

Abstract
Covalent modification of therapeutic compounds is a clinically proven strategy to devise prodrugs with enhanced treatment efficacies. This prodrug strategy relies on modified drugs that possess advantageous pharmacokinetic properties and administration routes over their parent drug. Self-assembling prodrugs represent an emerging class of therapeutic agents capable of spontaneously associating into well-defined supramolecular nanostructures in aqueous solutions. The self-assembly of prodrugs expands the functional space of conventional prodrug design, providing a possible pathway to more effective therapies as the assembled nanostructure possesses distinct physicochemical properties that can be tailored to specific administration routes and disease treatment. In this review, we will discuss the various types of self-assembling prodrugs in development, providing an overview of the methods used to control their structure and function and, ultimately, our perspective on their current and future potential.
Graphical Abstract
1. Introduction
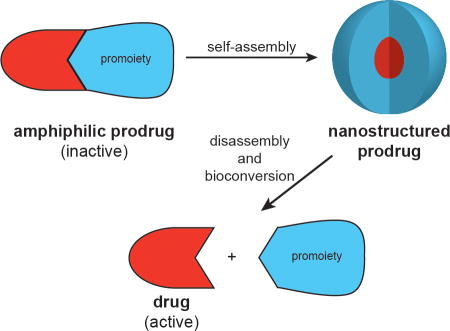
The successful delivery of a drug to its site(s) of action is a challenging prospect in light of the many physicochemical, biopharmaceutical, and pharmacokinetic barriers it may face. The use of prodrugs can address these issues by altering the absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties of the parent drug. The term ‘prodrug’, introduced by Albert in 1958,1 refers to the bioreversible modification of a drug into a masked, inactive form with more desirable physicochemical properties (Figure 1a). Subsequent in vivo bioconversion is then required to generate the active drug and exert a pharmacological effect.
Fig. 1.

Illustration of (a) conventional prodrugs and (b) self-assembling prodrugs.
The design of prodrugs requires consideration of the following: (1) can the drug be modified, (2) what effect will the modification have on the ADMET, and (3) can the parent drug be regenerated efficiently without producing toxic by-products?2 Chemical modification of a drug requires it to possess a suitable functional group that allows reaction with the selected promoiety, the element that bestows the desired physicochemical changes. Suitable groups include hydroxyls, amines, carboxylic acids, thiols, and carbonyls.
The purpose of the promoiety generally dictates its chemical identity. For example, increased membrane permeability of a hydrophilic drug can be accomplished through lipidation,3 typically via ester bond formation. In doing so, the prodrug offers improved bioavailability through enhanced absorption from the gastrointestinal (GI) tract into systemic circulation or via topical application. Other routes to improve ADMET properties include the addition of ionisable groups to increase solubility,4 the masking of metabolically labile groups to prevent their premature breakdown,5 the conjugation of peptidic epitopes for the active targeting of specific cell surface receptors,6–8 and the site-sensitive activation of the prodrug for selective release.9, 10
Effectively releasing the parent drug is perhaps the most critical step as failure to do so would nullify the strategy. Furthermore, the by-products should possess no significant bioactivity of their own and undergo safe metabolic degradation and excretion. The key features in the design of a suitable promoiety, therefore, are its linkage to the drug and the mechanism by which that link is cleaved. Simple esters can be both passively and actively hydrolysed by water and esterases,11 respectively, whereas amide linkages require the action of a specific peptidase/protease. Some promoiety designs can introduce greater site-selectivity by being responsive to a particular stimulus that is more prevalent at the target site(s) compared with non-targeted areas, thus reducing the occurrence of off-target effects. Examples of this would include acid-sensitive and reduction-sensitive disulphide linkers. To achieve this enhanced selectivity, however, often requires a more involved degradation mechanism, relying on self-immolative spacers that connect the drug to the stimuli-responsive element. Subsequent exposure to the desired stimulus triggers a linear or cyclic12 elimination pathway that releases the active drug.
The conversion of drugs into prodrugs is an effective strategy, with an estimated 10% of all small molecule pharmaceuticals13 and 15 of the top 100 blockbuster drugs14 being prodrugs. However, while they may have improved ADMET properties over their parent drug, they are still subject to the same disadvantages of rapid renal clearance and hepatic metabolism associated with their small molecule nature. As with unmodified drugs, the formulation of prodrugs can provide an effective means to further improve their ADMET properties. Much work has been performed to address this through the use of drug carriers (e.g. polymers,15, 16 liposomes,17 and dendrimers18) that can shield the cargo drug and transport it to the disease site. This approach is particularly prevalent in the field of cancer therapy, arguably taking advantage of the enhanced permeation and retention (EPR) effect to passively target a tumour’s leaky vasculature.19, 20 An increasingly important variation on this has been the development of nano-sized delivery vehicles formed from suitably derivitised drugs that can self-aggregate into well-defined nanostructures—so-called self-assembling prodrugs (Figure 1b).
The key characteristic of self-assembling prodrugs (SAPDs) is that they are amphiphilic in nature, possessing both hydrophilic and hydrophobic domains that enable aqueous assembly—either spontaneously or via kinetic trapping (e.g. nanoprecipitation). The drug itself typically constitutes one of these two domains, most commonly the hydrophobic, with the promoiety forming the other. Consequently, the drug also performs a structural role in addition to eliciting a therapeutic effect. By self-assembling into well-defined nanostructures, the resultant assemblies have a distinct, often improved, pharmacokinetic profile and may possess unique properties in tuning drug release rates and addressing multidrug resistance. First, the relatively larger size of the nanostructures can reduce rapid renal clearance, extending the circulation time and allowing for increased accumulation at the disease site. Second, sequestration of the drug within the nanostructure can protect it from the degradative process such as hydrolysis and metabolism. Third, in addition to breakdown of the promoiety to release the drug, disassembly of the nanostructure could provide an additional means to tune drug release rate. Last, further decoration and tuning of the nanostructure’s surface and physicochemical properties can lead to improved tumour targeting, cellular uptake, and intracellular trafficking that further enhance the drug’s activity compared to its free form.
Self-assembling prodrugs as a whole represent a new paradigm in drug delivery as the modified drugs essentially become single-component medicines that are self-delivering and self-formulating. In the case of the former, assembly of the SAPD into a nanostructure creates its own delivery vehicle; one that can be decorated with epitopes for targeting and that can be selectively degraded through considered use of cleavable linkages. For the latter, the nanostructure provides protection for the drug during storage and administration, fulfilling the role that excipients perform in a conversional drug formulation process.
Two broad classes of SAPDs exist, defined by their molecular nature—macromolecular and small molecule. Macromolecular SAPDs are polymeric systems formed by the conjugation of multiple drugs to a polymer of synthetic or biological origin. In this review, we will make a distinction between synthetically or biologically derived polymer–conjugates as each can lend their own unique characteristics to the resulting assemblies. These two conjugate types, polymer–drug conjugates and biopolymer–drug conjugates, respectively, both undergo microphase separation to form micellar structures that are frequently spherical in nature.
Small molecule SAPDs, on the other hand, comprise a discrete number of drug molecules (typically between one and four) connected to a single promoiety to create a homogeneous conjugate (monodisperse in molecular weight). These can then self-assemble into a range of well-defined nanostructures, with the molecular design influencing the morphology adopted. Like macromolecular SAPs, small molecule SAPDs can be sub-divided into two groups—drug conjugates and drug amphiphiles. Drug conjugates undergo aggregation in response to rapid aqueous dilution (kinetic trapping) or a chemical modification such as removal of a solubilizing group. Drug amphiphiles can spontaneously self-assemble by simple dissolution in water or physiological fluids.
Herein, we will discuss the development of the four self-assembling prodrug types, provide an overview of the guiding principles in the molecular design of each prodrug group, and offer our perspective on the future development of the field. A particular focus will be on examples in which the drug plays an active structural role in the assembly process.
2. Macromolecular SAPDs
2.1 Polymer–Drug Conjugates
2.1.1 Historical development and basic design considerations
Self-assembling polymer–drug conjugates (PDCs) emerged from the development of their non-assembling counterparts. The covalent attachment of a drug to a hydrophilic polymer provided a means to overcome the physicochemical and pharmacokinetic challenges associated with small molecule therapeutics.21 Through this conjugation, the drug’s pharmacokinetic profile becomes that of the polymer (with a relatively large radius of gyration), thereby improving the solubility, preventing rapid renal clearance, and altering the tissue distribution characteristics. For significantly hydrophobic drugs, however, the attachment of large numbers to a polymer often led to unwanted aggregation, or even precipitation.22, 23 Rather than viewing this as a negative, Kataoka and co-workers saw the potential opportunity to utilise the observed self-aggregation behaviour to create micelle-forming PDCs.24
In their early reports,24 the Kataoka group described the direct conjugation of multiple doxorubicin (DOX) molecules to a poly(ethylene glycol)-poly(aspartic acid) block copolymer, giving PEG-b-P(Asp(DOX)) (Figure 2). When dispersed in aqueous solution, these conjugates associated into spherical micelles with average diameters of 50 nm. The hydrophobic P(Asp(DOX)) block was presumed to comprise the micellar core, with the PEG block forming the outer shell. This arrangement was corroborated by an observed lack of interaction with serum albumins—known binders of drug molecules that can significantly influence their biodistribution and therapeutic effectiveness—indicating the drug is shielded within the core. Follow-up studies improved the synthesis,25 characterised the pharmacokinetics,26, 27 and assessed the in vivo effectiveness28 of this micellar platform.
Fig. 2.

Kataoka’s initial design of a self-assembling polymer–conjugate, PEG-b-P(Asp(DOX)).
This pioneering approach by Kataoka provides instructive insight into the various parameters to be considered when designing macromolecular SAPDs. A common issue with PDCs is the incomplete drug conjugation with the available reactive sites that leads to polydispersity in the drug loading. For self-assembling PDCs, the extent of drug coverage can be critical when the drug constitutes the hydrophobic component of the system and is thus able to impact the micellar stability. In PEG-b-P(Asp(DOX)), the initially reported coverage was 33% of the P(Asp) carboxylates24. Given that unreacted Asp carboxylates are negatively charged, a lower drug coverage would result in greater electrostatic repulsions. The consequence of this became apparent in a later study that showed a coverage of 40–50% was necessary for stable micelle formation.25 Thus, the efficiency of the drug loading is a crucial factor, especially since any unstable micelles may disassemble during circulation, giving individual polymer chains with significantly different pharmacokinetic profiles.26
Given the nature of PEG-b-P(Asp(DOX)) as a prodrug, the release of free DOX is also an important consideration. In this system, DOX is conjugated to the polymer through an amide bond that would require enzymatic action to cleave and release the drug. Accordingly, the conjugate required a much higher dose to achieve the same effect as free DOX, though this dosage was tolerated better than the lower free drug dose. Subsequent rigorous characterisation of PEG-b-P(Asp(DOX)), however, indicated that it’s in vivo activity derived from physically entrapped DOX, rather than the chemically conjugated drug.29 Since this discovery, PEG-b-P(Asp(DOX)) has been investigated as a carrier for DOX instead of as a prodrug,30, 31 undergoing clinical trials as NK911.32, 33 Later work introduced a new derivative in which DOX was conjugated via an acid labile hydrazone linker,34, 35 exhibiting high in vivo effectiveness and thus realizing the initial vision. These findings illustrate not only the importance of incorporating a drug release mechanism, but also the need to remove any unconjugated drug so that the true properties of the designed system can be accurately determined.
2.1.2 Growth of the field
Following Kataoka’s inspiring work, further expansion in the development of self-assembling PDCs was initially slow but has since grown to be an area of considerable interest. Much of this may be due to the increasing attention that nanotechnological approaches to biomedical challenges have received over time. Early work focused on the use of similar copolymers with a range of anticancer drug molecules. For instance, the Kataoka group incorporated cisplatin (CDDP) into PEG-b-P(Asp) through coordination bonds between the Asp caarboxylates and Pt.36–39 This conjugate formed micelles with average diameters of 20 nm in aqueous media,37 with the reduced size compared to PEG-b-P(Asp(DOX)) a consequence of the manner in which the smaller CDDP molecules bridge the Asp residues to produce a denser micelle. A later study showed that the micelle size can be controlled in a linear fashion through the P(Asp) feed ratio, allowing unimodal micellar diameters from 20 to 100 nm.38 CDDP release is based upon ligand exchange with free chloride ions, thus providing a decay mechanism in saline-rich physiological media. Despite exhibiting reduced nephrotoxicity, the low stability of the micelles led to increased accumulation within the liver and spleen that gave comparable toxicity to free CDDP.39 Replacement of the P(Asp) block with poly(glutamic acid) (P(Glu)) resulted in significant stabilization of the micelles, accompanied by an increase in the average diameter to ~30 nm (Figure 3).40 These conjugates, designated as NC-6004, sustainably released CDDP over 150 h—compared with 30 h for the P(Asp) analogue37—displayed reduced toxicity (both nephro-40, 41 and neurotoxicity41), and exerted a high in vivo activity due to increased drug accumulation within tumours. Moreover, NC-6004 was also found to reduce the occurrence of sentinel lymph node metastasis in an oral squamous cell carcinoma model.42 Clinical trials have followed these initial successes,43, 44 including a Phase III trial for the treatment of pancreatic cancer in combination with gemcitabine (ID# NCT02043288).
Fig. 3.

Examples of self-assembling PDCs using PEG-b-P(Glu) as the polymer backbone.
An analogue of NC-6004 in which cisplatin was replaced with dichloro(1,2-diaminocyclohexane)platinum(II) (DCHPt) (Figure 3), which is clinically administered as the oxalate complex oxaliplatin, has also been reported and is denoted as NC-4016.45 Incorporation of this more hydrophobic Pt drug in PEG-b-P(Glu) resulted in larger micelles of 40 nm that exhibited a 15 h induction period prior to the sustainable release of free drug. This delay may be due to the more hydrophobic interior of the micelle resisting the entry of water, initially preventing chloride ions from effecting ligand exchange to release the Pt drug. The greater hydrophobicity also led to enhanced micellar stability when compared to NC-6004 (240 vs 50 h) and to extended plasma and tumour retention times. In vivo studies showed no indication of any neurological damage, a common side effect of oxaliplatin and other Pt drugs. This observation was attributed to the absence of the oxalate ligand (known to bind Ca2+ and Mg2+) and the slow-release kinetics from the micelles.46
In another example, Matsumura and coworkers reported a variant that conjugated the topoisomerase I inhibitor SN-38 to the PEG-b-P(Glu) copolymer (Figure 3).47 SN-38 is the active metabolite of the clinically-relevant prodrug irinotecan (CPT-11), and the rationale for its conjugation was to potentially increase the potency by removing the need for carboxylesterase-induced activation. The conjugate, named NK012, contained ~20% (w/w) SN-38 and formed micelles with average diameters of 20 nm. Exemplifying its role as a prodrug, NK012 could release up to 74% of the conjugated SN-38 through simple ester hydrolysis, a significant improvement on the 2–8% metabolic conversion from CPT-11.48, 49 NK012 exhibited substantially greater activity than CPT-11 against hypervascularized tumours. Thus, tumours secreting higher levels of vascoendothelial growth factor (VEGF) were more sensitive to NK012 due to VEGF-induced hypervascularization and hyperpermeability that creates a stronger EPR effect. Furthermore, a later study suggested NK012 may have utility against malignant glioma, with the conjugate displaying improved accumulation, distribution, and retention within orthotopic human glioma xenografts compared to CPT-11.50 A significant extension in survival rates was subsequently observed, with over 60% of NK012-treated mice alive up to 40 d, compared with 25 and 15 d for the CPT-11 and control groups, respectively. NK012 is currently undergoing clinical evaluation, having completed Phase I trials that showed greatly reduced side-effects compared to the free drug.51
Paclitaxel (PTX), a very hydrophobic drug that has entered clinical use in both soluble polymer (PTX poliglumex, Xyotax®)52 and nanoparticle (Abraxane®)53 formulations, was conjugated to an amphiphilic triblock copolymer by Jing and coworkers.54 This PDC, based on a PEG block bookended by two blocks of PLGG (poly[(lactic acid)-co-P(glycolic acid)-alt-P(glutamic acid)]), assembled into nanoparticles ~119 nm in diameter (by DLS) with a PTX content of 16.5% (w/w).
It is clear from the above examples that the early work in this field was performed by a limited number of research groups, often working in collaboration with one another. However, the number of researchers exploring this area and developing new approaches has greatly expanded over the last six years as the field of nanomedicine gathered momentum. For instance, Zhong,55 Zhang,56 and Xie57 took advantage of improvements in polymer synthesis to construct their respective self-assembling PDCs. Both the Zhong and Zhang groups utilized reversible addition-fragmentation chain-transfer (RAFT) polymerization to produce polymer backbones with low polydispersity and controlled molecular weight. In Zhong’s example,55 a vinyl-containing moiety was grafted to a PEG-b-PAA (poly(acrylic acid)) polymer, followed by the Michael-type addition of PTX through its 2’-hydroxyl group (Figure 4a). Zhang, on the other hand, synthesized a hydrazide-bearing monomer forming the polymer MPEG-MBAH6 (methacrylamide Boc-hydrazide).56 Removal of the Boc protecting group then enabled DOX incorporation through hydrazone formation (Figure 4b). Xie and co-workers used anionic ring opening polymerization (ROP) to create a vinyl-functionalized polymer, PEG-b-PAGE (poly(allyl glycidyl ether)), which was then converted to a carboxylate-bearing polymer for the attachment of PTX (Figure 4c).57 These examples all led to polymers with polydispersity indices (PDI) close to unity.
Fig. 4.

