

Abstract
ACTB1-GLI1 fusions have been reported as the pathognomonic genetic abnormality defining an unusual subset of actin-positive, perivascular myoid tumors, known as ‘pericytoma with the t(7;12) translocation’. In addition, GLI1 oncogenic activation through a related MALAT1-GLI1 gene fusion has been recently reported in two unrelated gastric tumors, namely plexiform fibromyxoma and gastroblastoma. Triggered by unexpected targeted RNA sequencing results detecting GLI1-related fusions in a group of malignant neoplasms with round to epithelioid morphology, and frequently strong S100 protein immunoreactivity, we investigated their clinicopathologic features in relation to other known pathologic entities sharing similar genetics. Based on a combined approach of targeted RNA sequencing and FISH screening, we identified 6 cases with GLI1 gene fusions, including 4 fused to ACTB1, 1 with MALAT1 and 1 with PTCH1 gene. Patients had a mean age of 36 years at diagnosis (range: 16–79) and equal gender distribution; all except one tumor originated in the soft tissue. Microscopically, the tumors had a monomorphic epithelioid phenotype arranged in a distinctive nested or cord-like architecture, separated by thin septae and delicate capillary network. All except two cases were strongly positive for S100 protein, while being negative for SOX10, SMA, and EMA. Only one tumor showed focal Cytokeratin positivity in rare cells. Although the tumors showed some resemblance to pericytic/glomus tumors or myoepithelial tumors, the immunoprofile was not supportive of either lineage. Moreover, in contrast to the benign course of so-called pericytoma with t(7;12), 3 patients in this series developed metastatic disease to either lymph nodes or lung. In fact the only patient with lung metastases showed a novel PTCH1-GLI1 gene fusion. It remains to be determined whether these tumors represent a clinically and immunohistologically distinct subset of pericytoma, or an altogether novel soft tissue sarcoma. Our findings open new opportunities for targeted therapy, as tumors with GLI1 oncogenic activation, and subsequent PTCH1 overexpression, might be sensitive to SHH pathway inhibitors.
Keywords: GLI1, ACTB, MALAT1, PTCH1, SHH, pericytoma, gastroblastoma, S100 protein
INTRODUCTION
Recurrent ACTB-GLI1 fusions, resulting from a t(7;12)(p21–22;q13–15), have been described in a distinctive mesenchymal neoplasm with a pericytic phenotype composed of monomorphic spindle cells and immunoreactivity for smooth muscle actin and laminin1,2. Most of these lesions occurred in the tongue, with rare examples described in the stomach and bone; these tumors appear to pursue a benign clinical course1,3,4. Despite a pericytic phenotype, tumors with t(7;12) are considered distinct from the more common pericytic lesions, such as myopericytoma or glomus tumor, which have been associated with different genetic abnormalities, such as PDGFRB mutations5 or NOTCH gene rearrangements6, respectively. The mechanism of GLI1 (glioma-associated oncogene homologue 1) oncogenic activation occurs by promoter swapping with the ubiquitously expressed beta-actin gene (ACTB), leading to deregulation of GLI expression and its downstream targets. Importantly, the DNA-binding zinc finger domains of GLI1 are retained in the fusion transcript. A related gene fusion, MALAT1-GLI1, similarly resulting in GLI1 overexpression, was reported more recently in two seemingly unrelated tumors located in the stomach, namely plexiform fibromyxoma and gastroblastoma7,8. Importantly, these tumors lack any morphologic or immunoprofile overlap with the pericytoma with t(7;12).
The present study investigates a cohort of malignant mesenchymal neoplasms associated with a round to epithelioid phenotype, with frequent S100 protein expression, showing recurrent GLI1 gene fusions with ACTB1, MALAT1 or PTCH1. Although showing some histologic resemblance to pericytoma, such as nested growth and intricate capillary network, these lesions contain morphologic variability, lack actin immunoreactivity and have a propensity for locoregional lymph node and distant lung metastases.
