

Abstract
In 2011, the US Food and drug Administration (FDA) developed a strategic plan for regulatory science that focuses on developing new tools, standards, and approaches to assess the safety, efficacy, quality, and performance of FDA-regulated products. In line with this, the Division of Applied Regulatory Science was created to move new science into the Center for Drug Evaluation and Research (CDER) review process and close the gap between scientific innovation and drug review. The Division, located in the Office of Clinical Pharmacology, is unique in that it performs mission-critical applied research and review across the translational research spectrum including in vitro and in vivo laboratory research, in silico computational modeling and informatics, and integrated clinical research covering clinical pharmacology, experimental medicine, and postmarket analyses. The Division collaborates with Offices throughout CDER, across the FDA, other government agencies, academia, and industry. The Division is able to rapidly form interdisciplinary teams of pharmacologists, biologists, chemists, computational scientists, and clinicians to respond to challenging regulatory questions for specific review issues and for longer-range projects requiring the development of predictive models, tools, and biomarkers to speed the development and regulatory evaluation of safe and effective drugs. This article reviews the Division’s recent work and future directions, highlighting development and validation of biomarkers; novel humanized animal models; translational predictive safety combining in vitro, in silico, and in vivo clinical biomarkers; chemical and biomedical informatics tools for safety predictions; novel approaches to speed the development of complex generic drugs, biosimilars, and antibiotics; and precision medicine.
Keywords: drug development, drug safety, biomarkers, predictive modeling, computational modeling
Introduction
The United States Food and Drug Administration (FDA) has a mission to protect public health by assuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, our nation’s food supply, cosmetics, and products that emit radiation. In addition, FDA is responsible for facilitating innovations that make medicines more effective, safer, and more affordable.1 In order to accomplish these goals, FDA has stimulated advancement of regulatory science, which is defined as “the science of developing new tools, standards, and approaches to assess the safety, efficacy, quality, and performance of all FDA regulated products.”2 This was highlighted in FDA’s Regulatory Science Strategic Plan3 that prompted a new focus by FDA research staff to advance the state of science within the agency, publish their research findings in peer-reviewed scientific journals, and translate new science into the regulatory review of medical products.
The FDA Center for Drug Evaluation and Research (CDER) published a Science Needs Report in 2011 and Safety Research Priorities in 2015.4,5 These highlighted priorities span both extramural and intramural research, the latter being critical for rapid response to urgent regulatory questions and for ensuring that research is immediately transferrable to the review process. Within CDER, there are three dedicated laboratory research groups that meet this challenge: the Office of Pharmaceutical Quality’s Office of Testing and Research (OTR) and Office of Biological Products (OBP) (see Fisher et al6 for a review of research activities), and the Office of Translational Science’s Office of Clinical Pharmacology, Division of Applied Regulatory Science (DARS). This article focuses on the translational research division—DARS.
DARS tackles emerging regulatory questions and develops and implements new regulatory review tools and approaches across the spectrum of in vitro, in silico (computational), in vivo (animal models and clinical pharmacology/experimental medicine), and postmarket safety analyses (Figure 1). The vision of DARS is to facilitate the integration of new science into the process of evaluating the safety and efficacy of proposed and marketed products and to close the gap between scientific innovation and this process of drug review. To achieve this vision, DARS performs FDA and CDER mission-critical research to develop and evaluate tools, standards, and approaches to assess the safety, efficacy, quality, and performance of drugs and provides expert regulatory review consultations for mechanistic evaluation and other immediate regulatory needs. The Division forms teams around projects with individual researchers serving as team leaders or team members. This unique multidisciplinary group includes physicians, pharmacologists, pharmacists, veterinarians, pharmacokineticists, toxicologists, physiologists, immunologists, microbiologists, molecular and cellular biologists, biochemists, inorganic chemists, pharmaceutical scientists, computational biologists, bioengineers, biophysicists, and mathematicians.
Figure 1.

The FDA Division of Applied Regulatory Science (DARS) uses multiple avenues of research to close the gap between scientific innovation, translational research, and drug review. DARS applies in vitro and in vivo laboratory research, in silico computational modeling, and informatics and integrated clinical research to develop new regulatory science tools, perform post-market analyses, and assimilate innovative science into the drug review process to enhance drug safety and efficacy.
This article reviews the Division’s recent work, broadly grouping efforts into the development and validation of (1) safety biomarkers; (2) humanized animal models for detecting immune-mediated adverse events; (3) a translational cardiac safety paradigm combining in vitro and in silico predictions with clinical biomarkers in phase 1 trials; (4) chemical and biomedical informatics tools for assessing drug safety and mechanisms of action; (5) analytical methods, novel models, and new approaches to speed drug development, particularly of complex generic drugs, biosimilars, and antibiotics; and (6) novel approaches to advance precision medicine.
Biomarkers
Biomarkers are defined characteristics that are objectively measured and evaluated as indicators of normal biological processes, pathologic processes, or biological responses to a therapeutic intervention.7 Biomarkers can be used to select patients for clinical trials and treatment, identify safety problems related to a drug, or reveal pharmacological activity predictive of therapeutic response. CDER created a formal biomarker qualification process,7 through which biomarkers can be assessed and pronounced suitable for a particular context of use in drug development. This “qualification” allows use of the biomarker across multiple drug development programs without the need for repeated assessment and thereby promotes general use of these qualified biomarkers. DARS scientists have contributed to the development, characterization, and validation of multiple biomarkers and have been involved in the review of biomarker qualification packages submitted to the Agency. The following highlights safety biomarkers that DARS scientists have studied in FDA laboratories.
Biomarkers of Acute Pancreatic Injury
Following emergence of postapproval pancreatic injury safety signals for antidiabetic drugs targeting the glucagon-like peptide-1 pathway, DARS initiated a series of in vitro and in vivo studies on drug-induced acute pancreatitis to understand the mechanism, discover a viable model, and identify and characterize novel genomic (microRNA) biomarkers of acute pancreatic injury. In a project that generated 9 publications, molecular processes and components involved in early injury were verified8,9 and a role for glucagon-like peptide-1 drugs in injury was hypothesized.10 In addition, novel microRNA biomarkers of acute pancreatic injury were characterized in rodents11 and dogs.12 In ongoing work, cell specificity for the microRNAs in the pancreas is being studied using laser capture microscopy and leveraging Next Generation Sequencing to determine the prevalence of microRNA variants in cell types and to define the role of the microRNAs in the cell. Informed by this work, a public-private consortium has started the process to formally qualify these microRNA biomarkers for regulatory use.