Examples of self-assembling PDCs synthesized using alternative polymer backbones.
Conjugation to single component polymers, rather than block copolymers has also been investigated. For instance, Liu et al. conjugated docetaxel (DTX) to a medium molecular weight PEG (Mw > 2 kDa),58 generating spherical micelles ~46 nm in diameter. This conjugate was conceived as a delivery method for free DTX through encapsulation within the hydrophobic core, aided by PEG-DTX–DTX interactions. Fan et al. reported the synthesis of a P(BAsp-CPT) PDC, in which CPT was conjugated to the carboxyl groups of α,β-poly[(N-carboxybutyl)-l-aspartamide].59 The drug content was controlled through the CPT/P(BAsp) feed ratio, enabling a maximum CPT content of ~48% (w/w). An increase in CPT content was also seen to influence the micellar diameter, with a higher content leading to larger particles. To precisely control drug loading, one strategy to synthesize polymer-drug conjugates is to use hydrophobic drugs as initiators for the polymerization.60–66 Cheng and co-workers initially reported drug-initiated polymerization using a number of anticancer drugs such as paclitaxel and doxorubicin.61–63
2.1.3 Conjugation of hydrophilic drugs
The exploration of self-assembling PDCs has primarily focused upon the conjugation of hydrophobic drugs, a consequence of their high activity but poor solubility. While hydrophilic drugs generally do not suffer from solubility issues, they are subject to premature degradation and not immune to pharmacokinetic barriers that reduce their effectiveness. Nucleoside-based drugs in particular would benefit from strategies to increase their cellular uptake, prevent unwanted metabolic inactivation, and reduce their rapid clearance, especially given their first-line use against many cancer types. To accommodate hydrophilic drugs within the self-assembly strategy necessitates the use of additional hydrophobic elements to provide the required impetus for assembly. Gemcitabine (GEM), a first-line treatment for pancreatic cancer, is a nucleoside analogue often explored as a model for the self-assembly of hydrophobically-modified hydrophilic drugs. Mahato and co-workers, for example, conjugated Gem and dodecanol (C12H25OH) to a PEG-b-PCC (poly(2-methyl-2-carboxyl-propylene carbonate) polymer through amide and ester linkages, respectively (Figure 5a).67 This conjugate formed spherical micelles averaging 24 nm in diameter and with a drug loading of almost 13%. In vivo studies indicated a significant increase in effectiveness over free Gem. In another example, Kong and colleagues used RAFT polymerisation to grow a poly(methyl methacrylate) (PMMA) polymer from a trithiocarbonate-functionalized Gem (Figure 5b).68 While this PDC contains only one drug molecule per polymer chain, the use of a drug-based RAFT agent means that the drug content can be fine-tuned through control over the polymer length. For instance, conjugates with PMMA molecular weights of 500 and 1120 Da gave Gem contents of 43.7 and 21.5 % (w/w), respectively. Both conjugates could assemble into spherical nanoparticles with approximate diameters of 123 and 136 nm, respectively An initial in vivo investigation revealed that the Gem conjugates reduced tumour growth by 68% with little toxicity in an A549 cell derived xenograft murine model, whereas free Gem had no effect but significant toxicity.
Fig. 5.

Examples of hydrophilic drug containing PDCs.
In both Mahato’s and Kong’s examples, aggregation of the conjugate is induced through nanoprecipitation from an organic solution of the PDC (see Section 3.1.1 for more information on this technique), rather than direct self-assembly in water, suggesting limited aqueous solubility in the non-assembled state. Vingradov and co-workers synthesized a Gem-containing PDC that could undergo spontaneous aqueous self-assembly, utilizing polyvinyl alcohol (31 kDa) as the backbone with grafted cholesterol to provide hydrophobicity (Figure 6c).69 Furthermore, Gem is conjugated through a short polyphosphate linker, the negative charge of which provides greater solubility. Reversible protection of the exocyclic amine of Gem (as the isobutyric amide) also provides greater resistance to enzymatic deamination that forms the inactive deoxyuridine derivative. This conjugate, CPVC-p4GEMC, forms a nanogel in aqueous solution, sonication of which reveals small nanoparticles with hydrodynamic diameters of 30–44 nm. Vinogradov’s approach was initially demonstrated for the nucleoside drug, floxuridine.70
2.1.4 Mechanisms for improving drug release
The mechanism of releasing the bioactive, therapeutic moiety is an essential part in prodrug design. As previously noted, Kataoka’s initial demonstration of the self-assembling PDC strategy relied on enzymatic hydrolysis of the Dox–Asp amide linkage, a particularly ineffective method as was later discovered. Incorporation of an effective means to generate the free drug is, therefore, an essential requirement. Fortunately, a broad range of stimuli-specific cleavable linkers have been developed for use in drug delivery vehicles, with their applicability dependent on the available drug and polymer functionalities. Examples include pH-sensitive, enzyme-sensitive (e.g. protease, esterase, phosphatase), and redox-sensitive linkers.
Acid-sensitive linkers are often an effective method for releasing drugs within the intracellular environment as the lyosomal and endosomal compartments maintain a lower pH than the extracellular and cytosolic environments. This difference offers a means to selectively release the drug within the target cell rather than in circulation, though it offers no selectivity between healthy and diseased cells. An oral delivery route of administration is also ruled out due to the low pH of gastric acid. The most common examples of an acid-sensitive linker are the imine and hydrazone functionalities, used to connect a carbonyl group (aldehyde or ketone) to an amine or hydrazide, respectively. Hydrazones, in particular, are the most commonly used due to their greater intrinsic stability over imines.71 As previously mentioned, Kataoka used a hydrazone linkage to impart high activity to PEG-b-P(Asp(Hyd-Dox)),32, 33 and it was also used by Matsumura to create the epirubicin (Epi) analogue, PEG-b-P(Asp(Hyd-Epi)) (NC-6300) (Figure 6a).72 Both hydrazone conjugates exhibit excellent stability at pH 7 and demonstrate effective release of their respective anthracycline drugs in the pH 5–6 environment of the lysosome upon endocytotic entry. Interestingly, Howard et al. noted that their hydrazone-linked dexamethasone (DEX) PDC was stable at both pH 7.4 and 5.0,73 failing to appreciably release DEX under acidic conditions. They attributed this to the nature of the formed hydrazone bond, which would be in resonance with nearby π-bonds that would stabilize it to hydrolysis. By separating the hydrazone linkage from the drug via a short ester moiety, the acid-sensitivity was restored.
Fig. 6.

Examples of stimuli-sensitive linkers and their release mechanisms. (a) Acid-sensitive hydrazone linkers are cleaved under the acidic conditions found in the lysosomal and endosomal cellular compartments. (b) Reduction-sensitive disulphide linkers are cleaved in the presence of intracellular reductants such as glutathione (GSH) or cysteine.
Another acid sensitive linker is the acetal group (e.g. RCH(OR’)2), which can be hydrolyzed to release the OR’ groups. An example of this can be found in Zhong’s PEG-b-PAA-based PTX prodrug (Figure 4a),55 the various formulations of which sustainably released greater than 80% of the conjugated PTX over a 50 h period at pH 5.0, compared with under 35% at pH 7.4.
Acid-sensitivity can also be built into the polymer backbone. For instance, self-assembling PDCs with phosphoester backbones have been reported in which the acid-sensitive P–O bond can provide a potential means for breakdown and increased access to the drug–polymer linkage. Wooley and coworkers demonstrated this by synthesizing an alkyne-bearing PEG-PPE (poly(phosphoester)) polymer through organocatalyzed ROP, to which they attached an azide-functionalized PTX by copper-mediated azide-alkyne cycloaddition (CuAAC).74 These conjugates, PEO-b-(PPE-g-PTX), formed micellar nanoparticles with narrow size distributions (24 ± 7 nm by TEM) and a PTX loading capacity of 55% (w/w). However, while breakdown of the polymer occurred at pH 6, only ~5% of the conjugated PTX was released. This observation was attributed to increased aggregation and precipitation preventing access to C-2’ of the PTX-ester. Wooley suggested that the use of other sensitive linkers may lead to future generational improvements. Indeed, a DOX-containing PPE polymer synthesized by Sun et al. conjugated the drug through a hydrazone linkage displayed effective release of the drug under suitably acidic conditions.75 Whether the degradation of the PPE backbone under these conditions provided any additional benefit to drug release over a non-degradable polymer, however, was not explored.
Enzymes are the biology’s tools for specifically effecting molecular modifications to biomolecules and biopolymers. This functionality has been harnessed by the drug delivery community to liberate active drugs from their prodrug form, whether as small molecules or conjugated to delivery vehicles. Additionally, many diseases upregulate certain enzymes associated with disease progression and may also express them in locations they are not normally found (in the extracellular environment during tumour metastasis for example). As such, this abnormal expression can allow for a more selective targeting strategy.
The most popular enzymes utilized for the bioconversion of prodrugs are esterases and proteases, arising from the common strategy of lipidation through ester or amide formation, respectively. A prominent example that features heavily in polymer–drug conjugates is the use of proteases in the Cathepsin family, especially Cathepsin B (CatB). CatB is normally responsible for the lysosomal degradation of proteins but in tumours fulfils a variety of intra- and extra-cellular roles that aid disease progression. The GFLG (Gly-Phe-Leu-Gly) linker is a CatB substrate originally identified by Duncan, Kopecek, and coworkers, which as been used in numerous drug delivery and imaging systems.22, 76, 77 Gu and co-workers synthesized a triblock copolymer that incorporated GFLG as both a polymer backbone component and as a linker to conjugate DOX.78 The 90 kDa copolymer-DOX conjugate formed 100 nm micellar nanoparticles that upon treatment with CatB formed two smaller 44 kDa polymers, allowing for potential renal excretion once its purpose is served. In vivo studies showed a 62% increase in tumour growth inhibition over free DOX for a 4T1 murine breast cancer model.
The previously mentioned report of a Gem-containing PDC nanogel by Vinogradov69 (Figure 5c) relies upon alkaline phosphatase to generate the active tri-, di- and mono-phosphorylated forms of Gem (with concurrent hydrolysis of the amide protecting group). This release mechanism removes the need for further intracellular modification that free Gem requires. The intended application for Vinogradov’s nanogel is as an oral delivery vehicle and the conjugate exhibits negligible Gem release over a 12 h period in simulated gastric and intestinal fluids. Furthermore, this PDC demonstrated an ability to effectively cross a Caco-2 monolayer (an in vitro model of the GI barrier), a process aided by the PVA polymer.79 CPVC-p4GEMC was subsequently found to exhibit greater activity against both aggressive and drug-resistant cancer cell types in vitro and in vivo, with an up to three-fold extension in survival times.
Redox-sensitive mechanisms can provide an effective means to release a conjugated drug. Disulphide (S–S) bonds, in particular, are useful as they take advantage of the large difference in concentrations of glutathione (GSH) between the intra- and extracellular environments (mM vs. µM). This difference can be harnessed to furnish circulation stable vehicles that undergo rapid degradation upon internalization. As with pH cleavable linkages, the sensitivity of S–S is not necessarily selective for diseased tissues over healthy as GSH is an important cellular biomolecule, However, many cancer types exhibit elevated GSH levels as a means to counter the increased oxidative stress associated with cancer progression80 and a degree of selectivity may be possible. Accordingly, selectivity is primarily the responsibility of the carrier. Both the Chen81 and Zhang82 groups have created self-assembling PDCs that conjugate the drug via disulphide linkers. In Chen’s case,81 PTX is grafted to a PEG-b-PLL (poly-l-lysine) copolymer via a dithioproprionic acid linker. For Zhang’s,82 CPT is conjugated to a PEG-b-PMMA copolymer via an dithiolethylcarbonate linker (Figure 6b). Both conjugates exhibited greatly accelerated release in the presence of 10 mM GSH over aqueous hydrolysis alone. Additionally, Chen’s PTX conjugate also displayed pH-sensitivity, another property of their chosen linker.81 Adopting a different strategy, Zhu and co-workers incorporated the disulphide linkage into the polymer backbone of an ibuprofen (Ibu)-containing PEG-based copolymer.83 Assembly of this conjugate through nanoprecipitation gave 22 nm sized micelles that provided protection for the Ibu ester linkage at pH 7.4 (negligible release over 100 h). Treatment with 10 mM GSH, however, led to 35% IBU release within 30 h as breakdown of the micelle occurred to expose the conjugated drug.
A recent report by Matson and co-workers detailed the incorporation of a H2S-releasing prodrug onto a hydrophilic polymer to create a self-assembling PDC.84 H2S is a gasotransmitter that has shown anti-cancer activity, but its gaseous nature and high reactivity make delivery strategies crucial for its application. Conjugating S-aroylthiohydroxylamine (SATO), a H2S prodrug they developed,85 to PEG-b-P(FBEMA) (where FBEMA is 2-(4-formylbenzoyloxy)ethyl methacrylate) gave a PDC that could assemble into spherical micelles ~21 nm in diameter. Under reducing conditions, i.e. in the presence of GSH, the linked prodrug breaks down to release H2S. In vitro experiments indicated that the SATO-containing PDC possessed some selectivity towards cancer cells over healthy.
2.1.5 Fine-tuning of the shape
In the context of self-assembling PDCs, the shape here can refer to one of two properties: the architecture of the polymer itself (e.g. linear vs. branched) or the morphology of the assembled nanostructure. For the former, a change in polymer shape can alter how the drug is sequestered, leading to changes in protection and/or nanostructure dimensions. In the linear examples discussed thus far, the drug is more likely found at the centre of the micellar nanostructures and maximally protected from the external environment. Changing to a Y-shaped (branched) architecture, however, can result in relocation of the drug. For instance, Bensaid et al. conjugated cabazitaxel (CBZ), a member of the taxane family, at the junction between two PEG and PLA polymer chains (Figure 7a).86 In doing so, the CBZ position within the resulting micellar nanoparticles is at the interface between the hydrophilic PEG and hydrophobic PLA polymers. Comparison with the linear form, in which CBZ is conjugated at the PLA terminus, showed a more rapid drug release profile. This enhancement in the release rate was due to water having easier access to the CBZ–copolymer ester linkage, by penetrating only the hydrophilic PEG corona and not into the hydrophobic core. The drawback to this example, however, is that only a single CBZ molecule was conjugated, giving an 8% (w/w) drug loading.
Fig. 7.

Variations in the polymer architecture can affect the drug’s position and release characteristics. Panel (a) was adapted from reference [86] with permission from the American Chemical Society (ACS), copyright 2013. Panel (b) was adapted from reference [87] with permission from the Multidisciplinary Digital Publishing Institute (MDPI), copyright 2014. Panel (c) was adapted from reference [88] with permission from ACS, copyright, 2016.
A second example was reported by Sui et al., in which a branched DOX-based PDC was synthesized.87 Here, DOX was conjugated to a branched PEG-b-P(Glu)2 copolymer via hydrazone linkages (Figure 7b). Noted differences between the branched PDCs and their linear counterparts include smaller micellar diameters (166 vs 231 nm) and a more rapid drug release rate for the branched architecture. Both observations can be attributed to the shortened length of the hydrophobic drug-conjugated blocks that allow a more compact micelle to be formed. Furthermore, the branched orientation means that hydrophobic block occupies a greater volume near to the PEG block, reducing the density of PEG coverage in the assembled state. This more limited coverage facilitates easier access to the acid-labile hydrazine linkage that, when combined with less potential for polymer chain entanglement due to the shorter length, results in faster drug release. This manifests into improved cytotoxicity over their linear analogues.
Taking this concept further, Wei et al. demonstrated a stimuli-responsive hyper-branched HPMA–DOX conjugate in which each DOX-containing HPMA copolymer is covalently linked to other chains through enzyme-degradable GFLG peptide linkers (Figure 7c).88 The synthesized 165 kDa conjugates formed compact spherical micelles ~102 nm in diameter that, after treatment with papain, broke down to form 23 kDa polymers 8.6 nm in diameter. These smaller aggregates would allow subsequent renal clearance of the polymer. DOX was conjugated through hydrazone linkages for lyso- or endosomal release. A more complex example is provided by Liu and co-workers, who created a theranostic platform that can be used to simultaneously treat and image cancers. Their hyperbranched polymer drug conjugate contained disulphide-linked CPT for therapeutic applications and Gd-DOTA complexes for magnetic resonance (MR) imaging.89 Reductive cleavage of the disulphide not only releases CPT but turns on the MR imaging capability due to an almost 10-fold enhancement in the T1 relaxivity associated with the now hydrophilic Gd-containing polymer.
Nanostructure morphology is an important property of a self-assembled drug delivery. The shape of the nanostructure can have a significant influence on events such as circulation, excretion, and cellular uptake. The vast majority of self-assembled PDCs adopt spherical morphologies, in part due to the lack of directional intermolecular interactions that can influence the assembly process during hydrophobic collapse. Through variations in the processing conditions (co-solvent composition, polymer concentration, rate of water addition, and temperature),90 it is possible to influence the morphological outcome of copolymer assembly. Taking advantage of this approach, Liu and co-workers designed a camptothecin (CPT)-based vinyl monomer with a reducible disulphide linkage to create a PEG-b-PCPTM copolymer.91 This PDC, with a >50% (w/w) CPT content, could be induced to form a variety of morphologies, including spherical, disk-like, large compound vesicles (LCVs), and staggered lamellae (Figure 8). Subsequent studies showed that the morphology influenced the rate of cellular uptake through changes in the uptake mechanism. The most effective morphology was the staggered lamellae, which bypassed the endolysosomal compartment entirely to induce the greatest cytotoxic effect. Furthermore, this morphology also exhibited extended circulation times in vivo (t1/2 7.62 h) compared to the other morphologies, potentially due to some resemblance in shape to red blood cells.
Fig. 8.

