

Abstract
It is suggested that craniosynostosis is caused by a heterogeneous set of effects including gene mutations, teratogenic exposure during critical periods of development, and gene/environment interactions. Distinguishing between sufficient, additive, and interactive effects is important to the study of gene/environment interactions and allows for segregation of environmental exposures effecting susceptible populations. Through the identification of sufficient and interactive effects efforts in prevention of craniosynostosis may be successful. Here we provide a brief review focusing on defining these categorized exposures and relevant literature that has interrogated gene-environment interactions for craniosynostosis.
I. Craniosynostosis
The cranial suture is defined as the fibrous joint between the bony plates of the skull that allows for neuro-expansion during development. Functionally, the suture may dampen biomechanical stress upon the calvarial bones once neuro-cranial expansion is complete. Beginning about the 2nd decade of life bony fusion of the cranial sutures often begin. When the cranial suture undergoes bony infiltration (synostosis) prior to the completion of brain growth, deformation of the cranium and associated anomalies termed craniosynostosis can occur. Craniosynostosis occurs in 1 in every 1800–2500 live births (1) and affects males versus females at a ratio of 2:1. Often the co-morbidities associated with craniosynostosis or identification of aberrant growth trajectory allow for diagnosis. Many of the co-morbidities associated with craniosynostosis, including ocular proptosis and increased intracranial pressure, pose a threat to normal neurological development. Neurosurgery is often indicated in cases of craniosynostosis allowing for the “release” of the suture and hopeful reversion towards normal growth trajectories. Surgical intervention varies due to the heterogeneous nature of craniosynostosis but can include strip suturectomies, posterior expansion, fronto-orbital advancements, or partial and complete calvariectomies (1–3).
The fused suture drives alterations in morphological development associated with craniosynostosis. Normally the calvarium expands perpendicular to the patent sutures (Virchow’s law), however when premature fusion occurs, compensatory growth occurs in dimensions not restricted by fusion. The most common suture involved in clinical cases of craniosynostosis is the sagittal which, depending on timing of fusion, can lead to a scaphocephalic (elongated in the antero-posterior dimension) phenotype (1,4). The coronal is the second most commonly involved suture which when bilaterally affected can drive brachycranic (widening) of the cranium, or anterior plagiocephaly (asymmetry) when unilaterally compromised. Premature fusion of the metopic and lambdoid sutures are more rare accounting for only 15–20% of clinical cases.
Craniosynostosis is associated with more than 180 syndromes many of which present with limb abnormalities in addition to restricted cranial growth. The most commonly identified syndromes include those resulting from mutations in FGFr and TWIST genes. Syndromic craniosynostosis can be associated with multiple suture (e.g., Apert syndrome, Pfeiffer syndrome, Crouzon syndrome, Antley-Bixler syndrome), or single suture fusion (e.g., Saethre-Chotzen syndrome, Muenke syndrome) (1,3). However, greater than 85% of all craniosynostosis cases are classified as isolated non-syndromic occurrences where no genetic information is identified. From a clinical perspective, although non-syndromic cases are often less severe, clinical protocols are similar. Neurosurgical intervention is still indicated in cases of non-syndromic craniosynostosis; only strategies for genetic counseling differ.
In addition to the genetic factors associated with craniosynostosis, there have been several environmental exposures (e.g. teratogens) associated with craniosynostosis identified from case and surveillance studies. These exposures include maternal thyroid disorders (5,6), cigarette smoking (7–9), alcohol use (4,10), and maternal use of anti-depression drugs (11,12) although the identification of these exposures as causative is not conclusive. Overall it seems likely that these teratogens and others are sufficient to cause craniosynostosis. However, there are also likely genetic polymorphisms that exist allowing for a more complicated interpretation of the causation of craniosynostosis. Genetic factors (e.g., FGFr mutations), environmental factors (teratogen exposures), and gene/environment interactions, a concept we will develop below, have all been identified as causative for craniosynostosis.