Biomarkers of Acute Kidney Injury
The first biomarker package submitted to FDA for qualification was for a set of acute kidney injury biomarkers. The progression of this package through the process was an iterative learning experience for the submitters and the reviewers. Initial knowledge gaps included behavior of the biomarkers during recovery from injury (eg, whether they return to baseline levels). DARS provided early data on kidney injury biomarker behavior during recovery from injury and a template for further studies13 to be conducted by the submitters. DARS research supported the context-of-use criteria for those first kidney biomarkers qualified through the FDA process as well as those subsequently submitted in a second package, in part by demonstrating in a study with contrast media that the candidate biomarkers identified tubular injury and not primary glomerular impairment.14
Histopathology Methods for Biomarker Qualification Studies
In making determinations about the validity of a given toxicity biomarker to be used in regulatory decision making, FDA staff are interested in the quality, quantity, and consistency of histopathology data. DARS engaged with the reviewers to design and execute experiments to evaluate the impact of existing industry histopathology practices on data interpretation and to determine if alternative approaches might be more informative to the review process. Major sources of variation in standard industry histopathology methods and the impact of that variability on interpretation were defined by DARS.15 Follow-up studies indicated that more complex evaluation methods (Figure 2) did not outperform the existing standard methodology.16 Ultimately, FDA published the Guidance to Industry on Histopathology Methods for Biomarker Qualifications Studies that aligned with and was supported by DARS data.17
Figure 2.
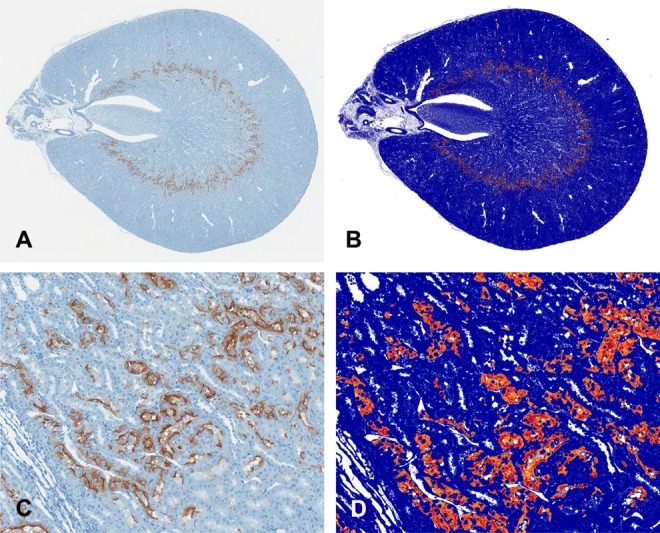
Digital image analysis of Kim-1 immunohistochemical staining of cisplatin-induced kidney injury used for quantitative injury assessment did not improve on pathologist semiquantitative evaluation. Figure shows 20× whole slide image (A) with magnified example (B) of Kim-1 positive staining (brown); corresponding positive pixel image analysis (B, D), with yellow, orange, and red representing weak positive, positive, and strong positive, respectively; reproduced from Shea et al.16
Noninvasive Biomarkers of Phospholipidosis
Phospholipidosis denotes an excessive accumulation of phospholipid in lysosome-derived vesicles that is not unusual to see in nonclinical studies with cationic amphiphilic drugs and aminoglycoside antibiotics. The actual clinical incidence and implications of phospholipidosis are unclear because of an inability to monitor the presence and progress of phospholipidosis in patients. The location and degree of phospholipidosis varies by drug and have generally been shown to be slowly reversible. However, because of morphological similarities with lysosomal storage disorders and undetermined toxicity potential from drug accumulation, the observation of phospholipidosis resulted in novel molecules being dropped from the drug development process. To allow further investigation of some of these potentially beneficial drugs, studies were conducted by DARS to examine candidate biomarkers for phospholipidosis, and a urinary biomarker was identified.18
Biomarkers of Drug Efficacy for Medical Countermeasures
Stimulating the development of treatments for diseases and toxins used as weapons against the public are another area in which FDA increasingly operates. Developing treatments called medical countermeasures (MCM) for these types of threats to the US public health is complicated by the inability to test for safety and efficacy in patients. These diseases and toxic exposures are too deadly to allow exposure and testing in healthy volunteers as in a routine drug development program and natural occurrences are too infrequent to provide adequate and timely data for efficacy testing. In response, DARS has worked to develop models and discover biomarkers to facilitate MCM development, including for high-mortality influenza,19 while continuing to explore opportunities to apply in vitro, in vivo, and in silico tools to facilitate MCM development.
Biomarkers of Drug-Induced Vascular Injury
Drug-induced vascular injury is a toxicity seen in nonclinical studies in rats, dogs, and primates.20–22 It is seen with significant frequency in drug development across many classes of small molecule drugs. The clinical occurrence and relevance of vascular injury are not well defined, and the mechanism of action is poorly understood, but appears to involve some dysregulation of nitric oxide production. For some drugs, surrogate biomarkers such as systemic blood pressure can be used to monitor for vascular injury, but for other drugs, the toxicity cannot directly be monitored. To overcome this hurdle, DARS engaged with a public-private consortium through the Critical Path Institute to perform research investigating the mechanism(s) of action in this toxicity as well as evaluating candidate biomarkers. A package of biomarkers derived from and supported by this consortium is presently in the FDA qualification process.
Novel Humanized Animal Models
While many drug-induced adverse events and safety biomarkers are transferable between species, the immune system is very species-specific and traditional animal models often fail to detect immune-mediated adverse events. This has become particularly important with the recent focus on biologic drugs and immunotherapies for cancer. To address this, DARS has taken immune23,24 and liver25 humanized mouse models (Figure 3) and enhanced them for application to the pharmacology and toxicity assessment of biologics, biosimilars, and small molecule drugs.
Figure 3.

DARS utilizes advanced “humanized” mouse models to improve drug safety assessment. These models, optimized in DARS laboratories, provide an enhanced predictive ability for human immune and liver responses to drugs, thereby improving the ability to recognize in an animal model potential adverse events that are specific to humans. These models can also inform the assessment process for determining the similarity between complex biological drugs (biosimilars). Scheme for immune-humanized mouse model is shown above.
