region of polio cDNA, which causes T7 RNA polymerase to stop. There should not be any visible smear beyond this low molecular weight band (Fig. 3, asterisk), as this would indicate RNA degradation. The RNA material between the high and the low molecular weight bands represents unfinished transcripts.
- If the transcription results are satisfactory, add 1μl of DNAse (provided in the transcription kit) to the remaining sample and incubate at 37 °C for 15 min, proceed to RNA purification.
-
RNA purification.
Add to the transcription reaction 380μl of DEPC-treated water with ~0.3% SDS (it is convenient to prepare SDS solution by adding 30μl of 10%SDS to 1 ml of DEPC-treated water, always make fresh).
-
Add 400μl of RNA phenol mix, vortex vigorously for ~1 min.
Make sure that phenol/water suspension is formed (the content of the tube should have milk-white appearance), rather than just swirling the content (in this case the material in the tube will be semi-transparent)
Centrifuge at max speed in a minifuge for 1 min. A thick white interphase of precipitated SDS should form between the upper (aqueous) and lower (organic) phases.
Transfer the upper (aqueous) phase into a new Eppendorf tube, paying attention not to grab white interphase material. Minimal amount of interphase carryover is acceptable.
Repeat steps b) and c). At this stage interphase should be minimal or totally absent.
Transfer upper (aqueous) phase to a new tube. Add 400μl of chloroform to the aqueous phase, vortex vigorously for 30 sec, centrifuge like in step c) and transfer aqueous phase in a new Eppendorf tube.
Add 800μl of ethanol, mix well, leave overnight at −20 °C.
The next day collect the RNA pellet by spinning down the tube for 15 min at max speed in a minifuge. Discard the supernatant. White RNA pellet should be clearly visible.
During centrifugation step h) start making 1% agarose gel with 1×TBE and 10 μg/ml ethidium bromide. Make sure to wipe the electrophoresis tank, comb and gel plate with RNAseZAP wipes and rinse with distilled water.
Being careful to not disturb the RNA pellet, add 800μl of 80% ethanol and carefully rotate the tube to wash the remnants of the previous supernatant form the walls. Centrifuge for 10 min at max speed.
During the centrifugation step j) start preparing Chroma Spin columns. Vortex the sealed column so that the gel matrix is thoroughly shaken, make sure it is not stuck to the lid.
Open the lid first and discard it, snap off the bottom seal second. Place the column in a 2 ml collection tube (provided with columns), place the whole assembly into a 15 ml polypropylene tube without lead.
Centrifuge the column at 700 g for 5 min at 4 °C in a tabletop centrifuge with a suitable bucket rotor.
In the meantime, at the end of centrifugation step j) carefully remove the supernatant and dry the RNA pellet in a Speedvac for 5 min (do not use heated drying regime).
Add 60μl of DEPC-treated water to the dry RNA pellet, leave at room temperature for 5 min.
-
Vortex the RNA-containing tube vigorously, spin down briefly in a minifuge. Repeat several times, making sure the RNA pellet is completely dissolved.
This is a critical step when RNA can be lost if it is not completely dissolved. When transferring RNA solution to the column (see step q), watch closely if there is any undissolved material (gelatinous-looking small blobs). Pipet the solution up and down several times with pipet tip if necessary.
Transfer the Chroma Spin column into a new 2 ml collection tube (provided with columns), apply RNA solution from step p) to the center of gel matrix, making sure it does not touch the walls.
Centrifuge the column at 700 g for 5 min at 4 °C in a tabletop centrifuge with a suitable bucket rotor.
Transfer the RNA solution (should be ~60–65 μl) from the collection tube into a new Eppendorf tube. Keep the RNA tube on ice.
Prepare sample for electrophoresis using 1μl of purified RNA solution, 4μl of DEPC-treated water and 1μl of loading dye (provided in the MEGA script transcription kit).
Run electrophoresis using 1x TBE buffer supplemented with 0.005% SDS (add 250μl of 10%SDS to 500ml of 1×TBE buffer).
-
In the meantime, measure RNA concentration using Nanodrop. Good RNA prep should have concentration of 500–1200ng/μl and ratio of OD260 to OD280 between 1.85 and 2.
The purified RNA sample usually appears on the gel as two bright high molecular weight bands and a low molecular weight band (Fig. 3, lane 2). Both high molecular weight bands are the full length replicon RNA, apparently different conformations are adopted during the purification process. There should not be any visible smear beyond the low molecular weight band (Fig. 3, asterisk).