II. Defining Gene/Environment Interactions
The literature can be unclear on how exactly to define a gene/environment effect, but is consistent in defining the necessity of polymorphisms (a variant occurring in greater than 1% of the population) or mutations to allow for appropriate genetic variability to be acted upon by an external factor (13–16). It is this variability that dictates where one genetic status responds differently to a stimulus (Figure 1). This concept should be distinguished from environmental only effects where a teratogen is sufficient to cause an anomaly. Segregation may also occur due to exposure versus non-exposure, e.g. hypervitaminosis A causing cleft lip/palate (16–18). Gene/environment interactions should also be distinguished from additive effects where a condition likely caused by a mutation is exacerbated phenotypically by an exposure. Thus, although gene mutations associated with disease states are amenable to gene/environment interactions it is likely that many more genetic polymorphisms are at work.
Figure 1. Theoretical Model of Craniosynostosis Causastion.
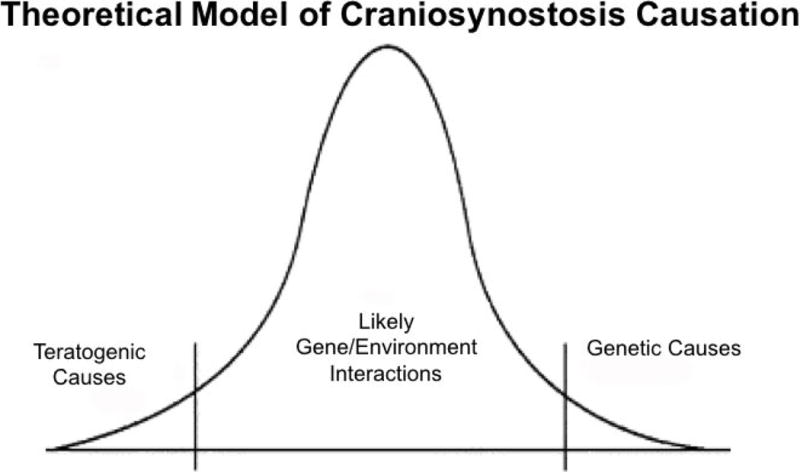
Gaussian curve represents cases of craniosynostosis of which the majority are likely caused by gene/environment interactions.
There are some well-defined gene/environment interactions for craniofacial development with respect to anomalies. This is not a surprise as research has long indicated environmental contribution or developmental plasticity of the craniofacial skeleton. For example, biomechanics and environment including temperature and diet effect final craniofacial form (19–23). One clear example of gene/environment effects is genetic variability in TGFα acted upon by cigarette smoking in causation of cleft lip/palate. Data suggests use of cigarettes acts upon variant TGFα alleles, which encode for a molecule important for proliferation and differentiation within tissues during both primary and secondary palate formation. Alteration of this homeostatic relationship can result in cleft lip/palate (8,16–18).
There are likely many more interactions between genetic polymorphisms and environmental exposures than may be detected. Craniosynostosis is heterogeneous in presentation, including age of presentation and severity (optimal age of repair is <1 year), and thus it is not as amenable to identification of gene/environment interaction as cleft lip/palate of which a majority of cases are identified at or before birth. The disjointed timeline between potential environmental or teratogenic exposure and identification of craniosynostosis may introduce further error (i.e. maternal/paternal self-report) to elucidation of these relationships. Craniosynostosis is also more rare than orofacial clefting leading to difficulty in large scale analyses such as genome wide associations (GWAS), discussed below.
Identification of gene/environment interaction relies on molecular epidemiology of DNA sequencing including GWAS, identification of candidate single nucleotide polymorphisms (SNPs), and linkage disequilibrium. A trait can be studied using GWAS, where common allele variants are interrogated for segregation of disease or phenotype, either through investigation of patients with or without disease, or phenotypic variants within disease states. Deletions, insertions, and single nucleotide polymorphisms are all amenable to analysis which is most powerful when a trait is normally distributed. This is to be contrasted with linkage studies which are amenable to heterogeneous genetic presentation and analysis of different alleles as well as allele states (13,14).