These immune-humanized mice can be used to test the functional effects of changes in therapeutic protein structure as a result of variations in drug-manufacturing processes. Immune-humanized mice can also be used to study adverse events associated with therapeutic proteins such as the loss of immune system modulation associated with the use of checkpoint inhibitors or the ability to induce excessive release of cytokines. Finally, therapeutic biologic drugs are made in living cells and can have differences from batch to batch, and between manufacturers, and thus “generic” products are called “biosimilars.” These mice can provide a useful nonclinical model system to compare responses between innovator therapeutic drugs and proposed biosimilar drugs. Ongoing research is being conducted using well-characterized protein drugs to determine the performance of humanized mouse models in these various regulatory review scenarios.
In addition to immune-humanized mice, DARS is evaluating liver-humanized mice. This model uses a mouse with liver-specific expression of viral thymidine kinase on an NOD/Shi-scid/IL-2Rγnull immune-deficient background. Treatment of these mice with ganciclovir results in loss of most mouse hepatocytes and allows engraftment with human hepatocytes that are injected into the spleen and which migrate to the liver. The mice become chimeras with mixed human and murine hepatocytes but with a majority of human hepatocytes. These mice have potential as a pharmacology model to assess aspects of human-specific drug metabolism in a nonclinical model. Finally, methods to create dual liver and immune humanized mice are being established. This is expected to result in a valuable research tool that, in combination with single liver- and immune-humanized mice, will allow critical investigations into the role and contribution of the immune system to drug-induced liver toxicity. The humanized mouse work is providing the drug development and regulatory communities with novel and valuable tools to evaluate the effects of pharmaceuticals on human cells and tissues.
Translational Approaches to Predictive Safety
While prior biomarker work has focused on in vivo markers that could be applied during the drug development process, ideally, drug safety predictions could be utilized even earlier during development to help drug developers select the safest and most effective compounds to move forward. With this in mind, a new predictive safety paradigm is being pursued that combines in vitro cell assays with in silico modeling of human cells to predict cardiac safety early in development, followed by a check in early phase 1 clinical trials with electrocardiographic (ECG) biomarkers.
In the 1990s to early 2000s, multiple drugs were removed from the market because they exhibited an increased risk of torsade de pointes, an arrhythmia that can lead to sudden cardiac death. A regulatory paradigm was put in place in 2005 by the International Council for Harmonisation for assessing the proarrhythmic risk of new drugs that focuses on determining whether a drug blocks the hERG potassium channel and prolongs repolarization time (the QT interval) on the ECG.26–28 Testing drugs for hERG potassium channel block and QT prolongation is sensitive for identifying drugs that may cause torsade de pointes, but is not specific as some drugs block hERG and prolong QT without causing torsade de pointes.
To improve on the current paradigm, DARS working together with FDA/CDER’s Office of New Drugs and an international consortium of drug regulators, industry, and academia developed the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative.29–33 The CiPA initiative is developing an in vitro and in silico paradigm for cardiac safety evaluation of new drugs that provides a more accurate and comprehensive mechanistic-based assessment of proarrhythmic potential. CiPA involves 4 components (Figure 4): (1) in vitro assessment of drug effects on multiple cardiac ion channels; (2) integration of the multi-ion channel effects in an in silico computer model of the human ventricular cardiomyocyte to output a proarrhythmic risk score; (3) assessment for unanticipated effects in vitro using human-induced pluripotent stem cell-derived cardiomyocytes; and (4) phase 1 clinical studies with exposure-response analysis. DARS is leading applied research across all 4 components to develop and validate this novel regulatory paradigm.
Figure 4.
The 4 components of the Comprehensive in vitro Proarrhythmia Assay (CiPA)
First, drug effects on multiple ion channel currents are assessed. Second, a proarrhythmic score is computed using an in silico model of the human ventricular cardiomyocyte, which integrates the individual ion channel effects. The third component is a confirmatory in vitro study using human stem cell–derived ventricular cardiomyocytes. The goal of the fourth component is to use human phase 1 ECG data to determine if unexpected ion channel effects are observed in humans compared to preclinical ion channel data predictions; reproduced from Vicente et al.32

For the in vitro assessment of drug effects on multiple cardiac ion channels, DARS is assessing 28 CiPA test drugs that were selected by an external group of experts, the CiPA Compound Selection Group. Drugs are being assessed using manual patch clamp techniques to assess block potency34 and with a protocol to elicit the dynamic interactions between the drug and hERG potassium channel.35,36 The effect of the 28 drugs on the multiple potassium, sodium, and calcium ion channels is integrated into the in silico model. DARS developed this in silico model by incorporating a novel dynamic hERG model37 into the O’Hara-Rudy ventricular cardiomyocyte cell model.38 An initial set of 12 of the 28 CiPA drugs has been classified into low, intermediate, and high risk for arrhythmias, corresponding with the categories assigned by the Compound Selection Group.37 In addition, the 28 CiPA drugs are being assessed in induced pluripotent stem cell-derived cardiomyocytes in an international, multisite validation study funded by FDA. Two different laboratory device platforms are being studied: multi-electrode arrays and voltage-sensitive optical technologies. While that study is ongoing, multiple preliminary studies have demonstrated the potential of these technologies, including a large study completed at FDA.39
Clinical Biomarkers for Cardiac Safety in Early Phase 1 Studies
The last component of CiPA involves an ECG assessment in early clinical phase 1 studies (single or multiple ascending dose studies) to determine if there are unexpected ion channel effects compared to the preclinical ion channel data, as might occur with human-specific metabolites or differences in protein binding. In order to identify novel clinical ECG biomarkers that could differentiate drug-induced multichannel block, an analysis of 500,000 digital ECGs from 34 clinical thorough QT studies was performed and compared to nonclinical ion channel data. An ECG biomarker (J-Tpeak interval) was identified that could differentiate drugs that selectively block hERG (high torsade risk) from those that block hERG and late sodium or L-type calcium currents (low torsade risk) (Figure 5).40 In order to further assess this biomarker, two prospective clinical trials were performed assessing a total of 8 drugs and 3 drug combinations.41,42 In an additional analysis, 12 potential biomarkers were evaluated and J-Tpeak was found to be the best biomarker at discriminating multichannel block.43,44 In order to facilitate widespread use of this new method, the algorithm was released as open-source software (https://github.com/FDA/ecglib),45 which has already been integrated into ECG analysis software by commercial vendors. The complete CiPA initiative was recently endorsed by the Pharmaceutical Science and Clinical Pharmacology Advisory Committee46 with a goal of revising the International Council for Harmonisation guidelines (E14 and S7B)26,27 to incorporate this new mechanistic, model-informed approach into drug safety assessment.