-
Store purified RNA at −80C. RNA samples prepared this way can be stored and freeze-thawed repeatedly for years without losing their quality.
If more RNA is required than can be obtained from a single standard 20μl transcription reaction, it is better to increase the number of transcription reactions, not its volume to maintain high quality of RNA.
Figure 3.

An example of agarose electrophoresis image of replicon transcription reaction (lane1) and purified replicon RNA (lane2). The small molecular weight band generated because of a sequence present in the poliovirus cDNA which causes T7 RNA polymerase to stop is indicated by an asterisk. Note that the images are from separate gels, so the mobility of the bands cannot be compared directly.
BASIC PROTOCOL 2: Transfection of HeLa cells using purified polio replicon RNA and in situ measurement of the replication signal
Since poliovirus is a (+)RNA virus, its RNA is directly translated by the ribosomes and initiates the replication cycle without the requirement for any additional viral proteins. Transfection of the replicon RNA into cells can serve both for direct measurement of replication (this protocol), as well as for trans-encapsidation of the replicon by poliovirus capsid proteins expressed from a recombinant Newcastle Disease virus (basic protocol 3). Depending on the purpose of the replication experiment, i.e. assessment of an anti-viral drug, or replication in cells defective in expression of certain proteins, it could be desirable to be able to observe the cells under the microscope to evaluate toxic effect of the treatment. In this case using white 96-well plates with transparent bottom is recommended. If no toxic effect on cells is anticipated, one can use solid white 96 well plates. They have minimal cross talk between wells and better reflect luciferase-generated light to the sensors, thus producing a stronger signal.
Materials
DMEM high glucose modification cell culture medium
Fetal bovine serum (FBS), heat inactivated
OPTI-MEM medium
EnduRen cell permeable Renilla luciferase substrate, 60mM (see recipe) (Promega cat# E6482)
TransIT mRNA transfection reagent (Mirus cat# MIR 2225)
96 well plates, white, flat bottom, cell culture treated
15 ml sterile polypropylene tubes with snap-on lids (Nunc cat# 362694)
Multichannel pipette reservoirs
Optical clear adhesive sealing tape for 96 well plates (VWR cat# 60941-078 or equivalent tape for real-time PCR plates)
Cell culture CO2 incubator
Water bath set at 37 °C.
Roller for applying sealing tape to a 96 well plate.
Cell counter
Vortex
Multichannel pipette (8 or 12 channels)
Multiwell plate reader with heated chamber, capable of reading luminescence (Tecan Infinite M1000 or equivalent)
Protocol steps
- The day before the experiment, seed HeLa cells into white tissue culture-treated 96 well plate at 40000 cells/well.
- The cell plating density may need to be adjusted depending on the growth properties of a particular cell line. It is possible to transfect cells with plasmids or siRNA before performing replication experiments. The goal is to obtain ~90% confluent but not overgrown monolayer of cells by the time of replicon RNA transfection.
Turn on the plate reader, set up the incubation chamber temperature at 37 °C.
- Prepare incubation medium.
- Pre-warm 10 ml of DMEM with 10% FBS in a 15 ml tube in a water bath at 37 °C.
- Add 5μl of 60mM EnduRen solution, place the tube back to 37 °C.
- Prepare RNA transfection mix
- Thaw replicon RNA sample, keep the tube on ice
- Add 1350 μl of OPTI-MEM medium into a 15 ml sterile tube.
- Add 1.5 μg of replicon RNA to OPTI-MEM in 15 ml tube, vortex briefly.
- Add 9 μl of Boost reagent from the RNA transfection kit to the RNA/OPTI-MEM mix, vortex briefly.
-
Add 9 μl of TransIT reagent from the RNA transfection kit, vortex briefly, incubate for 2 min.The total incubation time of the transfection mix before adding it to cells should not exceed 5 min.
-
Add 9 ml of pre-warmed EnduRen/DMEM mix from step 3 to the transfection mix, mix thoroughly by inverting the tube several times, transfer to a multichannel pipette reservoir.This protocol is set up to deliver the same transfection mix to each well in a 96 well plate, however it is possible to split the mix at this stage into separate samples and, for example, add different anti-viral compounds to assess their effect on replication. It is advisable to have at least 2 columns on a 96 well plate (16 wells) allocated for each experimental condition.