Varying experimental parameters to influence assembly during nanoprecipitation of a PDC. Reproduced from reference [91] with permission from ACS, copyright 2013.
2.1.6 Approaches to combination therapy
Multi-drug treatments are often used as a first-line therapy for various cancers, the rationale being that two or more drugs with differing action mechanisms may induce a synergistic effect that can enhance treatment efficacy and/or prevent the emergence of multi-drug resistance (MDR). This combination therapy approach is of interest within the drug delivery community as the potential for simultaneous delivery of multiple drugs exists. By co-delivering the drugs in a nanocarrier, the pharmacokinetics are determined by the physicochemical properties of the nanostructure. Accordingly, it can be rather simpler to determine the relative accumulation of each drug at the tumour site without having to worry about the individual pharmacokinetics of the drugs (differing biodistribution, cellular uptake, and excretion properties for instance). In PDCs, there are three possible approaches to consider: (1) co-assembly of two PDCs with different conjugated drugs (Figure 9a), (2) conjugation of multiple drug types to a single polymer chain (Figure 9b), and (3) the encapsulation of a second drug within a PDC-based nanostructure (Figure 9c).
Fig. 9.

Strategies for combination therapy using self-assembling PDCs.
The first method is a seemingly simple approach but co-assembly is not guaranteed to lead to a homogeneous mixture as the drug–drug interactions may lead to preferential assembly of like with like. Even if a homogeneous mixture can be prepared, uncertainty in the ratio of the two PDCs among the micelles formed will still exist. An example of this method is the co-assembly of PEG-b-(Asp(Hyd-DOX)) and PEG-b-(Asp(Hyd-GBM)) by Bae et al.92 Here, GBM is a hydroxyl-functionalized derivative of geldanamycin. Mixed micelles were prepared either by direct mixing of the two micellar solutions (AMM) or co-mixing of the two PDCs in DMSO prior to assembly (OMM). In vitro experiments appeared to show that there was no statistical difference between AMM and OMM, suggesting that simple mixing of preformed micelles is a suitable strategy.
The second method of conjugating multiple drugs to a single polymer chain can be synthetically more challenging, but offers a measurable drug content and a more homogeneous system. Satchi-Fainaro and co-workers demonstrated this approach by conjugating both PTX and DOX to poly(glutamic acid) (PGA) in a synergistic ratio.93 The nano-sized conjugate exhibited superior activity and safety over the free drugs in mice bearing orthotopic mammary adenocarcinomas. An interesting example of this strategy is provided by Zhou et al.94 Rather than conjugating the two drugs—cisplatin (CIS) and demethylcantharidin (DMC)—to individual sites on the polymer chain, they took advantage of the redox activity of Pt to oxidise the four-coordinate Pt(II) to six-coordinate Pt(IV) and then complex it with DMC. This tandem drug complex, Z-DMC-CIS, was then conjugated to a PEG-b-P(LA-co-MCC/OH) copolymer via the DMC’s second carboxylate group, creating a PDC that assembled through nanoprecipitation into micelles 200–240 nm in diameter. DMC release was effected through hydrolysis of the ester bond, while CIS was generated through reduction of Pt(IV) to Pt(II) with concomitant cleavage of the axial Pt–O bond to DMC. Polymer-Z-DMC-CIS exhibited significantly higher in vitro toxicity than either of the two individual drug conjugates, with a combination index (CI) determination indicating a synergistic effect of the dual drug PDC (CI < 1). In vivo studies also indicated a similar difference in activity, with polymer-Z-DMC-CIS-treated H22 xenografts showing little growth over a 28 day period. This method was also used to deliver oxaliplatin and DMC.95
A variation on this strategy is the coupling of two drugs at the termini of a bifunctional medium molecular weight PEG polymer (>2 kDa). One notable example is the conjugation of PTX and alendronate (ALN) to NH2-PEG4500-CO2H, a collaborative study by Pasut and Satchi-Fairnaro that gave drug loadings of 4.7 and 11% (w/w) for PTX and ALN, respectively.96, 97 In this conjugate, PTX forms the hydrophobic segment and the bisphosphonate-based ALN the hydrophilic (due its negatively charge phosphate groups), such that the micelles formed display ALN at the surface. Moreover, ALN possesses a high affinity for hydroxyapatite that allows its use as a targeting ligand for bone metastases. In vivo experiments revealed that the ALN can indeed allow increased accumulation of the nanostructured dual drug PDC within bones where leaky vasculature is present, i.e. at metastatic sites.97 More recently, Ye et al. utilized this bone-targeting strategy to co-deliver DOX and ALN.98 Other examples of conjugating two drugs to a bifunctional PEG polymer include a Gem/MTX conjugate99 and an AKBA/MTX conjugate,100 where MTX is the anti-metabolite methotrexate and AKBA is the anti-angiogenic acetyl-11-keto-β-boswellic acid.
The last method, in which a second drug is encapsulated within a self-assembled PDC, is a relatively straight forward approach to incorporating the second drug, representing a logical extension of drug-loaded polymeric micelles that have been studied for decades.15, 16 However, the same disadvantages are shared, including reduced control over drug content and loading efficiency, and the potential for undesired release through passive diffusion. Jung et al. reported the synthesis of phytosphingosine (PHS)-based PDC that could encapsulate DOX and exhibit a synergistic effect against the HeLa cervical cancer cell line.101 In this report, PHS was conjugate to a modified poly(2-hydroxyethyl-l-aspartamide) (PHEA) polymer and, using a nanoprecipitation method (see Section 3.1.1), assembled in the presence of DOX to form DOX-loaded micelles. The PHS content was deliberately maintained below 10% (w/w) as a morphological transition from spherical to worm-like structures was seen. Since PHS constitutes the hydrophobic portion of the PDC and is grafted via an acid-sensitive carbamate linker, the lower pH of the endosomal environment facilitates breakdown of the micelle to release both PHS and DOX. Furthermore, a folate ligand was utilized to allow targeting of the folate receptor and enhance cellular uptake. Other examples include the encapsulation of DTX in a DOX-based HPMA copolymer by Jager et al.102 and the loading of PTX into a floxuiridine-based PVA polymer by Senanayake et al.103 This encapsulation strategy has also been adopted for mono-drug therapies to increase the overall drug content of the PDC,30, 57, 58, 104, 105 whereby the free form of the conjugated drug is loaded within the PDC micelle during nanostructure formation.
2.2 Biopolymer–Drug Conjugates
2.2.1 Polypeptide–drug conjugates
Nature has created a wide variety of polymeric entities to fulfil numerous important biological roles, including structural components (cellulose, chitin, collagen, elastin), energy storage (starch, glycogen), and genetic information storage and replication (DNA, RNA). These biopolymers are composed of various monomeric species that include amino acids, monosaccharides, and nucleic acids. Of these, amino acids have found perhaps the greatest utility in drug delivery, with polymeric chains of single charged amino acids the most common, e.g. P(Asp), P(Glu) and P(Lys). However, these polymers have no natural counterpart or inspiration and are purely synthetically-derived. Natural polypeptides, on the other hand, can possess functions beyond providing solubility and a means by which to avoid rapid clearance. For instance, elastin is a highly elastic protein that is an important component of connective tissues, helping to confer shape recoverability to tissues under stress. This property arises from highly hydrophobic domains containing repetitive sequences of five amino acids, Val-Gly-Pro-X-Gly in tropoelastin for instance (where X is a variable amino acid). Polypeptides based on these elastin-derived sequences, termed elastin-like polypeptides (ELPs),106, 107 exhibit interesting thermoresponsive behaviour associated with a change in peptide conformation. At ambient temperatures, ELPs adopt a random conformation that maximises the solubility. At higher temperatures, however, a shift to a β-spiral conformation occurs that reduces the solubility and induces the aggregation of multiple chains. This reversible inverse temperature transition (ITT) has been utilized for protein purifications and is now finding use in drug delivery.
The Chilkoti group has pioneered the use of self-assembling recombinant ELP–drug conjugates for cancer therapy, entities they term chimeric peptides (CPs). Building on earlier work into DOX-based CPs, Chilkoti raised the number of DOX molecules conjugated to a VPGVG-based ELP from one110 to eight,108 giving CP-DOX (Figure 10a). The increased hydrophobicity this conferred allowed spontaneous aqueous self-assembly into nanoparticles ~40 nm in diameter. This hydrophobicity was found to be important for the conjugate’s self-assembly propensity, with a later study implicating the conjugated entity’s octanol–water distribution coefficient, logD, as a critical determinant.111 In CP-DOX, DOX was conjugated through acid-labile hydrazone linkers, thus allowing effective drug release. Both high in vitro and in vivo activity was observed for CP-DOX. Moreover, a single injection of the conjugate could abolish tumours in a murine model for C26 colon cancer. Eight of nine mice treated with CP-DOX showed a curative effect after 66 days compared to the control and DOX-treated groups, which displayed survival times of 21 and 27 days, respectively. Hu et al. extended the approach by attaching a tumour-homing F3 peptide ligand to the periphery of CP-DOX.112 By targeting the conjugate directly to tumours, an apparent improvement in anti-tumour activity and reduction in side-effects was seen.
Fig. 10.

Biopolymer–drug conjugates. (a) Chilkoti’s DOX-containing polypeptide–drug conjugate based on recombinant elastin-like peptides.108 (b) A Cellax-based carbohydrate–drug conjugate synthesized by Ernsting et al.109
More recently, Chilkoti has shown how harnessing of CP-DOX’s thermoresponsive nature allows for increased intratumoral accumulation of the conjugate through thermal cycling.113, 114 Externally raising the temperature of the tumour to 42°C triggered aggregation of CP-DOX, essentially trapping it within the tumour and establishing a concentration gradient with the external environment that encourages further uptake.114 Furthermore, in combination with resection of a primary tumour, CP-DOX was used to greatly increase survival time in two murine metastatic cancer models (4T1 and Lewis lung cancers), with a curative effect in 60% of the treated mice.115 This activity was attributed to the delayed dissemination of viable cells from the primary tumour that inhibited formation of metastases. The approach was also applied to the delivery of PTX to demonstrate its potential broader applicability for other drug types.116
2.2.2 Other biopolymer–drug conjugates
Beyond polypeptides, only a few other biopolymer types have been explored in the context of self-assembling prodrugs. A great deal of attention has focussed on polysaccharides as drug delivery vectors117 and it is from this research that new self-assembling systems appear to be emerging. One of the earliest examples was reported by Kwon and co-workers,118 who demonstrated the assembly of DOX-conjugated glycol chitosan (GC-DOX) into nanoparticles ~250 nm in diameter that could encapsulate free DOX, giving a drug loading of 38% (w/w). They showed it could accumulate within tumours via the EPR effect, though the antitumour activity was only comparable to free DOX.119 A later report detailed the incorporation of an acid labile cis-aconityl linker,120 though no in vivo data was presented to show if this modification offered any improvement in activity. Glycol chitosan was also used by Quiñones and colleagues as a basis for the conjugation of drugs used to treat non-cancerous diseases, including vitamin D2,121 vitamin E,122 and testosterone.123
Ernsting et al. reported a DTX-conjugated carboxymethylcellulose conjugate, designated as Cellax (Figure 10b).109 Carboxymethylcellulose was chosen as it is an approved excipient in parenteral formulations, exhibiting no bioactivity and possessing many carboxylic acid functionalities as attachment points. PEG chains were also attached to further improve the pharmacokinetic properties. Through nanoprecipitation, the Cellax polymer formed monodisperse nanoparticles 118 nm in diameter with a DTX content of 37%. Biodistribution studies indicated a 5.5-fold greater tumour accumulation of Cellax relative to the clinically administered DTX formulation (Taxotere), with low DTX levels in the heart, lungs and kidneys and only transient uptake in RES-associated organs (liver and spleen). Consistent with the enhanced tumour accumulation, Cellax exhibited a two-fold improvement in anti-cancer activity in a murine EMT-6 breast cancer model compared to Taxotere. Improved effectiveness was also observed in a metastatic model, whereby EMT-6 cells were injected into the tail vein. Liu et al. reported a similar approach in which DTX was conjugated to carboxymethylchitosan (CMCS), doing so via a succinyl linker.124 Comparable improvements in anti-tumour activity and tolerance were seen. The conjugation of methotrexate to CMCS and subsequent self-assembly was also reported, though no toxicity data was presented.125
2.3 Perspective
It is clear that the development of macromolecular SAPDs has benefitted greatly from the wealth of knowledge garnered from decades of research into polymer–drug conjugates21 and polymer-micelle/nanoparticles-encapsulated drugs.15, 16 With this in mind, it is not surprising that there are many examples currently in clinical trials, with a notable few at the Phase III stage. There are, however, still a number of issues to be resolved with regard to their inherent polydispersity in drug content and molecular weight that can influence the nanostructure’s stability, as well as an over-reliance on passive targeting. Synthetic advances may address the former, phasing out the “post-translational” modification of the polymer in favour of continuing the development of drug-based monomers for use in polymer synthesis. Active targeting strategies can of course improve tumour accumulation, but could also introduce additional complexity into the polymer design that may complicate the synthesis. Biopolymer–drug conjugates offer a potential avenue for expanding the range of biomedical applications as a number of polysaccharides are known to form hydrogels, possessing interesting properties such as antimicrobial.126 Such materials could be harnessed to produce self-assembling drug conjugates suitable for use as wound dressings or medical device coatings.
3. Small Molecule SAPDs
Small molecules SAPDs offer a number of advantages over their polymeric counterparts, the key characteristic being they are precisely defined, homogeneous entities. The drug content can be tuned through the molecular design and, unlike PDCs, is not a function of conjugation efficiency as purification will be carried out to produce a pure compound. Furthermore, the maximum theoretical drug content is 100%, either for single or multiple drugs, and there will be less extraneous material to be removed after its function has been fulfilled. In terms of self-assembly, small molecule SAPDs potentially offer more versatility as their smaller nature means that any structurally-directing intermolecular interactions incorporated will be able to exert an influence, offering greater control over morphology. On the downside, however, is the possibility for more cumbersome synthetic procedures with their associated purification steps and concerns with nanostructure stability in circulation.
As previously discussed, small molecule SAPDs can be divided into two categories—drug conjugates and drug amphiphiles. While there is a great deal of structural similarity and overlap between the two groups, the distinction we are making here is in the context of their aggregation and assembly behaviour. Drug conjugates are defined based on their need for a significant change in conditions and/or molecular structure to induce aggregation/assembly, such as rapid dilution of an organic solvent solution of drug conjugate into water or the removal of a solubilizing group. Despite the amphiphic design of drug conjugates, the initial ratio of hydrophobicity to hydrophilicity does not play a central role in the aggregation behaviour of these molecules. Without the appropriate treatment (nanoprecipitation) or enzymatic processing, drug conjugates could be either too hydrophobic or too hydrophilic, lacking the ability to spontaneously assemble in an aqueous environment and so require some external inducement. Drug amphiphiles, on the other hand, possess a suitable, often optimized, hydrophilic–hydrophobic balance and can self-assemble upon being dissolved in aqueous solution into well-defined supramolecular nanostructures of various sizes and shapes. Subtle changes, such as pH and temperature, could influence the behaviour but on the whole little is required to induce assembly. In other words, the self-assembly potential, the physicochemical properties of resultant assemblies, as well as their pharmacokinetic profiles are all coded in the chemical structures of the designed amphiphic prodrugs.
3.1 Drug Conjugates – Assembly via Nanoprecipitation
3.1.1 Nanoprecipitation
The term kinetic trapping refers to the process of placing a system in a thermodynamically metastable or instable state.129 This process has been seen in nature where platelets progress into branched structures of variant shapes and sizes to help clot an area of injury.130 For nanostructure formation, a solution of a hydrophobic amphiphilic entity in a water-miscible organic solvent is added dropwise to a much larger ratio of water, the system forms spherical nanoparticles (Figure 11). Subsequent removal of the organic solvent from the solvent mixture, typically by dialysis or evaporation, stabilizes these assemblies through further hydrophobic collapse.129, 131, 132 Removal of the organic solvent is important for controlling the size of the emergent nanostructures as its presence reduces the stability of the particles to Ostwald ripening,131 in which smaller structures dissociate in order to feed the formation of larger structures. This method is referred to as nanoprecipitation and is often used as the method of choice to encapsulate free drugs within polymer carriers.
Fig. 11.