Figure 5.

An illustration of the relation of the body surface electrocardiogram (ECG) to the underlying ventricular action potentials. Arrows pointing into the action potential are inward currents (calcium and late sodium) and arrows pointing out denote outward currents (hERG potassium). Blocking the calcium or late sodium current primarily shortens the early parts of repolarization (J-Tpeak), whereas hERG potassium channel block prolongs both early (J-Tpeak) and late repolarization (Tpeak-Tend). Thus, the J-Tpeak interval can differentiate drugs that only block hERG (eg, dofetilide, high torsade risk) from those that block hERG and inward currents (eg, ranolazine, low torsade risk). Drug-induced changes in heart rate-corrected QT (QTc), heart rate-corrected J-Tpeak (J-Tpeakc), and Tpeak-Tend are shown for dofetilide (left) and ranolazine (right). Both drugs prolong QTc; however, only dofetilide prolongs J-Tpeakc, and the absence of J-Tpeakc prolongation suggests that a drug is an hERG potassium channel block that has “balanced” inward current block and is not associated with torsade; reproduced from Johannesen et al.41
Chemical and Biomedical Informatics and Systems Pharmacology
As with the CiPA initiative, application of other in silico approaches can speed drug development by enabling pharmaceutical applicants to apply predictive models early in development and for regulators to perform rapid analyses in the premarket or postmarket phase. DARS develops and validates chemical and biomedical informatics tools and models for these applications.
Chemical Informatics Models for Improved Safety Assessment
The ability to apply knowledge about chemical structures and their relationship to biological activity has become an increasingly important part of drug development and regulatory review. The term chemical informatics describes the interface between chemistry and computational-based information science and includes the compilation, organization, and analysis of chemical information to develop knowledge bases and predictive models. Chemical informatics tools and approaches developed by DARS research activities are now routinely being used across CDER to inform regulatory decisions on product safety. They include databases of nonclinical toxicology and clinical adverse effect findings linked through chemical structures to facilitate structure-based searching, and (quantitative) structure-activity relationship ((Q)SAR) models that predict biological activity based on chemical structure (Figure 6). For example, a combination of a similarity search and a (Q)SAR evaluation on a drug structure is used to identify structural comparators and their corresponding clinical adverse effect profiles to determine whether a safety signal is typical of that structural class and can be attributed to a specific chemical feature.
Figure 6.

DARS chemical informatics projects are focused on endpoints of regulatory interest, linked through chemical structure. Research activities include (1) database development for model training and validation; (2) development of web-based applications to enable direct structure-based searching by CDER staff; and (3) (Q)SAR model development to support drug safety review. Databases and models are used to respond to regulatory review consultation requests.
DARS chemical informatics research efforts focus on the development, use, and improvement of chemical informatics tools to provide structure-based safety assessments. Emphasis is placed on nonclinical and clinical endpoints of regulatory significance, including genetic toxicity, carcinogenicity, hepatotoxicity, and cardiotoxicity.47 Models are constructed from public data to ensure transparency and to facilitate the application of “expert knowledge” to predictions to obtain the best overall conclusions. The steady improvement in (Q)SAR predictive performance in recent years has culminated in the implementation of the first harmonized pharmaceutical regulatory guidance where (Q)SAR models can be used as a replacement for experimental testing.48 The International Council for Harmonisation M7 guideline49 describes how to assess and control DNA-reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. This represents a state-of-the-art method for impurity qualification50 and is a major milestone in the regulatory acceptance of (Q)SAR modeling.51
In addition to developing new models and structure-linked databases, DARS chemical informatics scientists perform regulatory reviews for new and generic drug products. In 2016, 582 compounds were analyzed in 232 consultations. This includes (Q)SAR assessments performed in-house and expert reviews of (Q)SAR data submitted by pharmaceutical applicants for quality and completeness. In both situations, lessons can be learned from the real-time application of models and this knowledge can then be applied to the next generation of models, to build on strengths and address weaknesses. Newly emerging research and development areas for the application of chemical informatics at CDER include the evaluation of a strategy using (Q)SAR predictions in combination with structure-based searching to support the development of new drugs for severely debilitating and life-threatening diseases, where certain safety studies could potentially be deferred to the postmarket setting or waived entirely. Additionally, 3-dimensional (Q)SAR models are being developed to predict the receptor binding of newly identified synthetic street drugs (eg, opioids) to assess their abuse potential and subsequent risk to public health.
Biomedical Informatics Tools for Improved Safety Assessment
Clinical trials may not identify serious adverse events that subsequently come to light in the postmarket setting when drugs are administered to broader and larger patient populations. A number of approaches attempt to predict these adverse events with varying degrees of success.52–55 DARS is developing and testing informatics-based approaches that link adverse events to drug targets,56,57 capturing therapeutic on-target and “off-target” effects (Figure 7). An integrated approach is utilized that involves pooling together all drugs known to “hit” a specific target in combination with information from (1) postmarket safety reports in the FDA Adverse Event Reporting System, (2) adverse events listed on all FDA drug labels and (3) databases that synthesize adverse events from reports in the peer-reviewed literature. Statistical analyses are applied that integrate these different data sources into a single risk assessment, which is then manually reviewed by an expert.
Figure 7.

DARS utilizes target-adverse event profiles to predict adverse events. DARS researchers utilize adverse event data from the FDA Adverse Event Reporting System, FDA drug labels, and peer-reviewed literature and databases containing drug and target information to connect adverse events to drug targets.
Validation of this approach is underway with a large number of previously approved drugs to optimize thresholds for potential application at different points in drug development. This includes potential application at the end of phase 2 drug development where an analysis could inform which adverse events to most closely monitor in phase 3 clinical trials, application at the time of new drug review to inform review of safety data observed in clinical trials prior to a new drug’s approval, and for postmarket safety monitoring to determine which adverse events deserve a more careful examination. This work is ongoing in collaboration with FDA/CDER’s Office of Surveillance and Epidemiology and Office of New Drugs.
