

Abstract
A nickel-catalyzed coupling of azaarylmethylamines with aryl chlorides has been achieved. NIXANTPHOS together with low cost NiBr2 was successfully developed and optimized to exhibit high reactivity at 2.5 mol % loading. Under optimized reaction conditions, aryl(azaaryl)methylamine products were afforded in good to excellent yields (22 examples, up to 98% yield).
Keywords: NIXANTPHOS, NiBr2, Arylation, Azaarylmethylamines, Aryl Chlorides
Graphical Abstract
Aryl(azaaryl)methylamines are widely found in pharmaceuticals, bioactive substances and as ligands for metal complexes.[1] They have attracted significant attention due to their diverse activity against tumors,[2] viruses,[3] HIV,[4] tuberculosis,[5] iron metabolic disorders,[6] and cortisol dependent diseases.[7] Some prominent examples of aryl(azaaryl)methylamines are illustrated in Figure 1. Classic approaches to the synthesis of aryl(azaaryl)methylamines include addition of organometallic reagents to aldimines,[8] ketimine reduction[9] and C–N coupling reactions.[10] An intramolecular aryl rearrangement to give aryl(azaaryl)methylamines was also reported by Clayden and coworkers.[11] These approaches usually required prefunctionalized starting materials or organolithium reagents, which can limit their scope.
Figure 1.
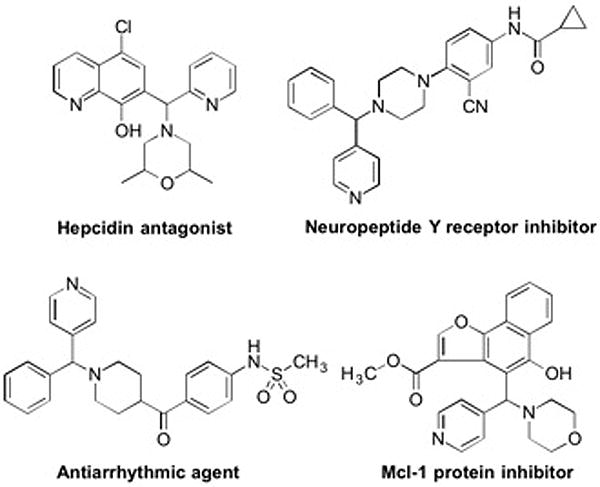
Examples of aryl(azaaryl)methylamines in pharmacologically active compounds.
Our group, among others, has strived to develop methods for the catalytic functionalization of weakly acidic sp3–hybridized C–H bonds via a deprotonative–cross–coupling process (DCCP).[12] These reactions involve reversible in-situ deprotonation of the substrates (pronucleophiles) to generate reactive nucleophiles that undergo transmetallation with the catalyst. Considering the value of aryl(azaaryl)methylamines, we recently introduced an efficient palladium-catalyzed approach for the synthesis of aryl(azaaryl)methylamines via in-situ deprotonation of (aminomethyl)pyridines followed by arylation with aryl bromides (Scheme 1). This umpolung approach directly provides the arylated tertiary amines without protecting groups or additional activating groups.
Scheme 1.

Pd-catalyzed arylation of azaarylmethylamines with aryl bromides involving reversible in-situ deprotonation of the substrate (aminomethyl)azaarenes.
The catalyst for this reaction was based on van Leeuwen’s NIXANTPHOS ligand,[13] which we have shown imparts enhanced reactivity to palladium catalysts under basic conditions where the ligand’s N–H is deprotonated.[14] We were curious if the enhanced reactivity of (NIXANTPHOS)Pd catalysts would translate to other transition metals. We therefore examined the arylation of 2-pyridylmethyl amine with bromobenzene and LiN(SiMe3)2 in the presence of 37 of the most common ligands in cross-coupling chemistry.[15] As shown in Scheme 2, the catalyst derived from Ni(COD)2 and NIXANTPHOS proved to be the most active, giving the arylation product in 93% isolated yield.[16] Although only one pyridylmethylamine was examined with the Ni(NIXANTPHOS)-based catalyst, the results appeared promising.
Scheme 2.

Preliminary result of arylation of 2-pyridylmethyl morpholine with bromobenzene using Ni(COD)2/NIXANTPHOS catalyst.