Chemical Structure of 4-(N)-trisnorsqualenoylgemcitabine (SQdFdC).127 After dropwise addition of the drug solution into a large body of water, successive removal of solvent will form stabilized nanoparticles. Transmission electron micrograph (TEM) of the resulting SQdFdC nanoassemblies is shown (scale bar 50nm). The TEM image was adapted from reference [128] with permission from ACS, copyright 2006.
The nanoprecipitation method, while an effective technique for the assembly of nanostructures, cannot strictly speaking be termed self-assembly since it is the environmental conditions that dictate the process rather than any intrinsic feature of the monomer chemistry. While both nanoprecipitation-formed and true self-assembled drug–conjugate nanostructures share an initial hydrophobic collapse step, the latter differs in that rearrangement of the internal structure will occur as intermolecular interactions between the monomer units begin to dominate and push toward a more thermodynamically stable structure. It is this absence of a propensity toward structural rearrangement that distinguishes the nanoprecipitation method from molecular self-assembly.
3.1.2 Conjugates of hydrophilic drugs
In the context of creating nanostructures made of drug conjugates, both Couvreur and Jin have been pioneers in the use of nanoprecipitation. Both groups’ initial concepts were based on the lipidation of hydrophilic drugs, many of which were nucleoside analogues. Couvreur’s approach was the conjugation of the nucleoside to squalenic acid (SQ), a flexible acyclic isoprenoid. The first demonstration showcased the strategy on three nucleosides, the most notable being the anticancer drug gemcitabine (dFdC), to attain an overall amphiphilic molecule.128 Nanoprecipitation stimulated these conjugates to form nanoassemblies (NAs) (nanoscale aggregates), 100–300 nm in diameter, which were seen to behave as colloids (Figure 11). A series of control molecules proved the necessity of the squalene segment in the formation of these NAs. Not only did this study prove the aggregation capability of nucleoside-squalene conjugates, but the conjugation of the lipid also enhanced the in vitro anticancer potency of SQdFdC and reduced its metabolism within the plasma. Furthermore, in vivo studies showed an increase in survival time, with some experiencing long-term survival, and reduced toxicity in comparison to free gemcitabine upon oral administration to leukaemia-afflicted mice. In a subsequent study by Couvreur and co-workers,127 SQdFdC additionally showed greater cytotoxic activity in drug-resistant leukaemia cells in comparison to free gemcitabine. Intravenous injection of SQdFdC particles into leukaemia-bearing mice prolonged survival time through the promotion of apoptosis by S-phase arrest. Couvreur has since demonstrated the broad utility of squalenoylation for a range of drug molecules and classes, including penicillin (antibiotic),133 paclitaxel134 and doxorubicin135 (anticancer), adenosine (neuroprotectant),136 and curcumin (antiparasitic).137
Jin’s strategy centred primarily on lipidation through the use of alkyl chains, with the first report concerning the lipidation of the anti-viral drug acyclovir.138 Here, stearyl-glycero-succinyl-acyclovir (SGSA), containing a single 18-carbon chain, was synthesized and induced to form nanostructures that were cuboid-like in shape with dimensions of 80 nm in width and 100–500 nm in length. Charge addition to SGSA promoted the formation of large vesicles due to a reduction in hydrogen bonding capabilities. Further studies show that these SGSA nanostructures were stable in both weak and neutral conditions but showed great sensitivity to alkaline conditions.139 Gemcitabine prodrugs were also developed by Jin and co-workers through the conjugation of lipid derivatives such as cholesterol-phosphonyl gemcitabine and N-octadecanonyl gemcitabine.140, 141 Li et al. also successfully applied this lipid conjugation technique adefovir for the treatment of hepatitis, overcoming the drug’s hydrophilic disadvantage and proving the system’s capability to target hepatocytes while exhibiting an enzyme-triggered drug release.142 Similarly, a lipidated Irinotecan prodrug was developed by Liang and co-workers that demonstrated increased cytotoxicity in cancer cells.143 A similar process was also used to develop cytarabine nanoparticles through the conjugation of hexanoic acid. This system exhibited an increased cellular uptake in comparison to the native drug, improving in vitro the cytotoxicity of the drug.144
3.1.3 Conjugates of hydrophobic drugs
In a similar but reversed manner to hydrophilic drugs, the self-assembly behaviour of hydrophobic drugs can be induced through the conjugation of hydrophilic moieties. Shen and coworkers demonstrated this strategy through the formation of DOX-loaded CPT-based nanocapsules.145 These nanocapsules were produced by conjugating the hydrophilic oligomeric ethylene glycol (OEG) to one or two hydrophobic CPT entities and subsequent nanoprecipitation from DMF in the presence of DOX (Figure 12). Shen’s multidrug capsules exhibited a linear release of CPT over 100 hours with rapid release of DOX over 20 hours. This combination therapy showed significant in vitro cytotoxic activity in comparison to the respective free drugs and OEG-diCPT capsules alone. In a similar manner, Kang and co-workers synthesized CPT nanoparticles through the conjugation of folic acid, demonstrating rapid cell penetration that enhanced the cytotoxicity in comparison to free CPT.146
Fig. 12.

Chemical structures of Shen’s diCPT-OEG and CPT-OEG prodrugs. These drug conjugates were capable of forming nanovesicles with the ability to encapsulate DOX (purple spheres). Figure was reproduced from reference [145] with permission from ACS, copyright 2010.
A wide range of drug-based functional nanosystems have been developed. One interesting, and rather unexpected, report by Couvreur and co-workers showed that squalenoylation of the hydrophobic drug PTX could also allow nanoparticle formation.134 This was attributed to the flexible nature of the polyunsaturated squalene chain granting an enthalpic benefit during aggregation, and is similar to an effect seen in a later study regarding lipidated SN-38 conjugates by Wang et al.147 In Couvreur’s report,134 a range of linkers were utilized to connect the two moieties, and all SQ-PTX conjugates showed the ability to aggregate into nanoparticles through nanoprecipitation in water. Their findings showed the ability to control release through molecular design: using a direct ester bond between the paclitaxel and squalenoyl moiety displayed the highest particle stability in comparison to using a succinate linker or diglycolate linker which showed a sequential decrease in stability. Furthermore, the addition of OEG chains (n = 3 or 11) between the squalenoyl and the PTX showed faster particle hydrolysis. Squalenoylation of the hydrophobic anti-parasitic drug curcumin was also demonstrated by Couvreur and colleagues, resulting in improved cytotoxic behaviour relative to free curcumin.137 In a similar manner to derivatisation with squalenic acid, the conjugated linoleic acid conjugate of PTX was found to be an effective strategy by Zhong et al.148 This suggests the use of unsaturated fatty acids may be a general approach for the creation of drug conjugate nanostructures by nanoprecipitation.
The aggregation potential of the bile acid, lithocholic acid (LA), has also found utility for creating drug conjugates. Patil et al., for example, conjugated LA onto three different cancer drugs: paclitaxel, doxorubicin, and PI103 (a PI3K inhibitor).149 In all cases, the hydrophilic nature of the acid combined with the hydrophobicity of the drug to produce an overall amphiphilic molecule, capable of generating nanoparticles with a slow and sustainable drug release.
3.1.4 Drug–drug conjugates for combination therapy
One advantage of small molecule prodrugs that was alluded to earlier is the potential for a high drug content of up to 100%. This can be achieved by conjugating two drugs together, either directly or through a linker. The difference in hydrophilicity between these two drugs can imbue the necessary amphiphilic nature needed for self-assembly. In prodrugs parlance, such systems are referred to as co-drugs, where one drug acts as the promoiety of the other.2 These heterodrug conjugates therefore provide a combination therapy that can enhance system potency and allows for a greater drug content.
A variety of co-drugs have been developed which demonstrate the ability to assemble into nanoparticles via nanoprecipitation. For example, the hydrophobic chlorambucil has been coupled to the hydrophilic drugs gemcitabine150 (Figure 13a) and irinotecan151 (Figure 13b) for the multi-therapeutic treatment of cancers. In another case, hydrophobic doxorubicin was chemically conjugated onto tocopherol succinate (Figure 13c), promoting a steady and long term release of drug.152
Fig. 13.

Examples of drug–drug conjugates in which two drugs are linked together, either directly or through a short linker.
The use of a linker between both agents was also studied by Yan, Zhu and colleagues where camptothecin (CPT) and floxuridine (FUDR) were combined using an anhydride linker (Figure 13d). This preserves the therapeutic activity of each agent until the linker is hydrolysed once inside the cell.153. Another self-assembling CPT co-drug was developed by He et al. through conjugation to the hydrophilic drug, Irinotecan.154 This combination therapy method has also been applied to HIV treatment were co-drugs consisting of both zidovudine and didanosine drugs were studied.155 In all cases, these drug cocktail systems displayed superior cytotoxic behaviour in comparison to the individual drug administration.
In some instances, prodrug dimers were produced using a hydrophilic linker to connect two molecules of the same hydrophobic drug. Sun and colleagues applied this to PTX, forming dimers via a glutamic acid linkage,156 whereas Jin and co-workers synthesized CPT dimers using a reducible disulphide-based linker.157 In both cases, stable nanoparticles were formed, while cytotoxic behaviour was maintained relative to the parent drug.
3.1.5 Perspective
The simplicity of nanoprecipitation has made it an attractive method for the synthesis of drug-based nanoparticles. Great strides have been made in this field of study from the design and synthesis of a variety of drug conjugates multidrug-bearing conjugates. However, despite extensive efforts in in vivo evaluation of these conjugates there are no examples close to clinical application nor any that appear to be currently in advanced clinical trials. A particular drawback of the nanoprecipitation method is the limited control over nanostructure size and morphology. In many cases, these drug conjugate systems consisted of spherical nanoparticles of various sizes, potentially causing problems during in vivo administration due to the need to overcome multiple biological barriers that require different physicochemical properties and biologically responsive features. Furthermore, the necessity of organic solvents in the synthesis process could result in undesirable side effects when applied to clinical trials. To some degree, these drawbacks have stimulated interest in developing new strategies for the production of self-assembling drug conjugates.
3.2 Drug Conjugates – Assembly via Chemical Modification
3.2.1 Prodrug-based prohydrogelators
Amphiphilic molecules exhibiting high hydrophobicity experience limited assembly behaviour and require methods such as nanoprecipitation to realize such characteristics. Particularly hydrophilic molecules, on the other hand, will also display a reduced capability to aggregate into NAs. In such cases, chemical modifications are necessary to moderate their hydrophilic nature and induce self-assembly. The advantage of this requirement is that it leads to stimuli-responsive materials with the potential for spatiotemporal control over their assembly behaviour.
A significant advancement in this area was developed by Xu and co-workers, who reported a range of supramolecular hydrogelators159 programmed to self-assemble only upon contact with certain enzymes, terming them prohydrogelators.158 By attaching a phosphotyrosine to a Nap-Phe-Phe-Lys(PTX) hydrogelator (where Nap is a naphthalene moiety) (Figure 14), the enhancement in hydrophilicity promoted solubility, yet preserved the self-assembly potential of this molecule. Upon contact with a phosphatase enzyme, dephosphorylation occurs and significantly reduces the solubility. Consequently, self-assembly takes place through spontaneous β-sheet interactions to form a 1D nanofiber-based hydrogel. After gel formation, the PTX conjugate is released at a linear rate and exhibits effective cytotoxicity against HeLa cells. This on-site gelation and release of drug was also applied by Li et al. to the platinum drug cisplatin, where it too was conjugated onto the phosphatase-peptide substrate.160 The enhanced activity of phosphatases in cancer cells limited self-assembly mainly to the tumour site. When systemically applied to an in vivo setting, an improved drug accumulation at the target site was observed which reduced molecular toxicity, in comparison to free cisplatin. This technique of enzyme instructed self-assembly (EISA) can therefore be used as a targeting method to enhance the antitumour efficacy of the respective prodrugs.
Fig. 14.

Enzyme-instructed self-assembly (EISA) strategy developed by Xu and co-workers. After enzyme-activated dephosphorylation, the taxol conjugates spontaneously self-assemble into nanofibers (A). The chemical structure of the corresponding taxol conjugate (B). A dense network of nanofibers contribute to the formation of a hydrogel (C, D). Adapted from reference [158] with permission from ACS, copyright 2009.
Interactions between the aromatic rings of phenylalanine amino acids greatly contribute to the gelation properties of these hydrogels.161 For this reason, numerous prohydrogelators have been developed where the drug acts as an aromatic moiety and is thus a key structural element. Xu and co-workers for example effectively utilized the aromatic ring found in common nonsteroidal anti-inflammatory drugs (NSAID) to enhance gelation properties. NSAIDs act to inhibit cyclooxygenase-2 (COX-2), locally reducing inflammation and relieving pain. However, adverse effects are associated with high dosages, making targeting a crucial aspect for their systemic application. By binding these NSAIDs to small peptide chains, these molecules can be programmed to self-assemble in water under certain conditions including pH changes, temperature and enzymatic reactions. Using the method of phosphorylation, these prohydrogelators were preset to self-assemble in the presence of phosphatase enzymes.161, 162
Another drug that can greatly contribute aromatic interactions is curcumin, an agent often used for the treatment of cancer. In an article by Yang and colleagues,163 a different approach to phosphorylation was used for the synthesis of a prohydrogelator. By attaching a hydrophilic RGD sequence, through a reducible disulphide bond linker to a curcumin hydrogelator, multiple accomplishments were made. The RGD peptide sequence increases binding to αv integrins that are expressed on the endothelium of the cancer cells and neovasculature at tumour sites.164 This allows enhanced localization of these curcumin conjugates at the tumour site after systemic administration. Furthermore, the hydrophilicity of the RGD sequence enhances solubility and prevents the spontaneous self-assembly of these molecules. After localization, the elevated intracellular glutathione (GSH) concentrations reduce the disulphide bond linker, cleaving the RGD sequence from the curcumin hydrogelator and allowing self-assembly to proceed. Furthermore, a linear in vitro release of curcumin was observed from these hydrogels and the cytotoxic activity of the prohydrogelator was not impacted relative to free curcumin. Additionally, in vivo application showed a significant tumour inhibition response by the prohydrogelator in comparison to free curcumin and the gelator itself. This result demonstrates the advancement of such a system in both tumour targeting and inhibition.
Combining both aromatic properties and enzymatically-stimulated self-assembly leads to the formation of multi-drug molecules as a means of providing a multi-mechanistic therapeutic method. In one study, the anti-inflammatory drug, dexamethasone (Dex) was accompanied by anti-cancer agents, PTX or 10-hydroxyCamptothecin (HCPT).165 The inspirational design of this molecule consisted of Dex bound to the peptide chain FFFK(PTX or HCPT)E-ss-EE. The purpose of the phenylalanines (FFF) was to effectively interact with the aromatic rings of Dex to stimulate self-assembly. The lysine (K) served as an anticancer drug carrier while the adjacent glutamic acid promoted solubility. The glutamic acid pair at the end of the molecule, conjugated to the main peptide through a disulphide bond linker, served the purpose of enhancing hydrophilicity and preventing spontaneous self-assembly. The reduction of the disulphide bond by GSH cleaves the glutamic acids from the original molecule, promoting the formation of a hydrogel. The resulting multi-component hydrogel demonstrated a sustainable release of drug while preserving the anti-cancer and anti-inflammatory activity of the corresponding drugs.
This reaction-necessary self-assembly method has been studied on a wide range of prodrugs. Another example can be seen by Yang and co-workers where a folic acid (FA)-PTX prohydrogelator was designed, capable of forming a hydrogel through the self-assembly into nanospheres.167 The design included PTX conjugated to folic acid via a phosphorylated and succinated tripeptide (GYK). As in previous cases, the dephosphorylation of this molecule resulted in an increased propensity for the FAs to form tetramers, with further associations producing nanospheres. The high density of these structures resulted in a stable and transparent hydrogel. Other prohydrogelators of taxol developed by Yang and colleagues include systems based on succinated taxol.168 In one example, succinylated PTX was further conjugated to an oxidized glutathione (GSSH) that showed the ability to form a hydrogel upon hydrolysis of the ester bond. In another study, the succinated taxol itself was shown to gelate upon sonication in PBS.169 Through the addition of more than 30% hyaluronic acid, the resulting hybrid gel demonstrated improved mechanical properties and a reduction in the release rate of PTX. In both cases, the PTX conjugate was shown to enhance in vivo tumour inhibition effects with regard to free PTX.168, 169 The former system further demonstrated its ability to reduce the metastasis of breast cancer.168 This technique has also been successfully applied to the treatment of eye diseases such as uveitis, minimizing potential side effects observed in commercial treatments.135
3.2.2 Enzyme-instructed intra- and extracellular assembly
An interesting extension of this approach is the intracellular or extracellular formation of filaments. Building on work that showed nanofilaments of Nap-FF can induce cellular apoptosis,170 Xu and colleagues extending this idea to develop a method where the soluble peptide is the prodrug and the assembled nanostructure is the drug. In this strategy, the enzyme-induced self-assembly plays the role of the activation step. To prove this, a combination therapy of the cancer drug cisplatin with enzyme-instructed intracellular self-assembly was evaluated on drug-resistant ovarian cells.171 Accordingly, Nap-FF was conjugated onto a succinate-taurine precursor through an ester bond. After cellular uptake of this monomer, carboxylesterase catalyses the hydrolysis of the ester bond to release the hydrogelators and in doing so induces self-assembly. The resulting intracellular nanofibers then interact with the cytoskeleton protein actin to induce cancer cell inhibition. The use of this system in combination with cisplatin magnified the cytotoxic activity of the drug and helped overcome drug resistance in ovarian cancer cells. This method was further improved by conjugating a mitochondria targeting agent to direct self-assembled fibres to the mitochondria, eliminating the potential for acquired drug resistance and further sensitizing cells with increased exposure.172 The mitochondria targeting agent, triphenyl phosphonium (TPP), was conjugated onto the phosphorylated FFYK tetrapeptide, with an environment-sensitive fluorophore (NBD) for self-assembly tracking taking the place of Nap. Upon contact with the overexpressed alkaline phosphatase (ALP) by cancer cells, dephosphorylation resulted in extracellular self-assembly. After internalization of the fibres, they were shown to localise in the mitochondria where they stimulated the release of cytochrome c to the cytosol, which in turn activated the caspase cascade and induced cellular apoptosis. Excitingly, this study demonstrated that the use of this system not only prevented the acquirement of drug resistance, but also that prolonged exposure made cells more sensitive to these TPP conjugates. While EISA has yet to be demonstrated in an in vivo setting, it was previously shown that the injection of self-assembled Nap-FF fibres into the peritumoural space resulted in an enhanced tumour regression effect.170 This demonstrated the potential cytotoxic nature of such a non-drug based system.
An alternative mechanism to apoptosis induction involved the extracellular self-assembly of prohydrogelators. This phenomenon was studied by Xu and colleagues through the use of an NDP-labelled Nap-FF phosphorylated peptide. ALP overexpression helped target the self-assembly of these molecules to within the tumour site.166 After fibres selectively formed around cancer cells, they interacted with extrinsic cell death ligands and receptors, prompting cellular apoptosis.173 An illustration of how EISA can produce a tumour inhibitory effect through the extracellular formation of nanofibrils can be seen in Figure 15. These studies all demonstrate the wide potential of EISA in targeting the tumour microenvironment and the ability of self-assembled structures alone to act as active cytotoxic agents.
Fig. 15.