-
Remove medium form the cells grown in 96 well plateMake sure the medium is removed fast so that the cells do not dry during this procedure. It is convenient to invert the plate and shake the medium off over the sink in one strong move.
- Dispense 75 μl of transfection mix from step f) per well using a multichannel pipette.
- Seal the plate with optical clear adhesive film using roller to ensure the wells are well sealed. Do not touch the clear film with fingers.
-
Place the sealed plate into the incubation chamber of the plate reader pre-heated at 37 °C, start the reading program.We usually use luminescence measurement mode with 1 sec integration time per well, readings repeat every hour for 16 hours, but the parameters can be adjusted depending on the requirements of a particular experiment. EnduRen takes about one hour to be imported and converted into a functional form inside the cell, so the interpretable data can be collected no earlier than 1 h after the beginning of incubation of cells with EnduRen. A representative replication curve of polio replicon is shown in Fig. 4.
Figure 4.
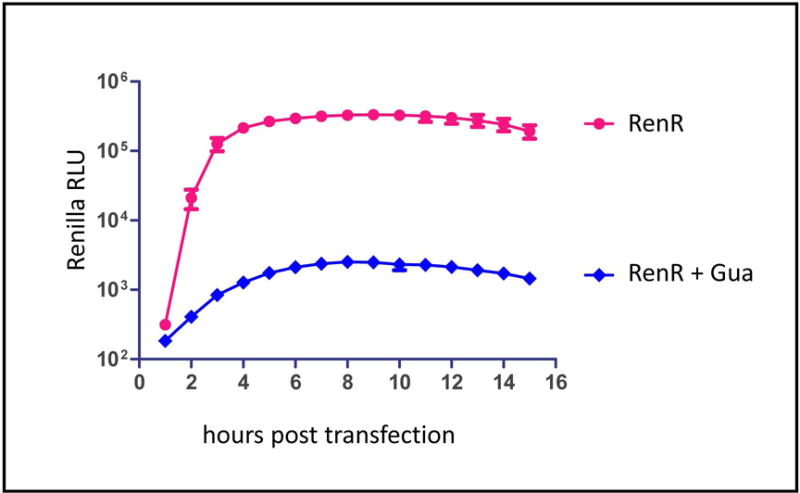
An example of a replication assay performed with replicon RNA transfection. The control sample (RenR) shows normal high level of replication; the sample incubated in the presence of 2mM guanidine-HCl (RenR+Gua) reflects translation of the input RNA. The data show average luciferase signal from 16 wells of a 96-well plate, bars represent standard deviation. The data were processed by GraphPad Prizm software.
BASIC PROTOCOL 3: Trans-encapsidation of polio replicon RNA using Newcastle Disease virus expressing poliovirus capsid proteins
While replicon RNA transfection described in the previous protocol bypasses the first steps of infection, such as virion attachment, penetration of the cellular membrane and release of the viral RNA in the cytoplasm, encapsidated replicons are bona fide virus particles that undergo all the natural steps of infection. Replication-competent poliovirus RNAs lacking the capsid coding region could be encapsidated by capsid proteins produced in trance (Nugent, Johnson, Sarnow, & Kirkegaard, 1999). An encapsidation system based on expression of poliovirus capsid proteins from vaccinia virus was developed previously (Nugent et al., 1999), however such system has significant limitations because the replicon preparations inevitably contain infectious vaccinia virus. We provide here a protocol for transencapsidation of poliovirus replicon using recombinant Newcastle Disease virus (NDV) expressing P1 capsid protein precursor and protease 3CD of poliovirus type I Mahoney (Fig. 5A). This recombinant NDV has been constructed as a vectored vaccine against poliomyelitis and will be described elsewhere (Viktorova et al. in preparation). The LaSota strain of NDV used as a backbone for recombinant construct expressing poliovirus proteins is propagated in embryonated chicken eggs (for propagation and quantitation of NDV please refer to (McGinnes, Pantua, Reitter, & Morrison, 2006) and cannot produce infectious particles in cell culture without addition of exogenous proteases, thus infectious NDV does not contaminate encapsidated poliovirus replicon preparations.
Figure 5.