An advantage of nickel catalysts over their palladium counterparts is the substantial decrease in cost of Earth-abundant nickel. That said, however, Ni(COD)2 is far from ideal, because it is both expensive and requires special handling precautions due to its air-sensitivity. To improve upon the palladium-based arylation of azaarylmethylamines in Scheme 1, we desired to develop a nickel-based catalyst using a readily available, air-stable, and inexpensive nickel source. Once identified, we planned to expand the scope beyond the singular nickel-catalyzed example in Scheme 1, and to focus on more economical aryl chlorides.
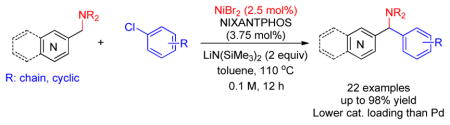
Herein, we report a highly efficient α-arylation of (aminomethyl)pyridines with aryl chlorides using a catalyst formed from NiBr2 and NIXANTPHOS (Scheme 3).
Scheme 3.

This work.
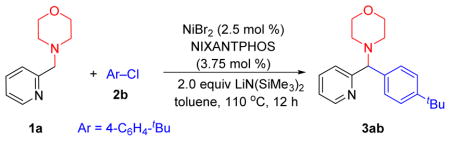
Our previous study in Scheme 2 used only bromobenzene as the electrophile. Therefore, using High Throughput Experimentation (HTE) techniques on a 10 umol scale, we initiated studies of the coupling between 4-(pyridin-2-ylmethyl) morpholine 1a and 4-tert-butyl chlorobenzene 2b with ligand screening on microscale. A total of 44 electronically diverse mono- and bidentate phosphines were examined using Ni(COD)2 as metal source, LiN(SiMe3)2 as base, and CPME (cyclopentyl methyl ether) as solvent at 110 °C for 12 h (see Supporting Information pg S2 for details). The two most promising ligands from this screen were NIXANTPHOS (L1) and JohnPhos (L2).[17] We continued with a second lab scale (0.1 mmol) screen examining nickel sources [NiBr2, Cp2Ni, Cl2Ni(PPh3)2 and Ni(COD)2 (5 mol % each)], NIXANTPHOS and JohnPhos, and 3 solvents [CPME, dioxane and toluene] with LiN(SiMe3)2, 110 °C for 12 h (see Supporting Information pg S5 for details). The top Ni/ligand/solvent combination from this screen was NiBr2/NIXANTPHOS/toluene, affording product 3ab in 97% assay yield (AY) (Table 1, entry 1). Reducing the catalyst loading to 2.5 and 1 mol % led to 95 and 43% AY (entries 2 and 3). Next, we examined the impact of temperature and found the AY dropped to 75% at 80 °C (entry 4). Reducing the base equivalents from 2 to 1 equiv. led to a significant drop in yield to 22% AY (entry 2). Changing the reaction concentration from 0.1 M to 0.2 M or 0.05 M resulted in a drop in the AY’s (76 and 80% yield, entries 5 and 6, respectively).
Table 1.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | NiBr2/L (mol %) | Conc. (M) | Temp (°C) | Assay yield (%) |
| 1 | 5/7.5 | 0.1 | 110 | 97 |
| 2 | 2.5/3.75 | 0.1 | 110 | 95(22c) |
| 3 | 1/1.5 | 0.1 | 110 | 43 |
| 4 | 2.5/3.75 | 0.1 | 80 | 75 |
| 5 | 2.5/3.75 | 0.05 | 110 | 76 |
| 6 | 2.5/3.75 | 0.2 | 110 | 80 |
Reactions conducted on a 0.1 mmol scale using 1 equiv of 1a, 2 equiv of LiN(SiMe3)2, and 1.2 equiv of ArCl.
Yields determined by 1H NMR spectroscopy of the crude reaction mixtures on a 0.1 mmol scale.
1 equiv. of LiN(SiMe3)2.