An illustration of the EISA mechanism in the pericellular space, where a chemical modification helps trigger the self-assembly of NDP1 into nanofibrils. (A) Dephosphorylation, by alkaline phosphatase (ALP), removes the phosphate group and in turn activates the NDP1 precursor. (B) The molecular structures of pNDP1 and NDP1 where the orange segment is the fluorophore NBD and red indicates the phosphate enzymatic trigger. (C) The overexpression of ALP by cancer cells limits dephosphorylation proximal to the cell membrane. This stimulates apoptosis. (D) Image of the fluorescent fibrils selectively forming around the HeLa cancer cells (white arrows) in a co-culture with Hs-5 stroma cells (white arrowheads). With time, the amount of self-assembled fibrils increases with the majority focused around HeLa cells. The left image was at time 0 h, middle at 6 h, and right at 24 h. Scale bar represents 10 µm. Adapted from reference [166] with permission from Elsevier, copyright 2016.
3.2.3 Perspective
Hydrogels have found use in a wide range of applications given their biocompatibility, biodegradability, and minimal and tunable immunogenicity.174–177 Furthermore, an extensive assortment of drugs have the necessary properties to allow their incorporation into prohydrogelators, broadening the range of diseases that can be treated using this method. However, despite having been in development for over 15 years, there is a lack of systematic in vivo studies involving prohydrogelators (e.g. specific and non-specific immunogenicity) that could allude to their future clinical success. Indeed, a major issue that needs to be addressed is being able to control the mechanical and physicochemical properties of the hydrogel in response to an in vivo environment. The release of the drug from the nanostructure is a key feature in the efficacy of the drug conjugate and it can be strongly influenced by the stability and the network features of the hydrogel.178 In in vivo settings, such as a resection cavity,179 the hydrogel may be subject to three-dimensional shear and strain stresses that could compromise its integrity thus leading to unexpected drug release profiles. Subsequent increased exposure of the drug conjugate caused by hydrogel breakdown can facilitate faster and unpredictable drug release. Furthermore, the retention of the hydrogel at the disease site will also be affected. Both factors will significantly affect the drug conjugate’s effectiveness to exert the desired therapeutic effect.
3.3 Drug Amphiphiles
3.3.1 Design aspects
Drug amphiphiles (DAs) are a class of self-assembling drug conjugates in which the hydrophobic–hydrophilic balance is sufficient for assembly to occur spontaneously, without the need for significant chemical or physical inducement.180–182 The term itself was introduced by Cui and coworkers, who demonstrated the rational design of a peptide–drug conjugate that could assemble into well-defined filamentous nanostructures.183, 184 The basic design of DAs includes a hydrophobic drug chemical conjugated onto a hydrophilic segment as shown in Figure 16. Given that peptides have been found to self-assemble into a variety of nanostructures in nature,185–187 Cui and co-workers utilized this property by using peptides to serve as the hydrophilic segments of these DAs.183 Many studies were conducted to understand the influence of molecular design on the various properties of these DAs. In their initial study, a β-sheet forming peptide sequence (CGVQIVYKK) derived from the microtubule stabilizing Tau protein was selected for the hydrophilic region. Connected to the peptide using a reducible disulfanyl butyrate (buSS) linker, different numbers of CPT molecules (one, two, or four denoted by mCPT, dCPT or qCPT, respectively) served as the hydrophobic region as a means to understand the effect of drug quantity on nanofilament assembly. In all cases, transmission electron microscopy (TEM) showed the molecules to self-assemble into filamentous nanostructures in water. In comparison to mCPT-buSS-Tau, dCPT-buSS-Tau showed wider yet shorter fibres. In both cases however, the fibre diameter was double the molecular length indicating a cylindrical geometry with core-shell micelles. CD data demonstrated the role of β-sheet hydrogen bonding in the self-assembly of these designs. By contrast, qCPT-buSS-Tau formed nanotubular structures, as indicated by the hollow central channel observed in TEM imaging. This behaviour points to the potential for gaining control over the nanostructure morphology through the molecular design. Furthermore, the stepwise increase in drug number also leads to an enhanced stability toward dissociation, with the conjugates exhibiting disassembly at 207, 74, and 54 nM for mCPT-, d-CPT-, and qCPT-buSS-Tau, respectively. In vitro cytotoxicity experiments showed that dCPT-buSS-Tau demonstrated the highest cytotoxicity followed by qCPT-buSS-Tau and mCPT-buSS-Tau. This observation was attributed to a more balanced hydrophobic–hydrophilic ratio in dCPT-buSS-Tau, which may allow a more effective cellular uptake, after which the CPT will be cleaved by GSH reduction.
Fig. 16.

Illustration of a CPT-based drug amphiphile that can spontaneously self-assemble into nanofibers upon ageing in water. Adapted from reference [183] with permission from ACS, copyright 2013.
Being prodrugs, the choice of linker plays a critical role in both the rate at which the drug is chemically cleaved from the peptide and into what form that drug is in—either the original form or a modified version requiring further degradation.182 Taking this into consideration, Cui and co-workers investigated the drug release mechanism from their DA platform using two disulphide linkers—buSS and a disulfanyl-ethyl carbonate etcSS linker.188 Results indicated that CPT-etcSS-Tau exhibited a faster release of free drug upon contact with GSH due to a more effective self-immolation mechanism upon reduction. This was reflected in the cytotoxic nature of the molecule as the CPT-etcSS-Tau demonstrated a much lower IC50 value than the CPT-buSS-Tau, one much closer to free CPT. Combined, this data revealed the enhanced potency and efficacy properties of the etcSS linker in comparison to buSS. In the case of the buSS linker, however, higher concentrations of conjugate led to a decreased efficiency of the reduction reaction and a reduced yield of free CPT. It was hypothesised that the nanostructure itself could influence the degradation pathway, as the presence of disulphide species were evident from LCMS analysis. Within the assembly, the linked drugs are in close proximity, such that cleavage of an S-S bond can generate a free SH group that could potentially attack another linked drug to generate the CPT-buSSbu-CPT species. These hydrophobic entities may then be protected within the nanostructure from hydrolysis that would otherwise release free CPT.
3.3.2 Influencing the morphology
Morphological characteristics are important determinants of how synthetic delivery vehicles interact with biological interfaces, in particular influencing the system’s pharmacokinetics and cellular uptake. Discher and co-workers observed the enhanced ability of polyethylene glycol (PEG)-coated, block copolymer filomicelles to circulate longer in the bloodstream than their spherical counterparts.189–191 In a study by Lock et al,192 the self-assembled arrangement of positively charged spherical nanobeacons exhibited a much higher cellular uptake ability compared to their monomeric form. Furthermore, it was shown that assembly into filamentous nanobeacons reduced their overall internalization capacity, regardless of the surface charge status. These studies all demonstrate the synergy between morphological changes and biological consequences.
The nanostructure morphology can be influenced in a number of ways such as drug type, number, and the auxiliary segment, or through the manipulation of the assembly kinetic pathways. Such changes alter the intra- and intermolecular interactions, affecting the sterics, packing density, and twisting abilities. For example, in Cui’s initial report,183 the quadruple conjugation of CPT resulted in unexpected nanotube formation. These structures possessed a wall thickness equal to that of the molecular length, indicating a monolayer arrangement. In comparison to mCPT and dCPT, a helical arrangement of CPT conjugates was identified within the nanotubes of qCPT. This differing arrangement of CPT molecules is a result of increased π–π interactions among individual CPT segments, as evidenced by excitonic coupling in the circular dichroism (CD) spectrum and molecular simulations.193, 194 A later study by the same group, however, also suggests that the peptide segment could have an influence in nanotube formation as nanotubes were observed for a number of dCPT-based DAs.195 By mixing of oppositely charged CPT drug amphiphiles (catanionic mixing), the Cui Lab reported on the formation of multiwalled nanotubes containing a fixed and high drug loading.196 They found that the molecular mixing ratio, the solvent composition, the overall drug concentrations, as well as the molecular design are all important experimental parameters contributing to the tubular morphology.
A change in the auxiliary segment or drug type, or even conjugation of multiple drugs onto the same molecule have also been shown to have a great impact upon the adopted morphology during self-assembly. To study the effect of the peptide segment on morphology, Cui and coworkers replaced the Tau peptide sequence with a more hydrophilic β-sheet forming peptide derived from the Sup35 yeast prion (NNQQNY).197 Similar nanofilaments were observed in both cases however, in comparison to dCPT-buSS-Tau, the nanofilaments of dCPT-buSS-Sup35 were slightly wider yet considerably longer. Moreover, a significant distinction was seen in the critical assembly concentration (CAC) where the more hydrophilic peptide showed a lower propensity to assemble into micelles, as evidenced by the higher CAC value, which could be a result of increased monomer solubility. Overall, the longer fibres observed in the dCPT-buSS-Sup35 design are more capable of intertwining, leading to higher viscosities and better candidates for hydrogel formation. On the other hand, the shorter fibres of dCPT-buSS-Tau are believed to make a stronger case for systemic delivery attributed to a reduced possibility of being trapped in the body’s filtration system, potentially increasing the circulation time. Another example of how the auxiliary segment can influence assembly is provided by Zhang and co-workers, who developed light-triggered nucleic acid–drug amphiphiles (Figure 17a).198 With CPT as the primary drug, these DNA drug amphiphiles were seen to form either spherical or cylindrical nanostructures depending on the length of the DNA strand. A short segment (5 base pairs) gave spherical structures, whereas a longer sequence (20 base pairs) led to the formation of rigid rods 53 ± 14 nm in length. Through light activation, the structures are programmed to disassemble into free drug which exerts its respective cytotoxicity. Moreover, their spherical nucleic acid-like nature means they possess a resistance to nuclease-induced degradation and can be rapidly uptaken into cells.
Fig. 17.