Transencapsidation of polio replicon RNA. A. Scheme of transencapsidation procedure. B. An example of a replication assay performed with transencapsdated replicon. HeLa cells grown in a 96-well plate were infected with either transencapsidated polio replicon, NDV, or mock-infected. Only those infected with transencapsidated replicon show replication signal. The data show average luciferase signal from 16 wells of a 96well plate, bars represent standard deviation. The data were processed by GraphPad Prizm software. C. Immunofluorescence assay showing poliovirus antigen 2B in HeLa cells infected with either transenacapsidated replicon (left panel), or poliovirus type I Mahoney (right panel), fixed at 6 hours post infection.
CAUTION: Newcastle disease virus LaSota strain is a Biosafety Level 2 (BSL-2) pathogen. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms. Permission for working with LaSota strain of Newcastle Disease virus should be obtained.
Materials
Purified polio replicon RNA (see protocol 1)
Recombinant NDV-polio expressing poliovirus proteins P1 and 3CD. The virus is available from the authors upon request.
DMEM high glucose modification
HeLa cells
TransIT mRNA transfection reagent (Mirus cat# MIR 2225)
Cell culture CO2 incubator
Vortex
Protocol steps
-
The day before the experiment seed HeLa cells into a 12 well plate at 450000 cells/well, so that the next day they form confluent but not overgrown monolayer
The cell seeding density may need to be adjusted depending on the growth properties of a particular HeLa line available.
- Infect cells with recombinant NDV expressing poliovirus proteins
-
Prepare 10x dilution of allantoic fluid containing recombinant NDV in DMEM. You will need 200 μl of the final solution per one well of the 12 well plate. Scale up as necessary if planning to infect more than one well.For propagation and quantitation of NDV please refer to (McGinnes et al., 2006)
- Remove growth medium from HeLa cell monolayer, add 200 μl of recombinant NDV solution, place the cells in an incubator at 37 °C, 5% CO2 for virus adsorption for one hour.
- Gently rotate the plate periodically making sure the virus solution is distributed evenly over the monolayer.
- Replace the virus solution with 1 ml of DMEM/2% FBS, leave the infected cells to incubate overnight in an incubator at 37 °C, 5% CO2.
-
- Transfection of NDV-infected cells with polio replicon RNA
-
Thaw the replicon RNA (see protocol 1), keep tube on ice.Make sure to maintain an RNAse-free environment by wiping all the equipment, pipets, etc. with RNAse wipes as described in Basic Protocols 1 and 2.
- Prepare an Eppendorf tube with 400 μl of OPTI-MEM.
- Add 1.5 μg of replicon RNA to OPTI-MEM, mix by brief vortexing.
- Immediately add 4 μl of Boost reagent from the RNA transfection kit to the RNA/OPTI-MEM mix, vortex briefly.
-
Add 4 μl of TransIT reagent from the RNA transfection kit, vortex briefly, incubate for 2 min.The total incubation time of the transfection mix before adding it to cells should not exceed 5 min.
- Add transfection medium dropwise to the cells infected with recombinant NDV, incubate for six hours in an incubator at 37 °C, 5% CO2.
- Freeze cells without removing incubation medium at −20 °C. Perform three freeze-thawing cycles to release intracellular packaged replicon.
- Transfer the medium with cell debris in a new Eppendorf tube and clarify by centrifugation at 1000g for 5 min.
-
Transfer supernatant containing packaged replicon in a new tube. This material can now be used to infect cells using poliovirus infection protocol (Burrill, Strings, & Andino, 2013).The packaged replicon can be used in replication assay, essentially as described in protocol 2, but obviously, using poliovirus infection protocol (Burrill et al., 2013) instead of RNA transfection. An example of a replication assay performed with packaged replicon is shown on Fig. 5B. The usual yield of packaged replicon after this procedure is 1E7–1E8 infectious particles per ml. To assess the efficiency of packaging infect HeLa cells with serial dilutions of the replicon (using regular poliovirus infection protocol (Burrill et al., 2013), fix cells at 6 h post infection and perform immunofluorescent staining for a virus antigen. Single infected cells should be clearly visible (Fig. 5C). It is convenient to perform such infection in parallel with a poliovirus prep of a known titer, this way the titer of the packaged replicon can be easily deduced by comparing the number of positive cells in a certain area of monolayer between the two samples. Otherwise you can calculate the packaged replicon titer taking into consideration the number of polio antigen-positive cells, the area of the monolayer scanned and the dilution used for infection.
-
REAGENTS AND SOLUTIONS
10×TBE
Dissolve 108 g Tris base and 55 g boric acid in 800 ml distilled water, add 40 ml 0.5M Na2EDTA (pH 8.0) and adjust volume to 1 liter with molecular grade water.