DARS also performs research into the application of systems pharmacology models to predict drug safety. Research efforts have included a systems pharmacology analysis of Stevens Johnson Syndrome and toxic epidermal necrolysis,58 along with identification of drug-specific pathways based on gene expression data with application to drug-induced lung injury.59 A postmarket analysis also studied trastuzumab-related cardiac toxicity.60
Facilitating Drug Development
Clinical Pharmacology Tools for Drug Development
DARS works with the other Divisions throughout the Office of Clinical Pharmacology and other CDER reviewers to enhance the drug review process by providing pharmaceutical applicants with tools and approaches that would yield information relevant to and required by the approval process as well as address mechanisms for adverse events. Collaborative projects have been undertaken to better define and characterize in vitro models for industry use in early drug discovery and development to assess absorption, distribution, metabolism, elimination, and the potential for drug-drug interaction61,62 as well as for investigating mechanisms for adverse events.63 These tools help bridge the gap between laboratory science that is the anchor for drug discovery, and clinical use, the ultimate goal of regulatory approval. DARS continues to bring new methods to inform drug development and approval by developing in vitro models of liver metabolism and drug transporters.
Complex Generic Drugs and Biosimilars
FDA can, in part, increase access to safe and effective therapies by advancing methods to more rapidly approve complex generic drugs and biosimilars. The standard process for approving small molecule generic drugs involves demonstrating bioequivalence based on similarity in physicochemical characteristics and pharmacokinetics. However, demonstrating similarity in physicochemical characteristics and/or pharmacokinetics may not be possible for some complex drugs (eg, iron colloid products, peptides) and locally acting drugs (eg, ophthalmic, inhalation, dermatologic) creating challenges for generic drug development. DARS works with CDER’s Office of Generic Drugs to assess the need for changes in FDA guidance or policy and to develop new tools for determining the bioequivalence of complex and/or nonsystemic products to overcome these barriers to generic drug approval. For example, the European Medicines Agency had required that generic iron colloid drugs be assessed in biodistribution studies, while FDA did not have this requirement. To assess the need for this, DARS completed in vivo biodistribution studies and cellular uptake studies and demonstrated no difference between brand and generic iron colloid products. This led to alignment of the European Medicines Agency and FDA requirements that now do not include biodistribution studies. In a separate effort to speed the development and approval of generic drugs, animal studies are being performed to validate a physiologically based pharmacokinetic (PBPK) model of topical drug distribution to different tissue compartments within the eye. Optimization and validation of a PBPK model of the eye could reduce the number of patients needed in comparative clinical trials, as well as the number of animals that would be required to demonstrate bioequivalence and support generic drug approval for locally acting ophthalmic drugs.
The recognized differences from batch to batch for a single manufacturer and between manufacturers of biosimilars also suggest a need for additional assessment beyond pharmacokinetic bioequivalence to approve a biosimilar drug.64 Submissions for biosimilars approved to date have all included clinical noninferiority outcome studies. However, with pharmacodynamic biomarkers, specific biosimilars could conceivably be approved without clinical outcome studies. DARS, together with the rest of the Office of Clinical Pharmacology, is evaluating which pharmacodynamic biomarkers may be appropriate for assessing biologic drugs that are expected to have biosimilar drug products. Combined in vitro and in vivo research is being planned to assess candidate biomarkers.
Antimicrobial Drug Development
The emergence and spread of antibiotic-resistant bacteria plagues modern medicine and helps disincentivize antibiotic drug development. The constantly evolving threat of antibiotic resistance has improved proper stewardship of new antibiotic products, which dictates minimizing their use and thereby limiting their sales potential.65,66 DARS is working with CDER’s Office of Medical Policy to examine approaches to minimizing antibiotic resistance by performing a series of in vitro and in vivo translational research studies to develop nonclinical tools to measure the rate at which antibiotic resistance appears. These methods could then be used to compare antibiotic treatment regimens or combinations for their ability to suppress the appearance of antibiotic resistance.
The approach uses an in vitro hollow fiber bioreactor system where bacteria are confined in the chamber outside of fibers with 20kD pores (Figure 8). Nutrients and drugs are pumped through the lumen of the fibers and freely exchange with the extra-fiber space. Computer-controlled peristaltic pumps are used to raise and lower the concentration of antibiotic to imitate human pharmacokinetic concentration profiles. The rate of emergence of antibiotic resistance is then studied with individual drugs and with 2- and 3-drug combinations. In parallel, the same bacteria and antibiotics are studied in a mouse model of urinary tract infection. Mice are infected and then treated with various concentrations of single and combination antibiotics to elucidate exposure conditions that lead to emergence of resistance in vivo. Collectively, this will determine whether antibacterial resistance studied in vitro has the same mechanisms as resistant strains identified in vivo and if the multidrug therapeutic strategies tested in vitro also apply in vivo. In addition, the genetic mutations that appear in the resistant populations are being examined with Next Generation Sequencing. This will establish the pattern of mutations that appear at different levels of selective pressure and also determine if the pattern differs in bacteria treated with combinations of antibiotics. The results of these studies will be used to inform the design of a clinical trial to the test ability of the in vitro- and in vivo-identified treatment regimens to suppress the emergence of antibiotic resistance in the clinical setting.
Figure 8.

DARS is using hollow fiber culture systems to investigate the factors critical to antibiotic resistance emergence and evaluate treatment strategies to suppress emergent resistance. This in vitro system allows culture of pathogenic bacteria and exposure of those bacteria to single and combination antibiotic treatments to discover the critical elements for evolution of resistance and how to best treat infections to suppress emergent resistance. Results will inform in vivo assessment of antibiotic resistance in mouse models and clinical trials.