With the optimized conditions in hand (Table 1, entry 2), the scope of 4-(pyridin-2-ylmethyl) morpholine 1a and different aryl chlorides 2 was explored (Table 2). The product 3aa was generated from chlorobenzene in 95% yield. Replacing aryl chloride 2a with its aryl bromide counterpart 2a’ led to a drop in the yield to 79%. It should be noted, however, that these conditions were optimized for aryl chlorides. Alkyl substituted aryl chlorides 4-tBu (2b) and 4-methyl (2c) also furnished products in excellent yields (90 and 89%, respectively, for 3ab and 3ac). Electron rich 4-chloroanisole (2d) was successfully coupled, delivering 3ad in 93% yield. 4-Fluorochlorobenzene (1e) also underwent coupling in 61% yield to provide 3ae, although the Ni loading had to be increased to 5 mol %. The sterically hindered 1-naphthyl chloride reacted in 62% yield to provide the expected product (3af). Aryl chlorides bearing either 3-methyl or 3-trifluoromethyl groups were suitable cross-coupling partners, furnishing tolyl (3ag) and trifluorotolyl (3ah) products in 88 and 98% yield, respectively. The substrate 4-chlorobenzophenone could undergo addition of the lithiated pyridyl amine to the carbonyl group. We were pleased to find, however, that the Ni(NIXANTPHOS)-based system exhibited excellent selectivity, providing the diarylmethylamine derivative (3ai) in 86% yield. Heteroaromatic 3-chloropyridyl participated in the coupling reaction with 1a to afford 3aj, albeit in 33% yield (despite substantial effort to optimize this substrate). Finally, coupling of 1a with aryl chloride bearing an acetamide group (2k) afforded products 3ak in 53% yield.
Table 2.
Substrate scope of aryl chlorides in the arylation of 4-(pyridin-2-ylmethyl)morpholine 1a.a

|
Reactions conducted on a 0.1 mmol scale using 1 equiv of 1a, 2 equiv of LiN(SiMe3)2, and 1.2 equiv of ArCl. Isolated yields after chromatographic purification.
5 mol % Ni.
10 mol % Ni.
4 equiv of LiN(SiMe3)2
Next, we examined the coupling reactions of various acyclic amines with aryl chloride 2b (Table 3). When the 2-pyridyl amine was changed from morpholine to N,N-dimethyl- and N,N-diethylamine the corresponding products 3bb and 3cb were obtained in 93 and 86% yield, respectively. Under the same conditions, pyrrolidine thiomorpholine and N-methylpiperazine were coupled, generating aryl(2-pyridyl)methylamines in 90, 73, and 82% yield, respectively. In order to achieve a broader scope for our protocol, we examined 4-pyridyl methylamines. N,N-Dimethyl-1-(pyridin-4-yl)methanamine (1g) and the morpholine analogue (1h) coupled with 4-tert-butyl chlorobenzene to form the desired products 3gb and 3hb in 60 and 89% yield, respectively. In addition, 1-methyl-4-(pyridin-4-ylmethyl)piperazine (1i) furnished product 3ib in 97% yield. Despite significant optimization, the 3-pyridylmethyl amine 4-(pyridin-3-ylmethyl)morpholine yield could not be raised above 61%. The isoquinoline derivative N,N-dimethyl-1-(quinolin-2-yl)methanamine (1k) furnished the coupling product 3kb in 80% yield. N-Methyl-N-(pyridin-2-ylmethyl)aniline (1l) underwent coupling to provide the product 3lb in 56% yield with 10 mol % catalyst.
Table 3.
Substrate scope of azaarylmethylamines in the arylation of aryl chloride 2b.a

|
Reactions conducted on a 0.1 mmol scale using 1 equiv of 1b–1i, 2 equiv of LiN(SiMe3)2, and 1.2 equiv of ArCl. Isolated yields after chromatographic purification.
10 mol % Ni.
5 mol % Ni.
In order to investigate the potential scalability of our protocol (Scheme 4), we conducted the arylation of 4-(pyridin-4-ylmethyl)morpholine 1h with 4-tert-buty chlorobenzene 2b on gram scale. The desire coupled product 3hb was isolated in 84% yield (2.09 g).
Scheme 4.

Gram scale synthesis of 3hb.
In conclusion, we report an efficient nickel-catalyzed arylation of azaarylmethylamines with aryl chlorides. Various aminomethyl azaarenes and aryl chlorides are easily coupled to provide attractive intermediates for the synthesis of biologically active compounds. The most significant advance is replacing palladium with an inexpensive and easily handled nickel source, NiBr2. In several cases, the loading of the nickel-based catalyst is lower than the palladium analogue.
Finally, a key finding of this work is that the combination of nickel and NIXANTPHOS outperformed other nickel ligand combinations in the coupling of azaarylmethylamines with aryl chlorides under the conditions examined. These results suggest that the high reactivity of the Pd(NIXANTPHOS) catalyst under conditions where the NIXANTPHOS N–H is deprotonated are translatable to nickel and perhaps other metals. We are currently exploring this hypothesis as well as attempting to develop an enantioselective version of this reaction.