60 mM EnduRen
Dissolve the entire content of EnduRen provided by the vendor in the tube (3.4mg) in 100 μl of DMSO (make sure DMSO is cell culture quality and is not saturated with water).
COMMENTARY
Background Information
Poliovirus is a fast replicating lytic virus. Its infectious cycle in HeLa cells takes only about six hours and by eight hours post infection the cells disintegrate releasing the progeny virus into environment. In spite of its small genome, poliovirus rapidly induces dramatic reorganization of multiple aspects of cellular metabolism, from mRNA translation to membrane synthesis. Cleavage of a cellular translation initiation factor eIF4G by the viral protease 2A results in inhibition of translation of capped cellular mRNAs so that by the middle of infectious cycle the infected cells synthesize virtually only viral proteins (Castello, Alvarez, & Carrasco, 2011). 2A is also implicated in cleavage of certain nucleoporins, thus destroying the barrier function of nuclear pores and inducing relocalization of nuclear proteins into the cytoplasm where they may facilitate viral RNA replication (Flather & Semler, 2015). Poliovirus genome replication, like that of all (+)RNA viruses of eukaryotes, is associated with specialized membranous structures, replication organelles. The development of these membranous compartments in infected cells requires rerouting of membrane trafficking and lipid synthesis pathways, creating membrane environment with unique lipid and protein composition, necessary to support viral replication and virion maturation (Belov, 2014). The importance of these changes of the cellular metabolism for the viral life cycle and their mechanisms are still a matter of active research. The replicon system expressing Renilla luciferase described here is very well suited to quantitatively assess the involvement of specific cellular proteins in the viral replication, and to evaluate the perspective antiviral intervention strategies. Moreover, the similarity of genome organization and expression strategy among other enteroviruses allows the development of similar replication monitoring systems for other members of this important genus, including such emerging pathogens as enteroviruses 71 and D68.
Critical Parameters
One cannot stress hard enough that the success of the replicon assays depends on the quality of RNA. Thus, all possible precautions should be taken to maintain RNAse-free environment during production and purification of replicon RNA. It is highly advisable to have a dedicated working place in the lab with assigned set of pipettors, glassware, electrophoresis tanks, etc. which are used only for RNA work. Purified RNA should always be stored at −80C, and when thawed for transfection, the tubes should be kept on ice. Good tissue culture techniques should also be maintained. It is highly recommended, if possible, to propagate cells without antibiotics in the medium, so that any contamination could be easily detected and the culture replaced if necessary. The cells should always be seeded without antibiotics for transfection experiments to increased efficiency of transfection.
Troubleshooting
If replication in control conditions (i.e. HeLa cells and wildtype replicon) gives unexpectedly low luciferase reading, quality of RNA should be assessed first. On agarose gel the RNA should run as discrete bands (see Fig. 3) without traces of degradation (smear) beyond the small molecular weight band (Fig. 3, asterisk). The ratio of OD260 to OD280 of the purified RNA should be within 1.85–2.0 interval. If the RNA quality is good, the other most common reason of failure is the incubation chamber conditions in the multiwall plate reader. Make sure that plate is properly sealed with optical clear film, so that there is no evaporation from the wells. Non-transfected cells after 16h of incubation in the device should look healthy and have many dividing cells. To test if Renilla luciferase activity detection parameters are adequate, perform luciferase assay with cells transfected with a plasmid expressing Renilla luciferase form a eukaryotic promotor (such as phRL-CMV (Promega)), optimize if necessary.
Statistical Analyses
The setup of a replication experiment in 96 well plate provides multiple parallel runs of the same conditions. We recommend allocating no less than 16 wells (2 columns) for any particular sample. Cells may grow differently in the wells located on the edge of the plate, leading to variability of the luciferase signal, in this case increase the number of wells allocated to a sample. Analysis of the results, including graphic representation of replication kinetics and statistical analysis is convenient to perform in GraphPad Prism or other similar statistical software package.
Understanding Results
It is important to keep in mind that Renilla luciferase reading is a cumulative measure of polio RNA replication and translation. In an infected cell these parameters are interconnected and cannot be separated. The RNA is translated generating replication proteins, and the replication process increases the number of RNAs available for translation. Moreover, expression of poliovirus protease 2A increases the efficiency of poliovirus RNA translation over the replication time course because of rerouting of cellular translation machinery towards poliovirus RNA (Castello et al., 2011). It is possible to study the efficiency of translation of the input RNA only, if replication process is inhibited (i.e. the cells are incubated in the presence of 2mM guanidine-HCl, a strong specific inhibitor of poliovirus replication, or if a replicon construct with defective polymerase gene is used).