Precision Medicine, Genomics, and Induced Pluripotent Stem Cells
The Precision Medicine Initiative67,68 seeks to move away from a one-size-fits-all approach for medical therapies and instead take into account specific characteristics of individual patients. The OCP’s Genomics and Targeted Therapy Group reviews all genomics data submitted in New Drug Applications or Biologics Licensing Applications and at times consults DARS to evaluate complex in vitro assays included in applicant submissions. A recent example is the expanded indication for the drug ivacaftor (Kalydeco) to treat additional mutations of cystic fibrosis. Because hundreds of rare mutations exist that can lead to cystic fibrosis, it is very difficult to enroll sufficient numbers of patients with each mutation in clinical trials. Previously ivacaftor was only approved for patients with one of 10 different mutations; however, FDA was able to expand approval to 33 total mutations based in part on an in vitro cell-based assay that expressed each mutation separately. As part of the review team, DARS carefully evaluated the raw data submitted by the applicant to be able to reproduce results and ensure data quality. These data are now included in the drug label.69 This is an example of the unique role DARS plays as active laboratory researchers and reviewers of complex laboratory assays to speed drug development.
DARS is also applying precision medicine to the prediction of drug-induced adverse events. As described earlier, drug-induced QT prolongation and torsade de pointes has resulted in multiple drugs being withdrawn from the market, and many drugs remain on the market with a rare risk of torsade de pointes. In order to investigate the contribution of common genetic variants to drug-induced QT prolongation and torsade de pointes risk, 61 common genetic variants previously associated with the QT interval were combined to generate a weighted risk score.70 The genetic risk score was then shown to explain 30% of the variability in individual subject QT prolongation in an FDA-sponsored clinical trial, and was found to be associated with drug-induced torsade de pointes in a case-control genome-wide association study. If confirmed in real-world collections of drug-exposed patients, this type of genetic risk score (updated as new variants are discovered) could potentially be used to personalize risk-benefit assessment of therapy for individual patients.
As an extension to this work, induced pluripotent stem cell-derived cardiomyocytes from the clinical trial subjects are being evaluated in laboratory assays to assess whether they can predict individual patient responses in the clinic. This “clinical trial in a dish” study is ongoing.71 Work has also been initiated to study induced pluripotent stem cell-derived hepatocytes for use in toxicity assessment and metabolism studies. These cell types also have the potential to be used in microphysiological systems. DARS staff has participated as FDA advisors to the microphysiological systems-funded programs of the National Center for Advancing Translational Sciences and the Defense Advanced Research Projects Agency and is expanding laboratory work into this area to determine how this technology can be used in the regulatory process.
Conclusion
New tools, standards, and approaches are needed to improve the scientific assessment of the safety, efficacy, quality, and performance of drugs. There is a unique role for FDA scientists to perform applied research to move new science into the drug regulatory review process and perform laboratory and computational research to address emergent regulatory needs such as assessing the mechanism of new drug safety signals. Better biomarkers and predictive models (in vitro, in vivo, and in silico) have the potential to identify safety concerns earlier in drug development and predict therapeutic effectiveness to decrease the burden of clinical trials, such as for complex generic drugs and biosimilars. The rapidly evolving fields of precision medicine and genomics require expert scientists in the Agency to determine how to bring targeted therapies to market when large clinical trials cannot be performed (eg, in vitro assays for patient selection for rare genetic diseases) and utilize genomic biomarkers (eg, microRNAs), human cell assays (eg, induced pluripotent stem cell assays), and humanized animal models to predict adverse events not detectable in traditional animal models. The Division of Applied Regulatory Science’s multidisciplinary and translational research staff continues to advance all these areas, working collaboratively with colleagues across CDER, FDA, other government agencies, industry, and academia.
Footnotes
Declaration of Conflicting Interests: No potential conflicts were declared.
Funding: No financial support of the research, authorship, and/or publication of this article was declared.
References
- 1. US Food and Drug Administration. Statement of FDA mission. https://www.fda.gov/downloads/aboutfda/reportsmanualsforms/reports/budgetreports/ucm298331.pdf. Accessed May 15, 2017.
- 2. US Food and Drug Administration. Science & research: advancing regulatory science. https://www.fda.gov/scienceresearch/specialtopics/regulatoryscience/. Accessed May 15, 2017.
- 3. US Food and Drug Administration. Science & research: strategic plan for regulatory science. https://www.fda.gov/ScienceResearch/SpecialTopics/RegulatoryScience/ucm267719.htm. Accessed May 15, 2017.
- 4. Identifying CDER’s science and research needs report, July 2011. The CDER Science Prioritization and Review Committee (SPaRC). https://www.fda.gov/downloads/Drugs/ScienceResearch/UCM264594.pdf. Accessed May 15, 2017.
- 5. Assessing CDER’s Drug Safety-Related Regulatory Science Needs and Identifying Priorities, March 2015. The CDER Safety Research Interest Group (SRIG). https://www.fda.gov/downloads/Drugs/ScienceResearch/UCM438138.pdf. Accessed May 15, 2017.
- 6. Fisher AC, Lee SL, Harris DP, et al. Advancing pharmaceutical quality: an overview of science and research in U.S. FDA’s Office of Pharmaceutical Quality. Int J Pharm. 2016;515:390–402. [DOI] [PubMed] [Google Scholar]
- 7. US Food and Drug Administration. Biomarker qualification program. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/default.htm. Accessed May 25, 2017.