Linkage disequilibrium, non-random association of alleles at different loci, suggests an association between variation found at a genetic locus and phenotypic expression of a trait, usually within the context of a disease state. As linkage disequilibrium relies on patterns of inheritance it is susceptible to the selected populations of study as well as rate of mutation and genetic drift within that population. Overall, SNPs found in coding regions are particularly susceptible to segregating differences, which results in different levels of susceptibility of disease and occurrence with environmental challenge (13,14). Cohort studies and genetic database sets are the most powerful tools to enhance identification of susceptible alleles, but case control studies are also used. Environmental variables that segregate by geographical location or condition are more easily recognized due to identification of cohort and proper controls. It is through these studies utilizing molecular epidemiology that better diagnosis and prevention of diseases, including craniosynostosis, can occur.
III. Examples from Human Condition
The United States Center for Disease Control Birth Defects Prevention Study has suggested that a focus of research and dissemination on craniosynostosis should be gene/environment interactions (24). Despite this call there are few examples in the clinical literature focusing on this topic. On example is a report on a familial case of FGFr3 mutation at Pro250Arg (Muenke syndrome), which revealed what was suggested to be a gene/environment interaction. The described environmental factor was maternal diabetes which appears to have exacerbated the paternally inherited craniosynostosis disorder. In this case, the child presented with laterality disorder and hepatoblastoma, conditions not previously associated with mutations at this locus. There was however, no interactive effects with respect to the craniosynostostic phenotype, which was described as presenting as typical Muenke syndrome. These associated effects were most likely additive, not interactive as the phenotype was severe likely due to a two-hit phenomenon. The gene mutation was sufficient to cause craniosynostosis and the hepatoblastoma was associated with maternal diabetes exposure (25). There is another recent report focusing on environmental variability of fetal constraint and a novel mutation in FGFr2 (Ala315Ser). It is interesting that this locus is on a gene well known to cause craniosynostosis and the patient presented as Crouzon-like. This locus has however, not previously been identified in human craniosynostosis cases or syndromes and thus may represent a gene/environment interaction, in this case fetal constraint being the environmental variable (breach) (26).
Research has indicated several genetic loci amenable to further study identified by GWAS that were not associated with traditional FGFr or Twist mutations. (14). For example, a study of non-syndromic cases of sagittal craniosynostosis found several susceptible loci including one down stream of BMP2, a gene which encodes for a potent bone development protein, and one within the gene BBS9. As neither of these genes has a known segregating population within craniosynostotic patients or is associated with a syndrome, these candidates may be amenable to gene/environment interactions. Research should continue to move forward to distinguish between additive versus gene/environment effects particularly for single suture isolated non-syndromic craniosynostosis cases.
Research has also focused on utilizing mechanistic targets of effect (mRNA expression) to identify likely gene markers or pathway in lieu of pure genetic markers that segregate in craniosynostosis cases. For example, calvarial osteoblast cells were cultured from single suture craniosynostosis cases and results identified SFRP4, FGF7, and VCAM1 as having segregating expression levels associated with craniosynostosis. These results are suggestive of FGF/IGF WNT signaling importance in non-syndromic craniosynostosis (27). These and associated genes (upstream) are likely candidates for gene polymorphism associated with disease. The identification of these genes also suggests that although the genes associated with syndromic craniosynostosis are less likely candidates for gene/environment interactions, the associated molecular pathways are rational targets for study.
IV. Examples of Modeling
Many preclinical models of human craniosynostosis syndromes exist; most of which are in vivo (transgenic) and in vitro (derived from transgenic or ex vivo transfected cells) murine models. Additional models of study concerning cranial suture biology and craniosynostosis may represent non-syndromic (although clearly genetic) craniosynostosis (e.g. lagomorph model) (1–3,28,29) or challenges to the normal developing mammalian skull by effecting genes and proteins important for suture patency, and finally the study of normal ontogenetic fusion (e.g. the murine posterior interfrontal suture) (30–34). It is difficult to model gene/environment interactions pre-clinically as there are exceedingly abundant possibilities for environmental variables and teratogens to act on polymorphisms that allow for susceptibility to craniosynostosis. Teratogenic studies alone are informative, but do not address interactive effects. Further, the addition of a teratogens within a genetic model of craniosynostosis have several drawbacks including the strong possibility of no effect as craniosynostosis is already predicted to occur (i.e. Twist 1 +/− mouse) (35). However, these studies in both the wild-type mammalian skull and genetic models of craniosynostosis may provide sufficient information on molecular pathways of effect (downstream of the gene) specific to an exposure. Further, as data becomes available from GWAS or linkage studies these preclinical investigations can proceed with greater likelihood of success.