METHODS
Patient Selection
The files of the senior authors (CRA, BCD) were searched for cases that harbored GLI1 gene abnormalities, either by FISH or targeted RNA sequencing. Hematoxylin and eosin stained slides were reviewed. The study group was analyzed for demographic information, anatomic site, tumor size, and morphologic features, including cell type (round, spindle, mixed), degree of cellularity, type and amount of stromal component, nuclear features, mitotic activity and presence of necrosis. Available immunohistochemical stains were reviewed, including S100 protein, SOX10, EMA, Cytokeratin, CD56, and SMA. The clinical follow-up information was obtained from review of the electronic medical records and from contacting referring pathologists and clinicians. The study was approved by the Institutional ethics Review Board.
Targeted RNA sequencing and analysis
RNA was extracted from FFPE tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA). Fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-seq libraries were prepared using 20–100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA). Each sample was subjected to targeted RNA sequencing on an Illumina MiSeq at 8 samples per flow cell (approximately 3 million reads per sample). All reads were independently aligned with STAR(ver 2.3) and BowTie2 against the human reference genome (hg19) for Manta-Fusion and TopHat-Fusion analysis, respectively. The mRNA expression levels of certain genes of interest (GL1, ACTB1, CBX5, PTCH1, SOX2, VEGF1, CCND1) were evaluated and compared to those of other samples analyzed on the same targeted RNA sequencing platform.
Fluorescence in situ hybridization (FISH)
Formalin-fixed paraffin-embedded tissues were available in each case for FISH analysis. FISH for GLI1 was performed on all cases and followed by subsequent FISH for various fusion partners, including ACTB1, MALAT1 and PTCH1. FISH was performed on 4 μm-thick formalin-fixed paraffin-embedded (FFPE) tissue sections. Custom probes were made by bacterial artificial chromosomes (BAC) clones (Supplementary Table 1) flanking the target genes, according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; http://bacpac.chori.org). DNA from each BAC was isolated according to the manufacturer’s instructions. The BAC clones were labeled with fluorochromes by nick translation and validated on normal metaphase chromosomes. The slides were deparaffinized, pretreated, and hybridized with denatured probes. After overnight incubation, the slides were washed, stained with DAPI, mounted with an antifade solution, and then examined on a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) controlled by Isis 5 software (Metasystems, Newton, MA).
RESULTS
Clinicopathologic Findings
The study cohort included 4 females and 2 males, all except one was younger than age of 40 (range 16–79, mean 36, median 32). Five tumors were located within soft tissue, one each in the thigh, foot, retroperitoneum, chest wall and head and neck (submandibular area). Only one tumor was located in the bone (C2 vertebral body). The gross description from the resection specimens available in 3 cases showed either a solid-cystic or fleshy cut surface. Histologically all tumors had a monomorphic appearance of round to epithelioid cells arranged in nests, cords, and reticular patterns (Fig. 1), often surrounding a rich capillary network (Fig. 2). Two tumors showed areas of tubulo-cribriform growth or sieve-like pattern admixed within solid sheets of monotonous epithelioid cells (Fig 3). Most tumors had a very delicate stromal component, mainly dividing the lesion into nests and compartments (Fig. 1). A myxoid stroma was noted in 2 cases (Fig. 1). All cases had a solid growth, except for one tumor which showed a distinctive cystic appearance, which included large, dilated malformed blood vessels (Fig. 1C, case 1). The neoplastic cells had a scant to moderate amount of amphophilic to light eosinophilic or clear cytoplasm, uniform round nuclei with fine chromatin and inconspicuous nucleoli (Figs. 1,2). No areas of spindling or pleomorphic cells were noted. The mitotic activity was relatively low, ranging from 1–5 MFs per 10 HPFs and necrosis was absent in all of the primary tumors. Focal geographic necrosis was noted in one of the local recurrences (case 4). A Ki-67 proliferative index available for review in 3 cases showed 5% nuclear labeling.
Figure 1. Morphologic spectrum of tumors positive for GLI1 gene rearrangements.

A nested growth pattern predominated, typically separated by a rich capillary network (A, Case 3; B, Case 4), with either scant (A) or more abundant clear cytoplasm (B). Most cases showed a solid growth pattern, with the exception of case 1 which showed large cystic spaces, including malformed blood vessels (C, case 1); A reticular or cord-like arrangement was noted in this case with conspicuous myxoid stroma (D, case 4).
Figure 2. Cytomorphology high power view of GLI1-fusion positive tumors.