Morphological changes induced by structural alterations. (a) Zhang’s nucleic acid–drug conjugates formed spherical or cylindrical nanostructures depending on the number of DNA base pairs in the molecular structure. (b) Cui and co-workers showed that the incorporation of two structurally different drug molecules, CPT and PTX in this case, can significantly alter the self-assembly pathway and the emergent nanostructure. Panel (a) is reproduced from reference [198] with permission from ACS, copyright 2015. Panel (b) is adapted from reference [200] with permission from the Royal Society of Chemistry (RSC), copyright 2014.
Changing the drug offers the possibility of influencing the morphology as the potential molecular interactions will differ between the various drug types. For example, Cui and colleagues replaced CPT with the bulkier PTX in their DA system to form PTX-buSS-Tau.199 This DA was seen to self-assemble into nanofilaments with a cylindrical micelle arrangement. In comparison to mCPT-buSS-Tau,183 however, wider filaments were observed and cytotoxic properties were revealed to be similar to that of free PTX. This latter observation may be due in part to the higher CAC concentration of this conjugate, though the more easily hydrolysable 2° ester will also aid with drug release (compared to 3° for CPT). As part of another study, Cui created as DA with double the number of PTX molecules attached to the β-sheet forming peptide, Sup35 in this instance.200 Unlike PTX-buSS-Tau, dPTX-Sup35 was unable to form well-defined nanostructures, instead producing a mixture of spherical and worm-like micelles. It is clear that the bulky PTX hindered self-assembly and indicates a possible limitation in controlling the drug content through multiple drug attachment for drug amphiphiles. Successful assembly is dependent on the molecular components being able to come together and interact in the prescribed manner. In cases where a slightly more hydrophilic drug, doxorubicin, is incorporated, the resultant drug conjugates are highly water soluble, unable to assemble into well-defined nanostructures under physiological conditions.201, 202
Drug amphiphiles have also been designed with potential combination therapy applications in mind. The attachment of two different drug molecules to a single hydrophilic entity can lead to interesting self-assembly behaviour, particularly if there are significant structural differences. For instance, Cui and colleagues conjugated both CPT and PTX to the Sup35 peptide using a statistical reaction that also produced dCPT-Sup35 and dPTX-Sup35.200 Upon dissolution of CPT-PTX-Sup35 in water, TEM revealed the initial formation of both small worm-like structures and long filaments (Figure 17b). After 24 hrs, only long twisted filamentous structures were observed which were determined to comprise short filaments associating with one another to give two-filament fibrils. On the other hand, dCPT-Sup35 was shown to assemble into typical filamentous structures whereas dPTX-Sup35 was seen to form the aforementioned ill-defined assemblies. These differences in self-assembly behaviour were explained using the fact that the dCPT conjugate can easily associate into filamentous structures yet the replacement of a CPT with a bulky PTX molecule slowed down the assembly process and resulted in the formation of twisted fibrils. This hindrance could be inferred from CD data that showed a gradual strengthening of β-sheet interactions, suggesting that the initial structure is the result of hydrophobic collapse with subsequent conformational rearrangements giving rise to the observed twisted fibrils. The effect of PTX inclusion is also seen in the stability of the respective nanostructures, with the dCPT conjugate being the most stable and the dPTX the least, based on dilution studies. An advantage of the heterodual drug conjugate CPT-PTX-Sup35 was its ability to effectively exert cytotoxic activity against PTX-resistant KBV1 cervical cancer cells.
A later study by Cui and co-workers replaced the PTX drug with a custom designed capecitabine (Cap).203 Cap is a prodrug of the anti-metabolite 5-fluorouracil (5-FU) that undergoes three enzymatic modifications to produce the free drug. Here, the alkyl carbamate tail of Cap was replaced with a disulphide-based carbamate moiety that could be reductively cleaved intracellularly. Unlike PTX, the Cap derivative is less bulky and possesses greater hydrophilicity due to its OH groups and smaller molecular weight. CPT-Cap-Sup35 was found to form filamentous structures wider than those formed by dCPT-Sup35. dCap-Sup35 formed spherical particles with an average diameter of 38 nm, suggesting an aggregate structure with an ill-defined internal packing arrangement since the molecular length is ~6 nm. As with the CPT-PTX DAs, a similar decrease in stability was seen as CPT was replaced by Cap. It is evident from these two studies that the CPT drug can imbue a greater stability through its self-association within the nanostructures π-π interactions, while inclusion of a second entity disrupts these interactions to give less stable structures. However, a trade off in stability can perhaps be made should there be additional benefits to the second drug’s inclusion. For CPT-Cap-Sup35, Cap was included on the basis of 5-FU’s observed synergism with CPT. Indeed, this proved to be the case as CPT-Cap-Sup35 exert a synergistic cytotoxic effect against esophageal cancer cells, being not only more potent than the other two DAs but also more effective than the free drugs—almost 8-fold greater than the free CPT/5-FU combination.203
Gaining control over the morphological and physicochemical outcome of DA self-assembly is, of course, only one step towards creating therapeutically effective DAs. What remains to be uncovered is how precisely the morphology affects the nanostructured drug’s bioactivity. This is particularly important since the morphology and surface chemistry will dictate how the nanostructure presents itself to any biological entities it encounters, including the therapeutic target. One complication in elucidating this stems from the fact that the majority of the various morphologies formed by DAs are done so by conjugates possessing differing chemical structures that make direct comparisons difficult. Some conjugates may contain the same drug molecules but then present different chemistries on their nanostructure surfaces and vice versa. Consequently, a particular element cannot be explicitly ascribed to being the sole cause of any observed difference in bioactivity between two morphologies. To do so would require an alteration in processing conditions such that two or more morphologies could be created from a single conjugate, as in Cui’s study on nanobeacons.192 An alternative strategy could encompass the study of isomeric species in which the spatial location of elements are varied, a method shown in the study of peptide-based amphiphiles to induce morphological or self-assembly behaviour variations through a simple switch in the amino acid order.204, 205 The rationale here is that the relocation of a particular amino acid can influence the intermolecular interactions that govern the self-assembly pathway.
3.3.3 Widening the field
Different molecular designs and drugs have been studied across many labs to prompt certain properties and further widen the application of DAs. For example, hydroxycamptothecin was converted into a PEGylated DA that demonstrated an ability to assemble into short individually dispersed fibers.206 Furthermore, Zhang and co-workers conjugated doxorubicin (DOX) onto a multi-functional peptide consisting of a tumour targeting RGD sequence and octaarginine (R8) sequence with membrane penetration capabilities.207 This system was able to effectively target the tumour microenvironment and release Dox for improved in vivo antitumour activity with reduced side effects. Nanoparticles were also observed when a PTX-conjugated cell penetrating peptide was aged in water.208 This system was seen to exhibit comparable cytotoxicity to free PTX.
In some cases, DAs can also act as hydrogelators.209 For example, Lock et al. converted an FDA-approved anticancer drug, Pemetrexed (Pem), to a molecular hydrogelator with inherent chemical exchange saturation transfer (CEST) MRI signals.209 The designed drug–peptide conjugate can spontaneously associate into filamentous assemblies under physiological conditions and consequently form theranostic supramolecular hydrogels for injectable delivery. The local delivery and distribution of Pem–peptide nanofiber hydrogels can be directly assessed using CEST MRI in a mouse glioma model. In another example, an aminoglycoside antibiotic hydrogelator of kanamycin was developed by attaching multiple phenylalanine residues.210 This design formed small fibrils upon placement in water, which subsequently entangled to form a hydrogel network. This hydrogel was seen to bind with bacterial 16S rRNA which can inhibit protein synthesis and act as bactericide. This platform therefore preserves the interaction of aminoglycoside conjugates with their respective macromolecular targets. In another study, the NSAID, naproxen, and the anti-HIV drugs AZT or 3TC were both conjugated onto a phosphorylated peptide chain, to provide both an anti-inflammatory and anti-HIV response.211 Upon encountering an acidic environment, these hydrogelators spontaneously self-assemble into a network of nanofibers, forming a weak hydrogel. After enzymatic cleavage of the phosphate group, by surrounding prostate acid phosphatase (PAP) enzymes, the resulting hydrogelator enhances interactions among the nanofibers to result in a robust gel. As a result, the gel treated with a phosphatase showed slower release kinetics compared to the acid-stimulated preformed hydrogel. More recently, Matson and colleagues demonstrated how their H2S prodrug85 can act as the hydrophobic segment in a peptide-based for the formation of H2S-releasing hydrogels.212 Chen et al. reported the creation of an (S)-ketoprofen-based DA,213 in which the anti-inflammatory was conjugated to a peptide with alternating hydrophobic and hydrophilic residues (Val-Glu-Val-Glu) that enabled formation of belt-like assemblies. These wide nanostructures were capable of forming a hydrogel at higher concentrations, making them suitable for local delivery to treat conditions such as rheumatoid arthritis. Indeed, it was demonstrated that they could be injected into the articular cavity of a rat knee joint and exhibit no significant burst release of the drug into systemic circulation.
3.3.5 Conjugation of drugs to self-assembling peptides
There are many reports wherein a drug has been conjugated to a self-assembling system in such a way that it plays no structural role, simply acting as cargo. Despite this, they can still be considered as prodrugs since in being covalently linked they are typically inactivated until being released. Peptide amphiphiles (PAs) developed by Stupp and co-workers provide numerous examples of this approach. PAs consist of a hydrophobic alkyl chain conjugated onto hydrophilic β-sheet forming peptide. The agent to be delivered can be easily incorporated by using a suitable reactive amino acid, such as lysine. In this manner, the Stupp lab created hydrazide containing peptide amphiphiles that could self-assemble into nanofiber gels with a long term release following near zero-order kinetics for the release of nabumetone214 and prodan.215 Gasotransmitters have also been incorporated into PAs, allowing their effective delivery in the form of hydrogels.216 For example, in one study by Stupp and co-workers, a nanofiber gel was developed from a peptide amphiphile with chemically conjugated carbon monoxide (CO) molecules.217 This gel spontaneously released CO with prolonged release kinetics in comparison to soluble CO donors. In a similar manner, nitric oxide was also delivered for the prevention of restenosis.218 A number of other hydrogelators have also been applied to various settings by conjugating different drug cargos. For example the formation of an antibacterial olsalazine hydrogel219 and a bortezomib-based hydrogel for local cancer treatment.220
3.3.6 Perspective
Drug amphiphiles represent a recent addition to the drug delivery toolkit that blurs the line between the prodrug and nanomedicine strategies. The ability to precisely control and alter drug content, properties and assembled morphology through molecular design enhances versatility, making them more compliant to both local and systemic administration. Homogeneity in terms of both drug loading (100% as the entire DA is considered as a prodrug) and morphology is also a great benefit of using such systems. The potential offered by drug amphiphiles is considerable as their design flexibility means that they can be tailored towards the treatment of many disease types. The majority of studies thus far tend to focus on cancer therapeutics since this disease affects so many, but drug amphiphiles could also be harnessed for regenerative medicine applications. Peptide amphiphiles,221, 222 from which drug amphiphiles draw their inspiration, have long been studied as scaffolds for the templated regrowth of damaged tissues (e.g. nerve,223 bone,224 cartilage225, and enamel226). These materials often present bioactive epitopes at their surface for the recruitment of essential growth factors and proteins, but possess a relatively inert core. It is certainly possible that drug amphiphiles could make an impact in the regenerative medicine area, as drugs beneficial to the regenerative process could be delivered while its nanostructure acts as a scaffold. For example, anti-inflammatory drugs could be used to reduce damaging inflammation at an injury site concurrently with nanostructure-influenced tissue growth.
The synthetic tractability of drug amphiphiles varies widely depending on how simple or complex the conjugate design is. The range of conjugated drug types explored thus far is also somewhat limited, focusing heavily on flatter, aromatic structures that can pack well within nanostructures. Those conjugates with bulkier drug types, such as the taxols, tend to exhibit stability issues resulting from the increased steric interactions within the assembly. Hydrophilic drugs too may also prove difficult to fully incorporate into the DA strategy, though prodrug strategies could be used to reversibly add hydrophobicity. Accordingly, the extent to which a wide range of drug amphiphile types can be created may be restricted. Furthermore, the biological roles of the peptide segment remain underappreciated in the DA design. Early research is biased towards harnessing the hydrogen bonding and assembly potential of peptide sequences to drive the formation of drug-based supramolecular nanostructures. There are many peptide-based drugs such as hormones and biologically active peptides with important roles in tumour targeting, tissue penetration, immune system evasion, enhanced cellular uptake, and subcellular uptake that could be considered in the DA design.227–231 Another concern is that of nanostructure stability, as relatively high concentrations (in comparison to polymer-based assemblies) may be necessary for self-assembly in some instances and could somewhat limit in vivo application. An emerging perspective is that the concentration-dependant and environment-responsive characteristics of DA assemblies are regarded as advantageous properties enabling programmed drug release and transformative physicochemical features. The large dependence of self-assembly behaviour on the drug itself may also limit design adjustments and application. Furthermore, some concerns have been raised with regard to the long-term stability of the nanostructures formed, particularly upon introduction to the in vivo physiological environment.178
4. Conclusions
The prevalence of prodrugs in clinical practice has had an enormous impact, both for the patients to whom they are administered in regard to the health benefits of a more effective treatment and for the manufacturers in terms of spurring investment into the next breakthrough drug. Despite these successes, prodrugs continue to suffer the same issues that they were created to solve, albeit to a lesser degree than their parent drug, and may create their own unique problems. Due to the many physicochemical, biophysical, and pathological barriers to be circumvented, a strategy that can address most or all of these is likely to be complex in nature. Self-assembling prodrugs as a potential solution are delicately poised at the moment, with the more well-developed polymeric systems in advanced clinical trials being the closest to providing true insight into whether their future will be bright. The number of research teams developing new and exciting approaches has grown rapidly in the last decade, and it is only a matter of time before the more recently created systems make it into advanced clinical trials and towards approved therapies. Whether these strategies will bear fruit remains to be determined, however, as there is much we have to discern about how these systems interact with complex, dynamic biological systems.
Self-assembled entities are generally governed by equilibrium processes, with their critical aggregation concentration indicating how strongly this equilibrium lies towards the assembled state. In solution by itself, concentration will be the deciding factor on the stability of the structure formed. In the body, however, interactions with other biomolecules could potentially disrupt the structure and lead to disassembly and unwanted metabolism. Take camptothecin, for instance, which exists in a pH-dependent equilibrium between the active lactone form and the inactive carboxylate. During circulation, interactions with serum albumins results in a shift towards the carboxylate form since this interacts more strongly with the protein.232 The same could be true for self-assembled prodrugs, with a strong interaction with plasma proteins drawing the monomers out of the assembled structures and leading to faster dissociation. Consequently, greater knowledge of how such assembled systems interact with biomolecules they are likely to encounter would be advantageous to inform future designs. In the interim, low CAC values would likely confer a greater degree of protection and self-immolative linkers can help overcome the associated lower rate of dissociation to release the drug at the target site.