V. Future Directions of Research
It is likely that within the context of gene/environment interaction there are thresholds levels at which teratogens act upon unknown genetic polymorphisms (Figure 2). Thus, testing these interactions either by amelioration of the contribution of that gene or loci alteration will likely prove to be quite an expensive and frustrating venture. If segregating populations are suggested from GWAS or linkage studies, then these may be the most useful directions for future analyses because of their ability to link genetic variants to a disease state. Overall, teratogenic studies are much easier from an implementation perspective, both for clinical identification and preclinical modeling. If identified teratogens are established to alter growth and development in a wild type pre-clinical model, those data may prove useful for loci specific testing via cellular transfection studies in vitro or in vivo modeling to inform the clinical practice of counseling, identification, and prevention.
Figure 2. Theoretical Model of Threshold Effects.
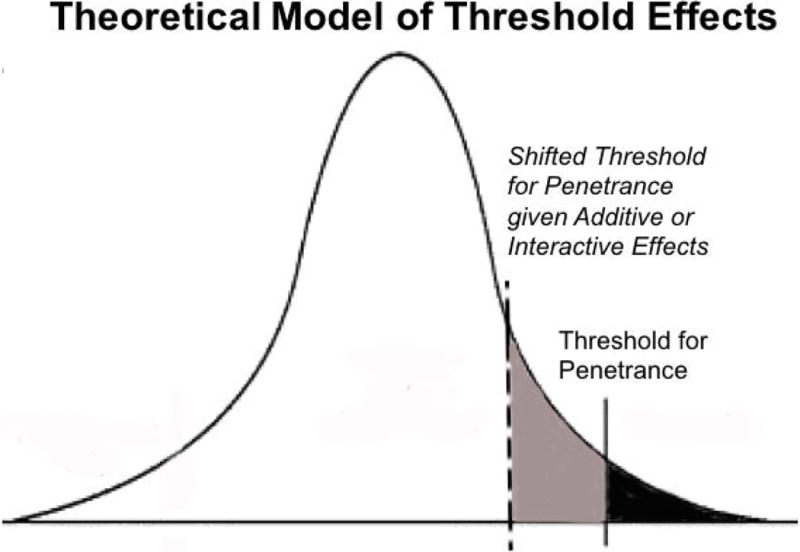
Gaussian curve highlighting a shifted threshold for penetrance of craniosynostosis in the general population due to additive or interactive effects of genes and environmental factors.
References
- 1.Johnson D, Wilkie AO. Craniosynostosis. European journal of human genetics : EJHG. 2011;19:369–76. doi: 10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grova M, Lo DD, Montoro D, Hyun JS, Chung MT, Wan DC, et al. Models of cranial suture biology. J Craniofac Surg. 2012;23:1954–8. doi: 10.1097/SCS.0b013e318258ba53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levi B, Wan DC, Wong VW, Nelson E, Hyun J, Longaker MT. Cranial suture biology: from pathways to patient care. J Craniofac Surg. 2012;23:13–9. doi: 10.1097/SCS.0b013e318240c6c0. [DOI] [PubMed] [Google Scholar]
- 4.Zeiger JS, Beaty TH, Hetmanski JB, Wang H, Scott AF, Kasch L, et al. Genetic and environmental risk factors for sagittal craniosynostosis. J Craniofac Surg. 2002;13:602–6. doi: 10.1097/00001665-200209000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Carmichael SL, Ma C, Rasmussen SA, Cunningham ML, Browne ML, Dosiou C, et al. Craniosynostosis and risk factors related to thyroid dysfunction. Am J Med Genet A. 2015;167A:701–7. doi: 10.1002/ajmg.a.36953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen SA, Yazdy MM, Carmichael SL, Jamieson DJ, Canfield MA, Honein MA. Maternal thyroid disease as a risk factor for craniosynostosis. Obstet Gynecol. 2007;110:369–77. doi: 10.1097/01.AOG.0000270157.88896.76. [DOI] [PubMed] [Google Scholar]
- 7.Carmichael SL, Ma C, Rasmussen SA, Honein MA, Lammer EJ, Shaw GM, et al. Craniosynostosis and maternal smoking. Birth Defects Res A Clin Mol Teratol. 2008;82:78–85. doi: 10.1002/bdra.20426. [DOI] [PubMed] [Google Scholar]