Time Considerations
Starting from the plasmid linearization, one can expect to have purified replicon RNA available for replication experiments in two days. The amount of RNA usually obtained from one transcription reaction is sufficient for transfection of ~40 96-well plates.
Significance Statement.
Reliable quantitation of biological processes is indispensable for the correct interpretation of experimental data and reproducibility of results. Here we describe protocols that will allow researchers easy and consistent assessment of replication of poliovirus RNA in live cells, over the entire time course of infection, using replicon RNA. Such a replicon expresses a reporter protein whose signal can be captured and quantified in semi-automatic settings. The replicon-based assays are extremely useful in screening of anti-viral compounds, studying virus-cell interaction, and investigating other aspects of molecular biology of the virus. The methods described here are easily translatable to other related viruses.
Acknowledgments
We are grateful for Raul Andino for his ideas on packaging poliovirus replicon RNA using recombinant NDV. This work was supported by the NIH grant R21AI15383A to GAB.
LITERATURE CITED
- Belov GA. Modulation of lipid synthesis and trafficking pathways by picornaviruses. Current Opinion in Virology. 2014;9:19–23. doi: 10.1016/j.coviro.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Belov GA, Altan-Bonnet N, Kovtunovych G, Jackson CL, Lippincott-Schwartz J, Ehrenfeld E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. Journal of Virology. 2007;81(2):558–567. doi: 10.1128/Jvi.01820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov GA, Fogg MH, Ehrenfeld E. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. Journal of Virology. 2005;79(11):7207–7216. doi: 10.1128/Jvi.79.11.7207.7216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrill CP, Strings VR, Andino R. Poliovirus: generation, quantification, propagation, purification, and storage. Curr Protoc Microbiol. 2013 doi: 10.1002/9780471729259.mc15h01s29. Chapter 15, Unit 15H 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Alvarez E, Carrasco L. The Multifaceted Poliovirus 2A Protease: Regulation of Gene Expression by Picornavirus Proteases. Journal of Biomedicine and Biotechnology. 2011 doi: 10.1155/2011/369648. doi:Artn 369648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flather D, Semler BL. Picornaviruses and nuclear functions: targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Frontiers in Microbiology. 2015;6 doi: 10.3389/fmicb.2015.00594. doi:ARTN 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Andino R. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. Journal of Virology. 2000;74(14):6394–6400. doi: 10.1128/Jvi.74.14.6394-6400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CA, Cobbs C, Peduzzi JD, Novak M, Morrow CD. Repetitive intrathecal injections of poliovirus replicons result in gene expression in neurons of the Central Nervous System without pathogenesis. Human Gene Therapy. 2001;12(15):1827–1841. doi: 10.1089/104303401753153893. [DOI] [PubMed] [Google Scholar]
- Kaplan G, Racaniello VR. Construction and Characterization of Poliovirus Subgenomic Replicons. Journal of Virology. 1988;62(5):1687–1696. doi: 10.1128/jvi.62.5.1687-1696.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnes LW, Pantua H, Reitter J, Morrison TG. Newcastle disease virus: propagation, quantification, and storage. Curr Protoc Microbiol. 2006 doi: 10.1002/9780471729259.mc15f02s01. Chapter 15, Unit 15F 12. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Johnson KL, Sarnow P, Kirkegaard K. Functional coupling between replication and packaging of poliovirus replicon RNA. Journal of Virology. 1999;73(1):427–435. doi: 10.1128/jvi.73.1.427-435.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy N, Barclay WS, Sullivan M, Almond JW. A Poliovirus Replicon Containing the Chloramphenicol Acetyltransferase Gene Can Be Used to Study the Replication and Encapsidation of Poliovirus Rna. Journal of Virology. 1992;66(8):5040–5046. doi: 10.1128/jvi.66.8.5040-5046.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DC, Ansardi DC, Wang J, McPherson S, Moldoveanu Z, Morrow CD. Demonstration of the specificity of poliovirus encapsidation using a novel replicon which encodes enzymatically active firefly luciferase. Virology. 1998;243(1):1–11. doi: 10.1006/viro.1998.9046. [DOI] [PubMed] [Google Scholar]