Experimental Section
1. General procedure for the arylation of azaarylmethylamines
An oven-dried microwave vial equipped with a stir bar was charged with LiN(SiMe3)2 (66.9 mg, 0.4 mmol, 2.0 equiv) under a nitrogen atmosphere in the glovebox. 4-(Pyridin-2-ylmethyl)morpholine (35.6 mg, 0.2 mmol, 1.0 equiv) and 4-t-Bu-chlorobenzene (40.5 mg, 0.24 mmol, 1.2 equiv) were added by syringe in the glovebox. Next, 1 mL stock solution containing NiBr2 (1.1 mg, 0.005 mmol) and NIXANTPHOS (4.2 mg, 0.0075 mmol) in toluene was added under a nitrogen atmosphere via syringe in the glovebox. The microwave vial was sealed with a cap and the reaction was stirred at 110 °C for the specified time then allowed to cool to room temperature. The reaction mixture was quenched with H2O (0.2 mL) and passed through a short pad of silica gel and eluted with ethyl acetate (1 mL x 3). The combined organics were concentrated in vacuo. The crude residue was purified by flash column chromatography to yield the monoarylated azaarylmethylamine derivatives 3.
2. Procedure for the gram scale synthesis
An oven-dried 100 mL Schlenk tube equipped with a stir bar was charged with 4-(pyridin-4-ylmethyl)morpholine 1h (1.44 g, 8.0 mmol). The Schlenk tube was sealed with a rubber septum and was connected to a Schlenk line, evacuated, and refilled with nitrogen (repeated three times). A solution (prepared in the glove box) of aryl chloride 2b (2.70 g, 16.0 mmol) in 5 mL anhydrous toluene was added to the Schlenk tube via syringe through the rubber septum. A solution (prepared in the glove box) containing NiBr2 (44 mg, 0.2 mmol) and NIXANTPHOS (168 mg, 0.3 mmol) in 5 mL anhydrous toluene was added to the Schlenk tube via syringe through the rubber septum. Next, a solution of LiN(SiMe3)2 (2.68 g, 16.0 mmol) in 30 mL anhydrous toluene was added by syringe through the rubber septum. The reaction mixture was stirred for 16 h in total at 110 °C, opened to air, and quenched with 10 mL of H2O. The layers were separated and the aqueous layer was extracted with DCM (3X10 mL). The combined organic layers were concentrated in vacuum. The crude material was loaded onto a deactivated silica gel column via pipette and purified by flash chromatography on silica gel (eluted with Methanol: DCM = 1:30) to give the product (2.09 g, 84% yield) a white solid. The 1H and 13C{1H} NMR data for this compound match the literature data.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-1464744) and National Institutes of Health (NIGMS 104349) for financial support. BZ thanks the Chinese Universities Scientific Fund (2017QC071) and National College Students Innovation Training Program (201610019084). GG thanks the National Key Technology Research and Development Program (No. 2015BAK45B01) for the financial support.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######. ((Please delete if not appropriate))
References
- 1.a) Lu J, Xu X, Wang C, He J, Hu Y, Hu H. Tetrahedron Lett. 2002;43:8367–8369. [Google Scholar]; b) Dong Y, Sun J, Wang X, Xu X, Cao L, Hu Y. Tetrahedron: Asymmetry. 2004;15:1667–1672. [Google Scholar]; c) Henry GD. Tetrahedron. 2004;60:6043–6061. [Google Scholar]; d) Michael JP. Nat Prod Rep. 2005;22:627–646. doi: 10.1039/b413750g. [DOI] [PubMed] [Google Scholar]; e) Wang S, Wang Y, Jiang J, Liu R, Li M, Wang Y, Su Y, Zhu B, Zhang S, Huang W. Catal Commun. 2009;10:640–644. [Google Scholar]; f) Chaudhary AR, Bedekar AV. Synth Commun. 