- 8.Hackshaw A, Rodeck C, Boniface S. Maternal smoking in pregnancy and birth defects: a systematic review based on 173 687 malformed cases and 11.7 million controls. Hum Reprod Update. 2011;17:589–604. doi: 10.1093/humupd/dmr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoyt AT, Canfield MA, Romitti PA, Botto LD, Anderka MT, Krikov SV, et al. Associations between Maternal Periconceptional Exposure to Secondhand Tobacco Smoke and Major Birth Defects. Am J Obstet Gynecol. 2016 doi: 10.1016/j.ajog.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 10.Richardson S, Browne ML, Rasmussen SA, Druschel CM, Sun L, Jabs EW, et al. Associations between periconceptional alcohol consumption and craniosynostosis, omphalocele, and gastroschisis. Birth Defects Res A Clin Mol Teratol. 2011;91:623–30. doi: 10.1002/bdra.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM, National Birth Defects Prevention S Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med. 2007;356:2684–92. doi: 10.1056/NEJMoa066584. [DOI] [PubMed] [Google Scholar]
- 12.Reefhuis J, Devine O, Friedman JM, Louik C, Honein MA, National Birth Defects Prevention S Specific SSRIs and birth defects: Bayesian analysis to interpret new data in the context of previous reports. BMJ. 2015;351:h3190. doi: 10.1136/bmj.h3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cummings AM, Kavlock RJ. Gene-environment interactions: a review of effects on reproduction and development. Crit Rev Toxicol. 2004;34:461–85. doi: 10.1080/10408440490519786. [DOI] [PubMed] [Google Scholar]
- 14.Justice CM, Yagnik G, Kim Y, Peter I, Jabs EW, Erazo M, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet. 2012;44:1360–4. doi: 10.1038/ng.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Willing MC, Marazita ML, Wendell S, Warren JJ, Broffitt B, et al. Genetic and environmental factors associated with dental caries in children: the Iowa Fluoride Study. Caries Res. 2012;46:177–84. doi: 10.1159/000337282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu T, Schwender H, Ruczinski I, Murray JC, Marazita ML, Munger RG, et al. Evidence of gene-environment interaction for two genes on chromosome 4 and environmental tobacco smoke in controlling the risk of nonsyndromic cleft palate. PLoS One. 2014;9:e88088. doi: 10.1371/journal.pone.0088088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–56. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- 18.Vieira AR. Genetic and environmental factors in human cleft lip and palate. Front Oral Biol. 2012;16:19–31. doi: 10.1159/000337521. [DOI] [PubMed] [Google Scholar]
- 19.Eng CM, Lieberman DE, Zink KD, Peters MA. Bite force and occlusal stress production in hominin evolution. Am J Phys Anthropol. 2013;151:544–57. doi: 10.1002/ajpa.22296. [DOI] [PubMed] [Google Scholar]
- 20.Paschetta C, de Azevedo S, Castillo L, Martinez-Abadias N, Hernandez M, Lieberman DE, et al. The influence of masticatory loading on craniofacial morphology: A test case across technological transitions in the Ohio valley. Am J Phys Anthropol. 2010;141:297–314. doi: 10.1002/ajpa.21151. [DOI] [PubMed] [Google Scholar]
- 21.Cray J, Jr, Mooney MP, Siegel MI. Cranial suture biology of the Aleutian Island inhabitants. Anat Rec (Hoboken) 2011;294:676–82. doi: 10.1002/ar.21345. [DOI] [PubMed] [Google Scholar]
- 22.Little BB, Buschang PH, Pena Reyes ME, Tan SK, Malina RM. Craniofacial dimensions in children in rural Oaxaca, southern Mexico: secular change, 1968–2000. Am J Phys Anthropol. 2006;131:127–36. doi: 10.1002/ajpa.20406. [DOI] [PubMed] [Google Scholar]