One clear advantage to self-assembling prodrugs as a whole is that there is a great deal of versatility offered by the various designs. Polymer–drug conjugates and drug–conjugates (via nanoprecipitation) are well suited to systemic administration routes due to their mostly spherical morphology. The hydrogelators that assemble through enzymatic modification are ideal for local delivery applications. Drug amphiphiles, while still in their infancy, straddle both administration routes owing to the flexibility in molecular design that can influence the morphological outcome. As already noted, however, only polymer–drug conjugates have so far progressed into advanced clinical trials and there is much work to be done assessing both the suitability and effectiveness of self-assembling small molecule prodrugs in an in vivo environment. Nevertheless, the broad potential on offer makes the investment of time and resources worthwhile. The knowledge and experience gained will no doubt be of benefit to not just drug delivery, but also to imaging and diagnostic applications since these can rely on the delivery of molecular agents to specific sites. We look forward to the exciting new opportunities that will present themselves in the coming years.
Acknowledgments
We acknowledge financial support from the National Science Foundation (DMR 1255281), the National Institutes of Health (NIH/R21CA191740), and the Johns Hopkins Catalyst Award.
References
- 1.Albert A. Nature. 1958;182:421–423. doi: 10.1038/182421a0. [DOI] [PubMed] [Google Scholar]
- 2.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Nature Reviews Drug Discovery. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 3.Kevin B, Robert W, Iain G, Kevin D. Current Drug Metabolism. 2003;4:461–485. doi: 10.2174/1389200033489253. [DOI] [PubMed] [Google Scholar]
- 4.Stella VJ, Nti-Addae KW. Advanced Drug Delivery Reviews. 2007;59:677–694. doi: 10.1016/j.addr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Simplício LA, Clancy MJ, Gilmer FJ. Molecules. 2008;13 doi: 10.3390/molecules13030519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han H-K, Amidon GL. AAPS PharmSci. 2000;2:48–58. doi: 10.1208/ps020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang C, Tirucherai GS, Mitra AK. Expert Opinion on Biological Therapy. 2001;1:159–175. doi: 10.1517/14712598.1.2.159. [DOI] [PubMed] [Google Scholar]
- 8.Giang I, Boland EL, Poon GMK. The AAPS Journal. 2014;16:899–913. doi: 10.1208/s12248-014-9638-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riber CF, Smith AAA, Zelikin AN. Advanced Healthcare Materials. 2015;4:1887–1890. doi: 10.1002/adhm.201500344. [DOI] [PubMed] [Google Scholar]
- 10.Choi KY, Swierczewska M, Lee S, Chen X. Theranostics. 2012;2:156–178. doi: 10.7150/thno.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liederer BM, Borchardt RT. Journal of Pharmaceutical Sciences. 95:1177–1195. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]
- 12.Gomes P, Vale N, Moreira R. Molecules. 2007;12 doi: 10.3390/12112484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clas S-D, Sanchez RI, Nofsinger R. Drug Discovery Today. 2014;19:79–87. doi: 10.1016/j.drudis.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Landis MS. Therapeutic Delivery. 2013;4:225–237. doi: 10.4155/tde.12.150. [DOI] [PubMed] [Google Scholar]
- 15.Croy SR, Kwon GS. Current Pharmaceutical Design. 2006;12:4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad Z, Shah A, Siddiq M, Kraatz H-B. RSC Advances. 2014;4:17028–17038. [Google Scholar]
- 17.Pattni BS, Chupin VV, Torchilin VP. Chemical Reviews. 2015;115:10938–10966. doi: 10.1021/acs.chemrev.5b00046. [DOI] [PubMed] [Google Scholar]
- 18.Medina SH, El-Sayed MEH. Chemical Reviews. 2009;109:3141–3157. doi: 10.1021/cr900174j. [DOI] [PubMed] [Google Scholar]
- 19.Matsumura Y, Maeda H. Cancer Research. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 20.Torchilin V. Advanced Drug Delivery Reviews. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Duncan R. Nature Reviews Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 22.Duncan R, Kopeckova-Rejmanova P, Strohalm J, Hume I, Cable HC, Pohl J, Lloyd JB, Kopecek J. British Journal of Cancer. 1987;55:165–174. doi: 10.1038/bjc.1987.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ulbrich K, Koňák Č, Tuzar Z, Kopeček J. Die Makromolekulare Chemie. 1987;188:1261–1272. [Google Scholar]
- 24.Yokoyama M, Miyauchi M, Yamada N, Okano T, Sakurai Y, Kataoka K, Inoue S. Cancer Research. 1990;50:1693–1700. [PubMed] [Google Scholar]
- 25.Yokoyama M, Okano T, Sakurai Y, Kataoka K. Journal of Controlled Release. 1994;32:269–277. [Google Scholar]
- 26.Kwon GS, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Pharmaceutical Research. 1993;10:970–974. doi: 10.1023/a:1018998203127. [DOI] [PubMed] [Google Scholar]
- 27.Kwon G, Suwa S, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Journal of Controlled Release. 1994;29:17–23. [Google Scholar]
- 28.Yokoyama M, Kwon GS, Okano T, Sakurai Y, Ekimoto H, Okamoto K, Mashiba H, Seto T, Kataoka K. Drug Delivery. 1993;1:11–19. [Google Scholar]
- 29.Yokoyama M, Fukushima S, Uehara R, Okamoto K, Kataoka K, Sakurai Y, Okano T. Journal of Controlled Release. 1998;50:79–92. doi: 10.1016/s0168-3659(97)00115-6. [DOI] [PubMed] [Google Scholar]
- 30.Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kataoka K. Journal of Drug Targeting. 1999;7:171–186. doi: 10.3109/10611869909085500. [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi T, Fukushima S, Okamoto K, Suzuki M, Matsumura Y, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Journal of Controlled Release. 2001;74:295–302. doi: 10.1016/s0168-3659(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 32.Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Watanabe N. Br J Cancer. 2004;91:1775–1781. doi: 10.1038/sj.bjc.6602204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsukioka Y, Matsumura Y, Hamaguchi T, Koike H, Moriyasu F, Kakizoe T. Jpn. J. Cancer Res. 2002;93:1145–1153. doi: 10.1111/j.1349-7006.2002.tb01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bae Y, Fukushima S, Harada A, Kataoka K. Angewandte Chemie International Edition. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]
- 35.Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Bioconjugate Chemistry. 2005;16:122–130. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
- 36.Yokoyama M, Okano T, Sakurai Y, Suwa S, Kataoka K. Journal of Controlled Release. 1996;39:351–356. [Google Scholar]
- 37.Nishiyama N, Yokoyama M, Aoyagi T, Okano T, Sakurai Y, Kataoka K. Langmuir. 1999;15:377–383. [Google Scholar]
- 38.Nishiyama N, Kataoka K. Journal of Controlled Release. 2001;74:83–94. doi: 10.1016/s0168-3659(01)00314-5. [DOI] [PubMed] [Google Scholar]
- 39.Nishiyama N, Kato Y, Sugiyama Y, Kataoka K. Pharmaceutical Research. 2001;18:1035–1041. doi: 10.1023/a:1010908916184. [DOI] [PubMed] [Google Scholar]
- 40.Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K. Cancer Research. 2003;63:8977–8983. [PubMed] [Google Scholar]
- 41.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. British Journal of Cancer. 2005;93:678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Endo K, Ueno T, Kondo S, Wakisaka N, Murono S, Ito M, Kataoka K, Kato Y, Yoshizaki T. Cancer Science. 2013;104:369–374. doi: 10.1111/cas.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ, Matsumura Y, Kataoka K, Nishiya T. British Journal of Cancer. 2011;104:593–598. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doi T, Hamaguchi T, Shitara K, Iwasa S, Shimada Y, Harada M, Naito K, Hayashi N, Masada A, Ohtsu A. Cancer Chemotherapy and Pharmacology. 2017;79:569–578. doi: 10.1007/s00280-017-3254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabral H, Nishiyama N, Okazaki S, Koyama H, Kataoka K. Journal of Controlled Release. 2005;101:223–232. doi: 10.1016/j.jconrel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 46.Ueno T, Endo K, Hori K, Ozaki N, Tsuji A, Kondo S, Wakisaka N, Murono S, Kataoka K, Kato Y, Yoshizaki T. International Journal of Nanomedicine. 2014;9:3005–3012. doi: 10.2147/IJN.S60564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S-i, Hamaguchi T, Matsumura Y. Cancer Research. 2006;66:10048. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 48.Slatter JG, Schaaf LJ, Sams JP, Feenstra KL, Johnson MG, Bombardt PA, Cathcart KS, Verburg MT, Pearson LK, Compton LD, Miller LL, Baker DS, Pesheck CV, Lord RS. Drug Metabolism and Disposition. 2000;28:423. [PubMed] [Google Scholar]
- 49.Rothenberg ML, Kuhn JG, Burris HA, Nelson J, Eckardt JR, Tristan-Morales M, Hilsenbeck SG, Weiss GR, Smith LS, Rodriguez GI. Journal of Clinical Oncology. 1993;11:2194–2204. doi: 10.1200/JCO.1993.11.11.2194. [DOI] [PubMed] [Google Scholar]
- 50.Kuroda J-i, Kuratsu J-i, Yasunaga M, Koga Y, Saito Y, Matsumura Y. International Journal of Cancer. 2009;124:2505–2511. doi: 10.1002/ijc.24171. [DOI] [PubMed] [Google Scholar]
- 51.Hamaguchi T, Doi T, Eguchi-Nakajima T, Kato K, Yamada Y, Shimada Y, Fuse N, Ohtsu A, Matsumoto S, Takanashi M, Matsumura Y. Clinical Cancer Research. 2010;16:5058–5066. doi: 10.1158/1078-0432.CCR-10-0387. [DOI] [PubMed] [Google Scholar]
- 52.Singer JW. Journal of Controlled Release. 2005;109:120–126. doi: 10.1016/j.jconrel.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 53.Gradishar WJ. Expert Opinion on Pharmacotherapy. 2006;7:1041–1053. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- 54.Xie Z, Guan H, Chen X, Lu C, Chen L, Hu X, Shi Q, Jing X. Journal of Controlled Release. 2007;117:210–216. doi: 10.1016/j.jconrel.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 55.Gu Y, Zhong Y, Meng F, Cheng R, Deng C, Zhong Z. Biomacromolecules. 2013;14:2772–2780. doi: 10.1021/bm400615n. [DOI] [PubMed] [Google Scholar]
- 56.Xu ZG, Zhang KL, Hou CL, Wang DD, Liu XY, Guan XJ, Zhang XY, Zhang HX. J Mater Chem B. 2014;2:3433–3437. doi: 10.1039/c4tb00128a. [DOI] [PubMed] [Google Scholar]
- 57.Hu X, Li J, Lin W, Huang Y, Jing X, Xie Z. RSC Advances. 2014;4:38405–38411. [Google Scholar]
- 58.Liu J, Zahedi P, Zeng F, Allen C. Journal of Pharmaceutical Sciences. 2008;97:3274–3290. doi: 10.1002/jps.21245. [DOI] [PubMed] [Google Scholar]
- 59.Fan N, Duan K, Wang C, Liu S, Luo S, Yu J, Huang J, Li Y, Wang D. Colloids and Surfaces B: Biointerfaces. 2010;75:543–549. doi: 10.1016/j.colsurfb.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 60.Tong R, Cheng JJ. Polym Rev. 2007;47:345–381. [Google Scholar]
- 61.Tong R, Cheng JJ. Angew Chem Int Edit. 2008;47:4830–4834. doi: 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]
- 62.Tong R, Cheng JJ. J Am Chem Soc. 2009;131:4744–4754. doi: 10.1021/ja8084675. [DOI] [PubMed] [Google Scholar]
- 63.Tong R, Gabrielson NP, Fan TM, Cheng JJ. Curr Opin Solid St M. 2012;16:323–332. doi: 10.1016/j.cossms.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tong R, Tang L, Ma L, Tu CL, Baumgartner R, Cheng JJ. Chem Soc Rev. 2014;43:6982–7012. doi: 10.1039/c4cs00133h. [DOI] [PubMed] [Google Scholar]
- 65.Liao LY, Liu J, Dreaden EC, Morton SW, Shopsowitz KE, Hammond PT, Johnson JA. J Am Chem Soc. 2014;136:5896–5899. doi: 10.1021/ja502011g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu JY, Liu WE, Weitzhandler I, Bhattacharyya J, Li XH, Wang J, Qi YZ, Bhattacharjee S, Chilkoti A. Angew Chem Int Edit. 2015;54:1002–1006. doi: 10.1002/anie.201409293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Bioconjugate Chemistry. 2013;24:1161–1173. doi: 10.1021/bc400032x. [DOI] [PubMed] [Google Scholar]
- 68.Wang W, Li C, Zhang J, Dong A, Kong D. J Mater Chem B. 2014;2:1891–1901. doi: 10.1039/c3tb21558j. [DOI] [PubMed] [Google Scholar]
- 69.Senanayake TH, Warren G, Wei X, Vinogradov SV. Journal of Controlled Release. 2013;167:200–209. doi: 10.1016/j.jconrel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Senanayake TH, Warren G, Vinogradov SV. Bioconjugate Chemistry. 2011;22:1983–1993. doi: 10.1021/bc200173e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalia J, Raines RT. Angewandte Chemie International Edition. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi A, Yamamoto Y, Yasunaga M, Koga Y, Kuroda J-i, Takigahira M, Harada M, Saito H, Hayashi T, Kato Y, Kinoshita T, Ohkohchi N, Hyodo I, Matsumura Y. Cancer Science. 2013;104:920–925. doi: 10.1111/cas.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Howard MD, Ponta A, Eckman A, Jay M, Bae Y. Pharmaceutical Research. 2011;28:2435–2446. doi: 10.1007/s11095-011-0470-1. [DOI] [PubMed] [Google Scholar]
- 74.Zhang S, Zou J, Elsabahy M, Karwa A, Li A, Moore DA, Dorshow RB, Wooley KL. Chemical Science. 2013;4:2122–2126. doi: 10.1039/C3SC50252J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun C-Y, Dou S, Du J-Z, Yang X-Z, Li Y-P, Wang J. Advanced Healthcare Materials. 2014;3:261–272. doi: 10.1002/adhm.201300091. [DOI] [PubMed] [Google Scholar]
- 76.Duncan R, Cable HC, Lloyd JB, Rejmanova P, Kopecek J. Makromolekulare Chemie-Macromolecular Chemistry and Physics. 1983;184:1997–2008. [Google Scholar]
- 77.Rejmanova P, Kopecek J, Pohl J, Baudys M, Kostka V. Makromolekulare Chemie-Macromolecular Chemistry and Physics. 1983;184:2009–2020. [Google Scholar]
- 78.Yang Y, Pan D, Luo K, Li L, Gu Z. Biomaterials. 2013;34:8430–8443. doi: 10.1016/j.biomaterials.2013.07.037. [DOI] [PubMed] [Google Scholar]
- 79.Yin Win K, Feng S-S. Biomaterials. 2005;26:2713–2722. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 80.Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C. Oxidative Medicine and Cellular Longevity. 2013;2013:10. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lv S, Tang Z, Zhang D, Song W, Li M, Lin J, Liu H, Chen X. Journal of Controlled Release. 2014;194:220–227. doi: 10.1016/j.jconrel.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 82.Xu Z, Wang D, Xu S, Liu X, Zhang X, Zhang H. Chemistry – An Asian Journal. 2014;9:199–205. doi: 10.1002/asia.201301030. [DOI] [PubMed] [Google Scholar]
- 83.Wang Y, Luo Q, Gao L, Gao C, Du H, Zha G, Li X, Shen Z, Zhu W. Biomaterials Science. 2014;2:1367–1376. doi: 10.1039/c4bm00065j. [DOI] [PubMed] [Google Scholar]
- 84.Foster JC, Radzinski SC, Zou X, Finkielstein CV, Matson JB. Molecular Pharmaceutics. 2017;14:1300–1306. doi: 10.1021/acs.molpharmaceut.6b01117. [DOI] [PubMed] [Google Scholar]
- 85.Foster JC, Powell CR, Radzinski SC, Matson JB. Organic Letters. 2014;16:1558–1561. doi: 10.1021/ol500385a. [DOI] [PubMed] [Google Scholar]
- 86.Bensaid F, Thillaye du Boullay O, Amgoune A, Pradel C, Harivardhan Reddy L, Didier E, Sablé S, Louit G, Bazile D, Bourissou D. Biomacromolecules. 2013;14:1189–1198. doi: 10.1021/bm400161g. [DOI] [PubMed] [Google Scholar]
- 87.Sui B, Xu H, Jin J, Gou J, Liu J, Tang X, Zhang Y, Xu J, Zhang H, Jin X. Molecules. 2014;19:11915–11932. doi: 10.3390/molecules190811915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei X, Luo Q, Sun L, Li X, Zhu H, Guan P, Wu M, Luo K, Gong Q. ACS Applied Materials & Interfaces. 2016;8:11765–11778. doi: 10.1021/acsami.6b02006. [DOI] [PubMed] [Google Scholar]
- 89.Hu X, Liu G, Li Y, Wang X, Liu S. J Am Chem Soc. 2015;137:362–368. doi: 10.1021/ja5105848. [DOI] [PubMed] [Google Scholar]
- 90.Mai Y, Eisenberg A. Chem Soc Rev. 2012;41:5969–5985. doi: 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- 91.Hu X, Hu J, Tian J, Ge Z, Zhang G, Luo K, Liu S. J Am Chem Soc. 2013;135:17617–17629. doi: 10.1021/ja409686x. [DOI] [PubMed] [Google Scholar]
- 92.Bae Y, Alani AWG, Rockich NC, Lai TSZC, Kwon GS. Pharmaceutical Research. 2010;27:2421–2432. doi: 10.1007/s11095-010-0234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Markovsky E, Baabur-Cohen H, Satchi-Fainaro R. Journal of Controlled Release. 2014;187:145–157. doi: 10.1016/j.jconrel.2014.05.025. [DOI] [PubMed] [Google Scholar]
- 94.Zhou D, Xiao H, Meng F, Li X, Li Y, Jing X, Huang Y. Advanced Healthcare Materials. 2013;2:822–827. doi: 10.1002/adhm.201200385. [DOI] [PubMed] [Google Scholar]
- 95.Wang E, Xiong H, Zhou D, Xie Z, Huang Y, Jing X, Sun X. Macromolecular Bioscience. 2014;14:588–596. doi: 10.1002/mabi.201300402. [DOI] [PubMed] [Google Scholar]
- 96.Clementi C, Miller K, Mero A, Satchi-Fainaro R, Pasut G. Molecular Pharmaceutics. 2011;8:1063–1072. doi: 10.1021/mp2001445. [DOI] [PubMed] [Google Scholar]
- 97.Miller K, Clementi C, Polyak D, Eldar-Boock A, Benayoun L, Barshack I, Shaked Y, Pasut G, Satchi-Fainaro R. Biomaterials. 2013;34:3795–3806. doi: 10.1016/j.biomaterials.2013.01.052. [DOI] [PubMed] [Google Scholar]
- 98.Ye W-L, Zhao Y-P, Li H-Q, Na R, Li F, Mei Q-B, Zhao M-G, Zhou S-Y. Scientific Reports. 2015;5:14614. doi: 10.1038/srep14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Das M, Jain R, Agrawal AK, Thanki K, Jain S. Bioconjugate Chemistry. 2014;25:501–509. doi: 10.1021/bc400477q. [DOI] [PubMed] [Google Scholar]
- 100.Li Y, Dong H, Li X, Shi D, Li Y. Medicinal Chemistry. 2014;4:672–683. [Google Scholar]
- 101.Jung B, Jeong Y-C, Min J-H, Kim J-E, Song Y-J, Park J-K, Park J-H, Kim J-D. Journal of Materials Chemistry. 2012;22:9385–9394. [Google Scholar]
- 102.Jäger E, Jäger A, Chytil P, Etrych T, Říhová B, Giacomelli FC, Štěpánek P, Ulbrich K. Journal of Controlled Release. 2013;165:153–161. doi: 10.1016/j.jconrel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 103.Senanayake TH, Lu Y, Bohling A, Raja S, Band H, Vinogradov SV. Pharmaceutical Research. 2014;31:1605–1615. doi: 10.1007/s11095-013-1265-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang D, Van S, Jiang X, Yu L. International Journal of Nanomedicine. 2011;6:85–91. doi: 10.2147/IJN.S15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu J, Chuan X, Zhang H, Dai W, Wang X, Wang X, Zhang Q. International Journal of Pharmaceutics. 2014;471:525–535. doi: 10.1016/j.ijpharm.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 106.MacEwan SR, Chilkoti A. Peptide Science. 2010;94:60–77. doi: 10.1002/bip.21327. [DOI] [PubMed] [Google Scholar]
- 107.Navon Y, Bitton R. Israel Journal of Chemistry. 2016;56:581–589. [Google Scholar]
- 108.MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Nature materials. 2009;8:993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ernsting MJ, Tang W-L, MacCallum NW, Li S-D. Biomaterials. 2012;33:1445–1454. doi: 10.1016/j.biomaterials.2011.10.061. [DOI] [PubMed] [Google Scholar]
- 110.Furgeson DY, Dreher MR, Chilkoti A. Journal of Controlled Release. 2006;110:362–369. doi: 10.1016/j.jconrel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 111.McDaniel JR, Bhattacharyya J, Vargo KB, Hassouneh W, Hammer DA, Chilkoti A. Angewandte Chemie International Edition. 2013;52:1683–1687. doi: 10.1002/anie.201200899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu J, Xie L, Zhao W, Sun M, Liu X, Gao W. Chemical Communications. 2015;51:11405–11408. doi: 10.1039/c5cc04035c. [DOI] [PubMed] [Google Scholar]