2012;42:1778–1785. [Google Scholar]; g) Baumann M, Baxendale IR. Beilstein J Org Chem. 2013;9:2265–2319. doi: 10.3762/bjoc.9.265. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wang Y, Yang Q, Yang L, Shi J, Zhang M. Tetrahedron Lett. 2013;54:5314–5317. [Google Scholar]; i) Chopde HN. Int J Pharma Bio Sci. 2014;5:541–548. 549. [Google Scholar]; j) Yang L, Yang Q, Shi J, Wang Y, Zhang M. Synth Commun. 2014;44:2468–2477. [Google Scholar]; k) Wang S, Zhang J, Jiang J, Liu R, Zhu B, Xu M, Wang Y, Cao J, Li M, Yuan Z, Zhang S, Huang W, Wu S. Microporous Mesoporous Mater. 2009;123:349–353. [Google Scholar]; l) Jain PP, Degani MS, Raju A, Anantram A, Seervi M, Sathaye S, Ray M, Rajan MGR. Bioorg Med Chem Lett. 2016;26:645–649. doi: 10.1016/j.bmcl.2015.11.057. [DOI] [PubMed] [Google Scholar]; m) McDaniel TJ, Lansdell TA, Dissanayake AA, Azevedo LM, Claes J, Odom AL, Tepe JJ. Bioorg Med Chem. 2016;24:2441–2450. doi: 10.1016/j.bmc.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Yang L, Xia S, Ma S, Zhou S, Zhao X, Wang S, Li M, Yang X. Mater Sci Eng C Mater Biol Appl. 2016;59:1016–1024. doi: 10.1016/j.msec.2015.10.037. [DOI] [PubMed] [Google Scholar]; o) Zhou B, Liu Z, Deng G, Chen W, Li M, Yang L, Li Y, Yang X, Zhang H. Org Biomol Chem. 2016;14:9423–9430. doi: 10.1039/c6ob01495j. [DOI] [PubMed] [Google Scholar]; p) Zhou Y, Duan K, Zhu L, Liu Z, Zhang C, Yang L, Li M, Zhang H, Yang X. Bioorg Med Chem Lett. 2016;26:460–465. doi: 10.1016/j.bmcl.2015.11.092. [DOI] [PubMed] [Google Scholar]; q) Zhang C, Liu Y, Liu Z, Duan S, Li M, Chen W, Li Y, Zhang H, Yang X. Bioorg Med Chem Lett. 2017;27:1808–1814. doi: 10.1016/j.bmcl.2017.02.053. [DOI] [PubMed] [Google Scholar]
- 2.a) Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Li Z, Dell J, Lipari P, Malkowski M, Prioli N, Rossman RA, Korfmacher WA, Nomeir AA, Lin CC, Mallams AK, Doll RJ, Catino JJ, Girijavallabhan VM, Kirschmeier P, Bishop WR. Cancer Chemother Pharmacol. 1999;43:50–58. doi: 10.1007/s002800050862. [DOI] [PubMed] [Google Scholar]; b) Yang L, Wang S, Zhou S, Zhao F, Chang Q, Li M, Chen W, Yang X. Mater Sci Eng C. 2017;76:1136–1145. doi: 10.1016/j.msec.2017.03.197. [DOI] [PubMed] [Google Scholar]; c) Errico JP, Mugrage B, Turchi I, Sills M, Ong J, Allocco J, Wines P. 20110301193. US Patents. 2011
- 3.Chern J, Shia K, Hsu T, Tai C, Lee C, Lee Y, Chang C, Tseng S, Shih S. Bioorg Med Chem Lett. 2004;14:2519–2525. doi: 10.1016/j.bmcl.2004.02.092. [DOI] [PubMed] [Google Scholar]
- 4.Summa V, Petrocchi A, Matassa VG, Gardelli C, Muraglia E, Rowley M, Paz OG, Laufer R, Monteagudo E, Pace P. J Med Chem. 2006;49:6646–6649. doi: 10.1021/jm060854f. [DOI] [PubMed] [Google Scholar]
- 5.Upadhayaya RS, Vandavasi JK, Vasireddy NR, Sharma V, Dixit SS, Chattopadhyaya J. Bioorg Med Chem. 2009;17:2830–2841. doi: 10.1016/j.bmc.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 6.Durrenberger F, Burckhardt S, Geisser PO, Buhr W, Funk F, Corden VA, Fryatt T, Jaeger S, Slack M, Yarnold CJ. 2011020886 A1. WO. 2011
- 7.a) Emmerich J, Hu Q, Hanke N, Hartmann RW. J Med Chem. 2013;56:6022–6032. doi: 10.1021/jm400240r. [DOI] [PubMed] [Google Scholar]; b) Liu F, Zhong J, Li S, Li M, Wu L, Wang Q, Mao J, Liu S, Zheng B, Wang M, Bian Q. J Nat Prod. 2016;79:244–247. doi: 10.1021/acs.jnatprod.5b00713. [DOI] [PubMed] [Google Scholar]
- 8.Robak MT, Herbage MA, Ellman JA. Chem Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 9.Chelucci G, Baldino S, Chessa S, Pinna GA, Soccolini F. Tetrahedron: Asymmetry. 2006;17:3163–3169. [Google Scholar]