- 8. Zhang L, Zhang J, Shea K, et al. Autophagy in pancreatic acinar cells in caerulein treated mice: immunolocalization of related proteins and their potential as markers of pancreatitis. Toxicol Pathol. 2014;42:435–457. [DOI] [PubMed] [Google Scholar]
- 9. Zhang J, Rouse R. Histopathology and pathogenesis of caerulein-, duct ligation-, and arginine-induced acute pancreatitis in Sprague-Dawley rats and C57BL6 mice. Histol Histopathol. 2014;29:1135–1152. [DOI] [PubMed] [Google Scholar]
- 10. Rouse R, Xu L, Stewart S, et al. Extended exenatide administration exacerbates pancreatic injury in mice on a high fat, high carbohydrate diet. PLoS One. 2014;9:e109477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goodwin D, Rosenzweig B, Thompson K, et al. Evaluation of miR-216a and miR-217 as potential biomarkers of acute pancreatic injury in rats and mice. Biomarkers. 2014;25:1–13. [DOI] [PubMed] [Google Scholar]
- 12. Rouse R, Rosenzweig B, Shea K, et al. MicroRNA biomarkers of pancreatic injury in a canine model. Exp Toxicol Pathol. 2017;69:33–43. [DOI] [PubMed] [Google Scholar]
- 13. Rouse R, Zhang J, Stewart S, Rosenzweig B, Espandiari P, Sadrieh N. Comparative profile of commercially available urinary biomarkers in preclinical drug-induced kidney injury and recovery in rats. Kidney Int. 2011;79:1186–1197. [DOI] [PubMed] [Google Scholar]
- 14. Rouse R, Zhang J, Stewart S, Thompson K. Kidney injury biomarkers in hypertensive, diabetic, and nephropathy rat models treated with contrast media. Toxicol Pathol. 2012;41:662–680. [DOI] [PubMed] [Google Scholar]
- 15. Rouse R, Min M, Francke-Carroll S, et al. Impact of pathologists, blind evaluations, and sampling methods on performance assessment of the kidney injury biomarker, Kim-1. Toxicol Pathol. 2015;43:662–674. [DOI] [PubMed] [Google Scholar]
- 16. Shea K, Stewart S, Rouse R. Comparison of histopathology, digital image analysis, and stereology for early detection of experimental cisplatin-induced kidney injury in rats. Toxicol Pathol. 2014;42:1004–1015. [DOI] [PubMed] [Google Scholar]
- 17. Rouse R. Regulatory forum opinion piece: blinding and binning in histopathology methods in the biomarker qualification process. Toxicol Pathol. 2015;43:757–759. [DOI] [PubMed] [Google Scholar]
- 18. Thompson K, Haskins K, Rosenzweig B, et al. Comparison of the diagnostic accuracy of bis (monoacylglycerol) phosphate and other urinary phospholipids for drug-induced phospholipidosis or tissue injury in the rat. Int J Toxicol. 2012;31:14–24. [DOI] [PubMed] [Google Scholar]
- 19. Chockalingam AK, Hamed S, Goodwin DG, et al. The effect of oseltamivir on the disease progression of lethal influenza a virus infection: plasma cytokine and miRNA responses in a mouse model. Dis Markers. 2016;2016:9296457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mikaelian I, Cameron M, Dalmas DA, et al. Nonclinical safety biomarkers of drug-induced vascular injury: current status and blueprint for the future. Toxicol Pathol. 2014;42:635–657. [DOI] [PubMed] [Google Scholar]
- 21. Frazier KS, Engelhardt JA, Fant P, et al. Scientific and Regulatory Policy Committee Points-to-consider Paper*: Drug-induced vascular injury associated with non-small molecule therapeutics in preclinical development, Part I: Biotherapeutics. Toxicol Pathol. 2015;43:915–934. [DOI] [PubMed] [Google Scholar]
- 22. Engelhardt JA, Fant P, Guionaud S, et al. Scientific and Regulatory Policy Committee Points-to-consider Paper*: Drug-induced vascular injury associated with non-small molecule therapeutics in preclinical development, Part 2: Antisense oligonucleotides. Toxicol Pathol. 2015;43:935–944. [DOI] [PubMed] [Google Scholar]
- 23. Theocharides APA, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato-lymphoid system mice. Haematologica. 2016;101:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crawford LB, Tempel R, Streblow DN, et al. Human cytomegalovirus induces cellular and humoral virus-specific immune responses in humanized BLT mice. Sci Rep. 2017;7:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dickie AP, Wilson CE, Schreiter K, et al. The pharmacokinetics and metabolism of lumiracoxib in chimeric humanized and murinized FRG mice. Biochem Pharmacol. 2017;135:139–150. [DOI] [PubMed] [Google Scholar]
- 26. International Conference on Harmonisation. ICH Topic S7B: the nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. 2005. [PubMed]
- 27. International Conference on Harmonisation. Guideline for Industry E14: Clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. 2005. [PubMed]
- 28. Stockbridge N, Morganroth J, Shah RR, Garnett C. Dealing with global safety issues: was the response to QT-liability of non-cardiac drugs well coordinated? Drug Saf. 2013;36:167–182. [DOI] [PubMed] [Google Scholar]
- 29. Gintant G, Sager P, Stockbridge N. Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov. 2016;15:457–471. [DOI] [PubMed] [Google Scholar]
- 30. Colatsky T, Fermini B, Gintant G, et al. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—update on progress. J Pharmacol Toxicol Methods. 2016;81:15–20. [DOI] [PubMed] [Google Scholar]
- 31. Fermini B, Hancox JC, Abi-Gerges N, et al. A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J Biomol Screen. 2016;21:1–11. [DOI] [PubMed] [Google Scholar]
- 32. Vicente J, Stockbridge N, Strauss DG. Evolving regulatory paradigm for proarrhythmic risk assessment for new drugs. J Electrocardiol. 2016;49:837–842. [DOI] [PubMed] [Google Scholar]
- 33. Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J. 2014;167:292–300. [DOI] [PubMed] [Google Scholar]
- 34. Crumb WJ Jr, Vicente J, Johannesen L, Strauss DG. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J Pharmacol Toxicol Methods. 2016;81:251–262. [DOI] [PubMed] [Google Scholar]
- 35. Li Z, Dutta S, Sheng J, et al. Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether-à-go-go-related gene) channel–drug binding kinetics and multichannel pharmacology. Circ Arrhythm Electrophysiol. 2017;10:e004628. [DOI] [PubMed] [Google Scholar]
- 36. Windley MJ, Abi-Gerges N, Fermini B, Hancox JC, Vandenberg JI, Hill AP. Measuring kinetics and potency of hERG block for CiPA [published online ahead of print February 10, 2017]. J Pharmacol Toxicol Methods. doi: 10.1016/j.vascn.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 37. Li Z, Dutta S, Sheng J, Tran PN, Wu W, Colatsky T. A temperature-dependent in silico model of the human ether-à-go-go-related (hERG) gene channel. J Pharmacol Toxicol Methods. 2016;81:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blinova K, Stohlman J, Vicente J, et al. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol Sci. 2017;155:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johannesen L, Vicente J, Gray RA, et al. Improving the assessment of heart toxicity for all new drugs through translational regulatory science. Clin Pharmacol Ther. 2014;95:501–508. [DOI] [PubMed] [Google Scholar]
- 41. Johannesen L, Vicente J, Mason JW, et al. Differentiating drug-induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin Pharmacol Ther. 2014;96:549–558. [DOI] [PubMed] [Google Scholar]
- 42. Johannesen L, Vicente J, Mason JW, et al. Late sodium current block for drug-induced long QT syndrome: results from a prospective clinical trial. Clin Pharmacol Ther. 2016;99:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vicente J, Johannesen L, Mason JW, et al. Comprehensive T wave morphology assessment in a randomized clinical study of dofetilide, quinidine, ranolazine, and verapamil. J Am Heart Assoc. 2015;4:e001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vicente J, Johannesen L, Hosseini M, et al. Electrocardiographic biomarkers for detection of drug-induced late sodium current block. PLoS One. 2016;11:e0163619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johannesen L, Vicente J, Hosseini M, Strauss DG. Automated algorithm for J-Tpeak and Tpeak-Tend assessment of drug-induced proarrhythmia risk. PLoS One. 2016;11:e0166925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. US Food and Drug Administration. March 15, 2017: Pharmaceutical Science and Clinical Pharmacology Advisory Committee meeting announcement. https://www.fda.gov/AdvisoryCommittees/Calendar/ucm535513.htm. Accessed May 15, 2017.