- 113.McDaniel JR, Macewan SR, Dewhirst M, Chilkoti A. Journal of Controlled Release. 2012;159:362–367. doi: 10.1016/j.jconrel.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McDaniel JR, MacEwan SR, Li X, Radford DC, Landon CD, Dewhirst M, Chilkoti A. Nano letters. 2014;14:2890–2895. doi: 10.1021/nl5009376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mastria EM, Chen M, McDaniel JR, Li X, Hyun J, Dewhirst MW, Chilkoti A. Journal of Controlled Release. 2015;208:52–58. doi: 10.1016/j.jconrel.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li X, Lin C-C, Chi J-TA, Chilkoti A. Nature Communications. 2015;6:7939. doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z. Advanced Drug Delivery Reviews. 2008;60:1650–1662. doi: 10.1016/j.addr.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 118.Son YJ, Jang J-S, Cho YW, Chung H, Park R-W, Kwon IC, Kim I-S, Park JY, Seo SB, Park CR, Jeong SY. Journal of Controlled Release. 2003;91:135–145. doi: 10.1016/s0168-3659(03)00231-1. [DOI] [PubMed] [Google Scholar]
- 119.Hyung Park J, Kwon S, Lee M, Chung H, Kim J-H, Kim Y-S, Park R-W, Kim I-S, Bong Seo S, Kwon IC, Young Jeong S. Biomaterials. 2006;27:119–126. doi: 10.1016/j.biomaterials.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 120.Park JH, Cho YW, Son YJ, Kim K, Chung H, Jeong SY, Choi K, Park CR, Park R-W, Kim I-S, Kwon IC. Colloid and Polymer Science. 2006;284:763–770. [Google Scholar]
- 121.Quiñones JP, Gothelf KV, Kjems J, Caballero ÁMH, Schmidt C, Covas CP. Carbohydrate Polymers. 2012;88:1373–1377. [Google Scholar]
- 122.Quiñones JP, Gothelf KV, Kjems J, Yang C, Caballero AMH, Schmidt C, Covas CP. Carbohydrate Polymers. 2013;92:856–864. doi: 10.1016/j.carbpol.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 123.Quiñones JP, Gothelf KV, Kjems J, Heras A, Schmidt C, Peniche C. Journal of Biomaterials and Tissue Engineering. 2013;3:164–172. [Google Scholar]
- 124.Liu F, Feng L, Zhang L, Zhang X, Zhang N. International Journal of Pharmaceutics. 2013;451:41–49. doi: 10.1016/j.ijpharm.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 125.Wang Y, Yang X, Yang J, Wang Y, Chen R, Wu J, Liu Y, Zhang N. Carbohydrate Polymers. 2011;86:1665–1670. [Google Scholar]
- 126.Upadhyaya L, Singh J, Agarwal V, Tewari RP. Carbohydrate Polymers. 2013;91:452–466. doi: 10.1016/j.carbpol.2012.07.076. [DOI] [PubMed] [Google Scholar]
- 127.Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. Journal of controlled release. 2007;124:20–27. doi: 10.1016/j.jconrel.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 128.Couvreur P, Stella B, Reddy LH, Hillaireau H, Dubernet C, Desmaële D, Lepêtre-Mouelhi S, Rocco F, Dereuddre-Bosquet N, Clayette P. Nano letters. 2006;6:2544–2548. doi: 10.1021/nl061942q. [DOI] [PubMed] [Google Scholar]
- 129.Lepeltier E, Bourgaux C, Couvreur P. Advanced drug delivery reviews. 2014;71:86–97. doi: 10.1016/j.addr.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 130.Yan Y, Huang J, Tang BZ. Chemical Communications. 2016;52:11870–11884. doi: 10.1039/c6cc03620a. [DOI] [PubMed] [Google Scholar]
- 131.Kumar V, Prud’homme RK. Chemical Engineering Science. 2009;64:1358–1361. [Google Scholar]
- 132.Ganachaud F, Katz JL. ChemPhysChem. 2005;6:209–216. doi: 10.1002/cphc.200400527. [DOI] [PubMed] [Google Scholar]
- 133.Sémiramoth N, Meo CD, Zouhiri F, Saïd-Hassane F, Valetti S, Gorges R, Nicolas V, Poupaert JH, Chollet-Martin S, Desmaële D, Gref R, Couvreur P. ACS nano. 2012;6:3820–3831. doi: 10.1021/nn204928v. [DOI] [PubMed] [Google Scholar]
- 134.Dosio F, Reddy LH, Ferrero A, Stella B, Cattel L, Couvreur P. Bioconjugate chemistry. 2010;21:1349–1361. doi: 10.1021/bc100154g. [DOI] [PubMed] [Google Scholar]
- 135.Maksimenko A, Dosio F, Mougin J, Ferrero A, Wack S, Reddy LH, Weyn A-A, Lepeltier E, Bourgaux C, Stella B. Proceedings of the National Academy of Sciences. 2014;111:E217–E226. doi: 10.1073/pnas.1313459110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gaudin A, Yemisci M, Eroglu H, Lepetre-Mouelhi S, Turkoglu OF, Dönmez-Demir B, Caban S, Sargon MF, Garcia-Argote S, Pieters G, Loreau O, Rousseau B, Tagit O, Hildebrandt N, Le Dantec Y, Mougin J, Valetti S, Chacun H, Nicolas V, Desmaële D, Andrieux K, Capan Y, Dalkara T, Couvreur P. Nature Nanotechnology. 2014;9:1054–1062. doi: 10.1038/nnano.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cheikh-Ali Z, Caron J, Cojean S, Bories C, Couvreur P, Loiseau PM, Desmaële D, Poupon E, Champy P. ChemMedChem. 2015;10:411–418. doi: 10.1002/cmdc.201402449. [DOI] [PubMed] [Google Scholar]
- 138.Jin Y, Qiao Y, Li M, Ai P, Hou X. Colloids and Surfaces B: Biointerfaces. 2005;42:45–51. doi: 10.1016/j.colsurfb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 139.Jin Y, Tong L, Ai P, Li M, Hou X. International journal of pharmaceutics. 2006;309:199–207. doi: 10.1016/j.ijpharm.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 140.Jin Y, Lian Y, Du L, Wang S, Su C, Gao C. International journal of pharmaceutics. 2012;430:276–281. doi: 10.1016/j.ijpharm.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 141.Li M, Qi S, Jin Y, Dong J. International journal of pharmaceutics. 2015;478:124–130. doi: 10.1016/j.ijpharm.2014.11.033. [DOI] [PubMed] [Google Scholar]
- 142.Du L, Wu L, Jin Y, Jia J, Li M, Wang Y. International journal of pharmaceutics. 2014;472:1–9. doi: 10.1016/j.ijpharm.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 143.Zhang C, Jin S, Xue X, Zhang T, Jiang Y, Wang PC, Liang X-J. J Mater Chem B. 2016;4:3286–3291. doi: 10.1039/c6tb00612d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu J, Zhao D, He W, Zhang H, Li Z, Luan Y. Journal of Colloid and Interface Science. 2017;487:239–249. doi: 10.1016/j.jcis.2016.10.041. [DOI] [PubMed] [Google Scholar]
- 145.Shen Y, Jin E, Zhang B, Murphy CJ, Sui M, Zhao J, Wang J, Tang J, Fan M, Van Kirk E. J Am Chem Soc. 2010;132:4259–4265. doi: 10.1021/ja909475m. [DOI] [PubMed] [Google Scholar]
- 146.Xu Z, Hou M, Shi X, Gao Y-E, Xue P, Liu S, Kang Y. Biomaterials Science. 2017 doi: 10.1039/c6bm00800c. [DOI] [PubMed] [Google Scholar]
- 147.Wang H, Xie H, Wang J, Wu J, Ma X, Li L, Wei X, Ling Q, Song P, Zhou L, Xu X, Zheng S. Advanced Functional Materials. 2015;25:4956–4965. [Google Scholar]
- 148.Zhong T, Yao X, Zhang S, Guo Y, Duan X-C, Ren W, Dan H, Yin Y-F, Zhang X. 2016;6:36614. doi: 10.1038/srep36614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Patil S, Patil S, Gawali S, Shende S, Jadhav S, Basu S. RSC Advances. 2013;3:19760–19764. [Google Scholar]
- 150.Fan M, Liang X, Li Z, Wang H, Yang D, Shi B. European Journal of Pharmaceutical Sciences. 2015;79:20–26. doi: 10.1016/j.ejps.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 151.Huang P, Wang D, Su Y, Huang W, Zhou Y, Cui D, Zhu X, Yan D. J Am Chem Soc. 2014;136:11748–11756. doi: 10.1021/ja505212y. [DOI] [PubMed] [Google Scholar]
- 152.Duhem N, Danhier F, Pourcelle V, Schumers J-M, Bertrand O, LeDuff CcS, Hoeppener S, Schubert US, Gohy J-Fo, Marchand-Brynaert J. Bioconjugate chemistry. 2013;25:72–81. doi: 10.1021/bc400326y. [DOI] [PubMed] [Google Scholar]
- 153.Hu M, Huang P, Wang Y, Su Y, Zhou L, Zhu X, Yan D. Bioconjugate chemistry. 2015;26:2497–2506. doi: 10.1021/acs.bioconjchem.5b00513. [DOI] [PubMed] [Google Scholar]
- 154.He D, Zhang W, Deng H, Huo S, Wang Y-F, Gong N, Deng L, Liang X-J, Dong A. Chemical Communications. 2016;52:14145–14148. doi: 10.1039/c6cc07595a. [DOI] [PubMed] [Google Scholar]
- 155.Jin Y, Xin R, Tong L, Du L, Li M. Molecular pharmaceutics. 2011;8:867–876. doi: 10.1021/mp100457d. [DOI] [PubMed] [Google Scholar]
- 156.Wang Z, Zhuang M, Sun T, Wang X, Xie Z. Bioorganic & Medicinal Chemistry Letters. 2017;27:2493–2496. doi: 10.1016/j.bmcl.2017.03.101. [DOI] [PubMed] [Google Scholar]
- 157.Pei Q, Hu X, Li Z, Xie Z, Jing X. Rsc Advances. 2015;5:81499–81501. [Google Scholar]
- 158.Gao Y, Kuang Y, Guo Z-F, Guo Z, Krauss IJ, Xu B. J Am Chem Soc. 2009;131:13576–13577. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 159.Du X, Zhou J, Shi J, Xu B. Chemical Reviews. 2015;115:13165–13307. doi: 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu H, Li Y, Lyu Z, Wan Y, Li X, Chen H, Chen H, Li X. J Mater Chem B. 2014;2:8303–8309. doi: 10.1039/c4tb01563k. [DOI] [PubMed] [Google Scholar]
- 161.Li J, Kuang Y, Shi J, Gao Y, Zhou J, Xu B. Beilstein journal of organic chemistry. 2013;9:908–917. doi: 10.3762/bjoc.9.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Li J, Kuang Y, Gao Y, Du X, Shi J, Xu B. J Am Chem Soc. 2012;135:542–545. doi: 10.1021/ja310019x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang C, Wang Z, Ou C, Chen M, Wang L, Yang Z. Chemical Communications. 2014;50:9413–9415. doi: 10.1039/c4cc03139c. [DOI] [PubMed] [Google Scholar]
- 164.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mao L, Wang H, Tan M, Ou L, Kong D, Yang Z. Chemical Communications. 2012;48:395–397. doi: 10.1039/c1cc16250k. [DOI] [PubMed] [Google Scholar]
- 166.Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, Xu B. Chem. 2016;1:246–263. doi: 10.1016/j.chempr.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang H, Yang C, Wang L, Kong D, Zhang Y, Yang Z. Chemical Communications. 2011;47:4439–4441. doi: 10.1039/c1cc10506j. [DOI] [PubMed] [Google Scholar]
- 168.Wang H, Wei J, Yang C, Zhao H, Li D, Yin Z, Yang Z. Biomaterials. 2012;33:5848–5853. doi: 10.1016/j.biomaterials.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 169.Yang C, Bian M, Yang Z. Biomaterials Science. 2014;2:651–654. doi: 10.1039/c3bm60252d. [DOI] [PubMed] [Google Scholar]
- 170.Kuang Y, Du X, Zhou J, Xu B. Advanced healthcare materials. 2014;3:1217–1221. doi: 10.1002/adhm.201300645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li J, Kuang Y, Shi J, Zhou J, Medina JE, Zhou R, Yuan D, Yang C, Wang H, Yang Z, Liu J, Dinulescu DM, Xu B. Angewandte Chemie International Edition. 2015;54:13307–13311. doi: 10.1002/anie.201507157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang H, Feng Z, Wang Y, Zhou R, Yang Z, Xu B. J Am Chem Soc. 2016 [Google Scholar]
- 173.Zhou J, Li J, Du X, Xu B. Biomaterials. 2017;129:1–27. doi: 10.1016/j.biomaterials.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Peppas N, Bures P, Leobandung W, Ichikawa H. European journal of pharmaceutics and biopharmaceutics. 2000;50:27–46. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 175.Anderson CF, Cui H. Industrial & Engineering Chemistry Research. 2017;56:5761–5777. doi: 10.1021/acs.iecr.7b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li Y, Wang F, Cui H. Bioengineering & Translational Medicine. 2016 doi: 10.1002/btm2.10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Su H, Wang Y, Anderson CF, Koo JM, Wang H, Cui H. Chinese Journal of Polymer Science. 2017;35:1194–1211. [Google Scholar]
- 178.Ma W, Cheetham AG, Cui H. Nano Today. 2016;11:13–30. doi: 10.1016/j.nantod.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chakroun RW, Zhang P, Lin R, Schiapparelli P, Quinones-Hinojosa A, Cui H. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2017 doi: 10.1002/wnan.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lin R, Cui H. Current Opinion in Chemical Engineering. 2015;7:75–83. [Google Scholar]
- 181.Su H, Koo JM, Cui H. Journal of Controlled Release. 2015;219:383–395. doi: 10.1016/j.jconrel.2015.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Advanced drug delivery reviews. 2017;110:112–126. doi: 10.1016/j.addr.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cheetham AG, Zhang P, Lin Y-A, Lock LL, Cui H. J Am Chem Soc. 2013;135:2907–2910. doi: 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lock LL, LaComb M, Schwarz K, Cheetham AG, Lin Y-a, Zhang P, Cui H. Faraday discussions. 2013;166:285–301. doi: 10.1039/c3fd00099k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang S. Nature biotechnology. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 186.Cui H, Webber MJ, Stupp SI. Peptide Science. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hu Y, Lin R, Patel K, Cheetham AG, Kan C, Cui H. Coordination Chemistry Reviews. 2016;320:2–17. [Google Scholar]
- 188.Cheetham AG, Ou Y-C, Zhang P, Cui H. Chemical Communications. 2014;50:6039–6042. doi: 10.1039/c3cc49453e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cai S, Vijayan K, Cheng D, Lima EM, Discher DE. Pharmaceutical research. 2007;24:2099–2109. doi: 10.1007/s11095-007-9335-z. [DOI] [PubMed] [Google Scholar]
- 190.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nature nanotechnology. 2007;2:249. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Christian DA, Cai S, Garbuzenko OB, Harada T, Zajac AL, Minko T, Discher DE. Molecular pharmaceutics. 2009;6:1343–1352. doi: 10.1021/mp900022m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lock LL, Reyes CD, Zhang P, Cui H. J Am Chem Soc. 2016;138:3533–3540. doi: 10.1021/jacs.6b00073. [DOI] [PubMed] [Google Scholar]
- 193.Kang M, Cui H, Loverde S. Soft Matter. 2017 doi: 10.1039/c7sm00943g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kang M, Zhang P, Cui H, Loverde SM. Macromolecules. 2016;49:994–1001. doi: 10.1021/acs.macromol.5b02148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Su H, Zhang P, Cheetham A, Koo J, Lin R, Masood A, Schiapparelli P, Quiñones-Hinojosa A, Cui H. Theranostics. 2016;6:1065–1074. doi: 10.7150/thno.15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lin Y-A, Cheetham AG, Zhang P, Ou Y-C, Li Y, Liu G, Hermida-Merino D, Hamley IW, Cui H. ACS nano. 2014;8:12690–12700. doi: 10.1021/nn505688b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cheetham AG, Lin Y-A, Lin R, Cui H. Acta Pharmacologica Sinica. 2017;38:874–884. doi: 10.1038/aps.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tan X, Li BB, Lu X, Jia F, Santori C, Menon P, Li H, Zhang B, Zhao JJ, Zhang K. J Am Chem Soc. 2015;137:6112–6115. doi: 10.1021/jacs.5b00795. [DOI] [PubMed] [Google Scholar]
- 199.Lin R, Cheetham AG, Zhang P, Lin Y-A, Cui H. Chemical Communications. 2013;49:4968–4970. doi: 10.1039/c3cc41896k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cheetham AG, Zhang P, Lin Y-A, Lin R, Cui H. J Mater Chem B. 2014;2:7316–7326. doi: 10.1039/C4TB01084A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chen Z, Zhang P, Cheetham AG, Moon JH, Moxley JW, Lin Y-a, Cui H. Journal of Controlled Release. 2014;191:123–130. doi: 10.1016/j.jconrel.2014.05.051. [DOI] [PubMed] [Google Scholar]
- 202.Lock LL, Tang Z, Keith D, Reyes C, Cui H. ACS Macro Letters. 2015;4:552–555. doi: 10.1021/acsmacrolett.5b00170. [DOI] [PubMed] [Google Scholar]
- 203.Ma W, Su H, Cheetham AG, Zhang W, Wang Y, Kan Q, Cui H. Journal of Controlled Release. 2017 doi: 10.1016/j.jconrel.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 204.Cui H, Cheetham AG, Pashuck ET, Stupp SI. J Am Chem Soc. 2014;136:12461–12468. doi: 10.1021/ja507051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ghosh A, Dobson ET, Buettner CJ, Nicholl MJ, Goldberger JE. Langmuir. 2014;30:15383–15387. doi: 10.1021/la5037663. [DOI] [PubMed] [Google Scholar]
- 206.Yang M, Xu D, Jiang L, Zhang L, Dustin D, Lund R, Liu L, Dong H. Chemical Communications. 2014;50:4827–4830. doi: 10.1039/c4cc01568a. [DOI] [PubMed] [Google Scholar]
- 207.Chen J-X, Xu X-D, Chen W-H, Zhang X-Z. ACS applied materials & interfaces. 2013;6:593–598. doi: 10.1021/am404680n. [DOI] [PubMed] [Google Scholar]
- 208.Tian R, Wang H, Niu R, Ding D. Journal of colloid and interface science. 2015;453:15–20. doi: 10.1016/j.jcis.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 209.Lock LL, Li Y, Mao X, Chen H, Staedtke V, Bai R, Ma W, Lin R, Li Y, Liu G, Cui H. ACS nano. 2017;11:797–805. doi: 10.1021/acsnano.6b07196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yang Z, Kuang Y, Li X, Zhou N, Zhang Y, Xu B. Chemical Communications. 2012;48:9257–9259. doi: 10.1039/c2cc34935c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li J, Li X, Kuang Y, Gao Y, Du X, Shi J, Xu B. Advanced healthcare materials. 2013;2:1586–1590. doi: 10.1002/adhm.201300041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Carter JM, Qian Y, Foster JC, Matson JB. Chemical Communications. 2015;51:13131–13134. doi: 10.1039/c5cc04883d. [DOI] [PubMed] [Google Scholar]
- 213.Chen Z, Xing L, Fan Q, Cheetham A, Lin R, Holt B, Chen L, Xiao Y, Cui H. Theranostics. 2017;7:2003–2014. doi: 10.7150/thno.19404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Matson JB, Stupp SI. Chemical Communications. 2011;47:7962–7964. doi: 10.1039/c1cc12570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Matson JB, Newcomb CJ, Bitton R, Stupp SI. Soft Matter. 2012;8:3586–3595. doi: 10.1039/C2SM07420F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Qian Y, Matson JB. Advanced Drug Delivery Reviews. 2016 doi: 10.1016/j.addr.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 217.Matson JB, Webber MJ, Tamboli VK, Weber B, Stupp SI. Soft Matter. 2012;8:6689–6692. doi: 10.1039/C2SM25785H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bahnson ES, Kassam HA, Moyer TJ, Jiang W, Morgan CE, Vercammen JM, Jiang Q, Flynn ME, Stupp SI, Kibbe MR. Antioxidants & redox signaling. 2016;24:401–418. doi: 10.1089/ars.2015.6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Li X, Li J, Gao Y, Kuang Y, Shi J, Xu B. J Am Chem Soc. 2010;132:17707–17709. doi: 10.1021/ja109269v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Pu G, Ren C, Li D, Wang L, Sun J. RSC Advances. 2014;4:50145–50147. [Google Scholar]
- 221.Hartgerink JD, Beniash E, Stupp SI. science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 222.Cui H, Pashuck ET, Velichko YS, Weigand SJ, Cheetham AG, Newcomb CJ, Stupp SI. Science. 2010;327:555–559. doi: 10.1126/science.1182340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, Stupp SI, Kessler JA. The Journal of Neuroscience. 2008;28:3814. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mata A, Geng Y, Henrikson KJ, Aparicio C, Stock SR, Satcher RL, Stupp SI. Biomaterials. 2010;31:6004–6012. doi: 10.1016/j.biomaterials.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shah RN, Shah NA, Del Rosario Lim MM, Hsieh C, Nuber G, Stupp SI. Proceedings of the National Academy of Sciences. 2010;107:3293–3298. doi: 10.1073/pnas.0906501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Huang Z, Newcomb CJ, Bringas P, Stupp SI, Snead ML. Biomaterials. 2010;31:9202–9211. doi: 10.1016/j.biomaterials.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cheetham AG, Keith D, Zhang P, Lin R, Su H, Cui H. Current cancer drug targets. 2016;16:489–508. doi: 10.2174/1568009616666151130214646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhang P, Cheetham AG, Lin Y-a, Cui H. Acs Nano. 2013;7:5965–5977. doi: 10.1021/nn401667z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zhang P, Cheetham AG, Lock LL, Cui H. Bioconjugate chemistry. 2013;24:604–613. doi: 10.1021/bc300585h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang P, Lock LL, Cheetham AG, Cui H. Molecular pharmaceutics. 2014;11:964–973. doi: 10.1021/mp400619v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lin R, Zhang P, Cheetham AG, Walston J, Abadir P, Cui H. Bioconjugate chemistry. 2014;26:71–77. doi: 10.1021/bc500408p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Burke TG, Mi Z. Journal of Medicinal Chemistry. 1994;37:40–46. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]