- 10.a) Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069–1094. doi: 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]; b) Kobayashi S, Mori Y, Fossey JS, Salter MM. Chem Rev. 2011;111:2626–2704. doi: 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]; c) Carey FA, Sundberg RJ. Advanced Organic Chemistry: Part B: Reaction and Synthesis. 5. Springer; New York: 2007. [Google Scholar]; d) Smith MB, March J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 7. John Wiley & Sons, Inc; Hoboken: 2013. [Google Scholar]
- 11.a) Clayden J, Hennecke U. Org Lett. 2008;10:3567–3570. doi: 10.1021/ol801332n. [DOI] [PubMed] [Google Scholar]; b) Maury J, Zawodny W, Clayden J. Org Lett. 2017;19:472–475. doi: 10.1021/acs.orglett.6b03603. [DOI] [PubMed] [Google Scholar]
- 12.For reviews of sp3–hybridized C–H bonds functionalization see, Bellina F, Rossi R. Chem Rev. 2010;110:1082–1146. doi: 10.1021/cr9000836.Tollefson EJ, Hanna LE, Jarvo ER. Acc Chem Res. 2015;48:2344–2353. doi: 10.1021/acs.accounts.5b00223.For our deprotonative sp3–hybridized C–H bonds functionalization see, Zheng B, Li M, Gao G, He Y, Walsh PJ. Adv Synth Catal. 2016;358:2156–2162. doi: 10.1002/adsc.201600090.Li M, Yucel B, Adrio J, Bellomo A, Walsh PJ. Chem Sci. 2014;5:2383–2391. doi: 10.1039/C3SC53526F.Li M, Berritt S, Walsh PJ. Org Lett. 2014;16:4312–4315. doi: 10.1021/ol502043j.Li M, Gutierrez O, Berritt S, Pascual-Escudero A, Yeşilçimen A, Yang X, Adrio J, Huang G, Nakamaru-Ogiso E, Kozlowski MC, Walsh PJ. Nat Chem. 2017 doi: 10.1038/nchem.2760.Yang X, Kim B, Li M, Walsh PJ. Org Lett. 2016;18:2371–2374. doi: 10.1021/acs.orglett.6b00815.
- 13.van der Veen LA, Keeven PH, Schoemaker GC, Reek JNH, Kamer PCJ, van Leeuwen PWNM, Lutz M, Spek AL. Organometallics. 2000;19:872–883. [Google Scholar]
- 14.a) Kim B, Jimenez J, Gao F, Walsh PJ. Org Lett. 2015;17:5788–5791. doi: 10.1021/acs.orglett.5b02898. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li M, Gonzalez-Esguevillas M, Berritt S, Yang X, Bellomo A, Walsh PJ. Angew Chem Int Ed. 2016;55:2825–2829. doi: 10.1002/anie.201509757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Hama T, Liu X, Culkin DA, Hartwig JF. J Am Chem Soc. 2003;125:11176–11177. doi: 10.1021/ja036792p. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; c) Li M, Yucel B, Jiménez J, Rotella M, Fu Y, Walsh PJ. Adv Synth Catal. 2016;358:1910–1915. doi: 10.1002/adsc.201600075. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) He T, Yu L, Zhang L, Wang L, Wang M. Org Lett. 2011;13:5016–5019. doi: 10.1021/ol201779n. [DOI] [PubMed] [Google Scholar]; e) DiRocco DA, Rovis T. J Am Chem Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Rossi R, Bellina F, Lessi M, Manzini C. Adv Synth Catal. 2014;356:17–117. [Google Scholar]; g) Liu F, Zhong J, Zheng B, Li S, Gao G, Wang Z, Li M, Hou S, Wang M, Bian Q. Tetrahedron: Asymmetry. 2015;26:961–965. [Google Scholar]; h) Shin K, Kim H, Chang S. Acc Chem Res. 2015;48:1040–1052. doi: 10.1021/acs.accounts.5b00020. [DOI] [PubMed] [Google Scholar]; i) Mastalir M, Stoeger B, Pittenauer E, Allmaier G, Kirchner K. Org Lett. 2016;18:3186–3189. doi: 10.1021/acs.orglett.6b01398. [DOI] [PubMed] [Google Scholar]
- 16.Cao X, Sha S, Li M, Kim B, Morgan C, Huang R, Yang X, Walsh PJ. Chem Sci. 2016;7:611–618. doi: 10.1039/c5sc03704b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aranyos A, Old DW, Kiyomori A, Wolfe JP, Sadighi JP, Buchwald SL. J Am Chem Soc. 1999;121:4369–4378. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.