- 23.Richardson ER. Racial differences in dimensional traits of the human face. Angle Orthod. 1980;50:301–11. doi: 10.1043/0003-3219(1980)050<0301:RDIDTO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 24.Rasmussen SA, Yazdy MM, Frias JL, Honein MA. Priorities for public health research on craniosynostosis: summary and recommendations from a Centers for Disease Control and Prevention-sponsored meeting. Am J Med Genet A. 2008;146A:149–58. doi: 10.1002/ajmg.a.32106. [DOI] [PubMed] [Google Scholar]
- 25.Baynam GS, Goldblatt J. A child with an FGFR3 mutation, a laterality disorder and an hepatoblastoma: novel associations and possible gene-environment interactions. Twin Res Hum Genet. 2010;13:297–300. doi: 10.1375/twin.13.4.297. [DOI] [PubMed] [Google Scholar]
- 26.Johnson D, Wall SA, Mann S, Wilkie AO. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? European journal of human genetics : EJHG. 2000;8:571–7. doi: 10.1038/sj.ejhg.5200499. [DOI] [PubMed] [Google Scholar]
- 27.Stamper BD, Park SS, Beyer RP, Bammler TK, Farin FM, Mecham B, et al. Differential expression of extracellular matrix-mediated pathways in single-suture craniosynostosis. PLoS One. 2011;6:e26557. doi: 10.1371/journal.pone.0026557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert JR, Cray JJ, Jr, Kreithen A, Marazita ML, Cooper GM, Losee JE, et al. Genetic Homozygosity and Phenotypic Variability in Craniosynostotic Rabbits. Cleft Palate Craniofac J. 2016 doi: 10.1597/15-226. [DOI] [PubMed] [Google Scholar]
- 29.Holmes G. The role of vertebrate models in understanding craniosynostosis. Childs Nerv Syst. 2012;28:1471–81. doi: 10.1007/s00381-012-1844-3. [DOI] [PubMed] [Google Scholar]
- 30.Cray JJ, Jr, Weinberg SM, Parsons TE, Howie RN, Elsalanty M, Yu JC. Selective serotonin reuptake inhibitor exposure alters osteoblast gene expression and craniofacial development in mice. Birth Defects Res A Clin Mol Teratol. 2014;100:912–23. doi: 10.1002/bdra.23323. [DOI] [PubMed] [Google Scholar]
- 31.Durham E, Jen S, Wang L, Nasworthy J, Elsalanty M, Weinberg S, et al. Effects of Citalopram on Sutural and Calvarial Cell Processes. PLoS One. 2015;10:e0139719. doi: 10.1371/journal.pone.0139719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James AW, Levi B, Xu Y, Carre AL, Longaker MT. Retinoic acid enhances osteogenesis in cranial suture-derived mesenchymal cells: potential mechanisms of retinoid-induced craniosynostosis. Plast Reconstr Surg. 2010;125:1352–61. doi: 10.1097/PRS.0b013e3181d62980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Jian Y, Cai B, Wang M, Chen M, Huang H. All-Trans Retinoic Acid-Induced Craniofacial Malformation Model: A Prenatal and Postnatal Morphological Analysis. Cleft Palate Craniofac J. 2016 doi: 10.1597/15-271. [DOI] [PubMed] [Google Scholar]
- 34.Cray JJ, Jr, Durham EL, Smalley MA, Finegold DN, Siegel MI, Losee JE, et al. The effects of testosterone on craniosynostotic calvarial cells: a test of the gene/environmental model of craniofacial anomalies. Orthod Craniofac Res. 2011;14:149–55. doi: 10.1111/j.1601-6343.2011.01520.x. [DOI] [PubMed] [Google Scholar]
- 35.Durham EL, Howie RN, Black L, Bennfors G, Parsons TE, Elsalanty M, et al. Effects of thyroxine exposure on the Twist 1 +/− phenotype: A test of gene-environment interaction modeling for craniosynostosis. Birth Defects Res A Clin Mol Teratol. 2016 doi: 10.1002/bdra.23543. [DOI] [PMC free article] [PubMed] [Google Scholar]