- 47. Kruhlak NL, Benz RD, Zhou H, Colatsky TJ. (Q)SAR modeling and safety assessment in regulatory review. Clin Pharmacol Ther. 2012;91:529–534. [DOI] [PubMed] [Google Scholar]
- 48. Stavitskaya L, Aubrecht J, Kruhlak NL. Chemical structure-based and toxicogenomic models In: Jacobson-Kram D, Graziano M, eds. Genotoxicity and Carcinogenicity Testing of Pharmaceuticals. New York: Springer;2015:13–34. [Google Scholar]
- 49. International Conference on Harmonisation. M7: Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk (2014). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M7/M7_Step_4.pdf. Accessed May 15, 2017.
- 50. Amberg A, Beilke L, Bercu J, et al. Principles and procedures for implementation of ICH M7 recommended (Q)SAR analyses. Regul Toxicol Pharmacol. 2016;77:13–24. [DOI] [PubMed] [Google Scholar]
- 51. Barber C, Amberg A, Custer L, et al. Establishing best practice in the application of expert review of mutagenicity under ICH M7. Regul Toxicol Pharmacol. 2015;73:367–377. [DOI] [PubMed] [Google Scholar]
- 52. Xu R, Wang Q. Automatic signal extraction, prioritizing and filtering approaches in detecting post-marketing cardiovascular events associated with targeted cancer drugs from the FDA Adverse Event Reporting System (FAERS). J Biomed Inform. 2014;47:171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cheng F, Li W, Wang X, Zhou Y, Wu Z, Tang Y. Adverse drug events: database construction and in silico prediction. J Chem Inf Model. 2013;53:744–752. [DOI] [PubMed] [Google Scholar]
- 54. Wang Z, Clark NR, Ma’ayan A. Drug-induced adverse events prediction with the LINCS L1000 data. Bioinformatics. 2016;32:2338–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abernethy DR, Bai JP, Burkhart K, Xie HG, Zhichkin P. Integration of diverse data sources for prediction of adverse drug events. Clin Pharmacol Ther. 2011;90:645–646. [DOI] [PubMed] [Google Scholar]
- 56. Burkhart KK, Abernethy D, Jackson D. Data mining FAERS to analyze molecular targets of drugs highly associated with Stevens-Johnson syndrome. J Med Toxicol. 2015;11:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moore PW, Burkhart KK, Jackson D. Drugs highly associated with infusion reactions reported using two different data-mining methodologies. J Blood Disord Transf. 2014;5:2. [Google Scholar]
- 58. Hur J, Zhao C, Bai J. Systems pharmacological analysis of drugs inducing Stevens Johnson Syndrome and toxic epidermal necrolysis. Chem Res Toxicol. 2015;28:927–934. [DOI] [PubMed] [Google Scholar]
- 59. Melas I, Sakellaropoulos T, Iorio F, et al. Identification of drug-specific pathways based on gene expression data: application to drug induced lung injury. Integr Biol. 2015;7:904–920. [DOI] [PubMed] [Google Scholar]
- 60. Chien H-C, Yang Y-H, Bai J. Trastuzumab-related cardiotoxic effects in Taiwanese women: a nationwide cohort study. JAMA Oncol. 2016;2:1317–1325. [DOI] [PubMed] [Google Scholar]
- 61. Brouwer KR, Ferguson SS, Lai Y, et al. The importance of in vitro liver models: experts discuss whole-cell systems, transporter function, and the best models for future in vitro testing. Appl In Vitro Toxicol. 2016;2:1–7. [Google Scholar]
- 62. Volpe DA. Transporter assays as useful in vitro tools in drug discovery and development. Expert Opin Drug Discov. 2016;11:91–103. [DOI] [PubMed] [Google Scholar]
- 63. Hartman N, Zhou H, Mao J, et al. Characterization of the methemoglobin forming metabolites of benzocaine and lidocaine. Xenobiotica. 2017;47:431–438. [DOI] [PubMed] [Google Scholar]
- 64. Christl LA, Woodcock J, Kozlowski S. Biosimilars: the US regulatory framework. Annu Rev Med. 2017;68:243–254. [DOI] [PubMed] [Google Scholar]
- 65. Leupke KH, Suda KJ, Boucher H, et al. Past, present, and future of antibacterial economics: increasing bacterial resistance, limited antibiotic pipeline, and societal implications. Pharmacotherapy. 2017;37:71–84. [DOI] [PubMed] [Google Scholar]
- 66. Marston HD, Dixon DM, Knisely JM, Palmore TN, Fauci AS. Antimicrobial resistance. JAMA. 2016;316:1193–1204. [DOI] [PubMed] [Google Scholar]
- 67. The Precision Medicine Initiative. https://obamawhitehouse.archives.gov/node/333101. Accessed May 25, 2017.
- 68. The Precision Medicine Initiative: What is it? https://syndication.nih.gov/multimedia/pmi/infographics/pmi-infographic.pdf. Accessed May 25, 2017.
- 69. Kalydeco FDA Drug Label for NDA 203188 (Action Date 05/01/2017). https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203188s019lbl.pdf. Accessed July 8, 2017.
- 70. Strauss DG, Vicente J, Johannesen L, et al. Common genetic variants predict drug-induced QT prolongation and torsade de pointes risk: a pilot study. Circulation. 2017;135:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Strauss DG, Blinova K. Clinical trials in a dish. Trends Pharmacol Sci. 2017;38:4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]