A. A tumor was composed of small blue round cells arranged in tight nests separated by a rich capillary network (Case 3); B. Ovoid to epithelioid cells with scant eosinophilic cytoplasm arranged in cords and sinusoidal pattern (case 2); C. Cuboidal to epithelioid cells with clear to vacuolated cytoplasm and round uniform nuclei with fine chromatin (Case 1); D. Diffuse and strong S100 protein reactivity was present in all except one case (Case 3).
Figure 3. Morphologic features of S100 protein-negative tumors harboring ACTB1-GLI1 fusion.
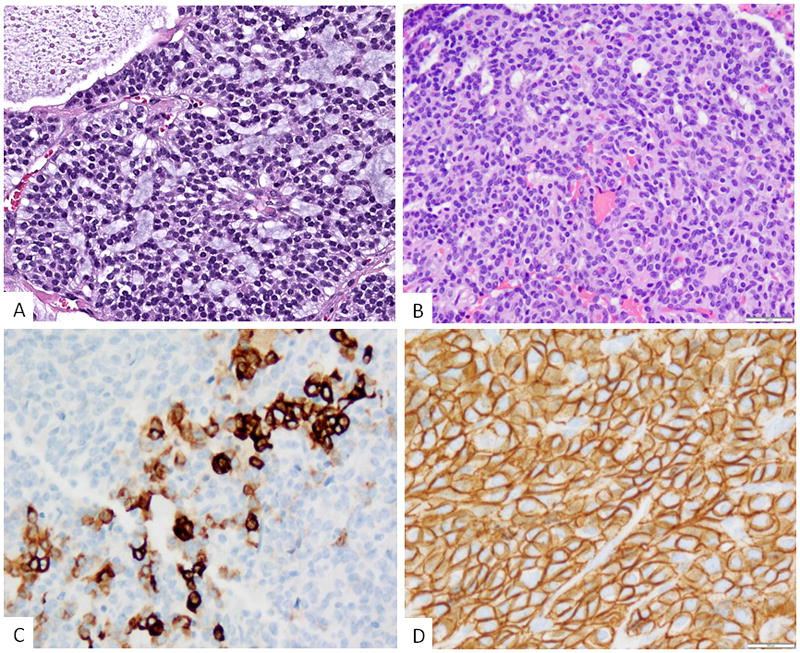
A. Epithelioid neoplasm showing a distinctive cribriform or sieve-like pattern with intervening myxoid stroma (Case 5). The tumor was negative for all immunomarkers tested; B. A second tumor revealed mainly solid sheets of epithelioid cells with scattered tubular structures (case 6); immunohistochemically the lesion showed (C) focal cytokeratin staining, mainly as single cells and small clusters, most of the tumor being negative; and (D) diffuse staining for CD56.
Immunohistochemically 4 of the 6 cases showed strong and diffuse reactivity for S100 protein (Fig 2), while none showed positivity for SOX10, EMA, SMA, and HMB45. In fact, most cases were tested for a much wider immunopanel, mainly to include myoepithelial markers, such as p63, calponin, GFAP, etc, all being negative. Two cases were negative for S100 protein, both showing an ACTB1-GLI1 fusion (cases #5 and #6), one of them metastasizing to locoregional lymph nodes. CD56 was diffusely positive in two cases, but negative for other neuroendocrine markers, such as synaptophysin or chromogranin. Cytokeratin was negative in all except one case (case 6); the latter showed mostly rare single cells and small clusters immunoreactivity (Fig 3), while most of the tumor being negative and not highlighting the tubular structures.
GLI1 gene fusions with either ACTB1, MALAT1, or PTCH1 define tumors with a distinct phenotype and frequent S100 protein positivity
Targeted RNA sequencing performed in 4 cases showed the presence of an ACTB1-GLI1 fusion in 3 and a novel PTCH1-GLI1 fusion in one case (Table 1, Fig 4). The GLI1 breakpoints illustrated in Fig. 4A revealed the fusion transcript included GLI1 exons 5–12 in case#1, exons 6–12 in case #5 and exons 7–12 in case #4. All 3 transcripts were predicted to retain the FOXP coiled-coil domain and the zinc finger double domains (zf-h2C2 and C2H2) of GLI1 protein (Fig. 4A). Validation by FISH was done in all cases confirming break-apart for GLI1, ACTB1 and PTCH1 (Fig. 4B, Supplem. Fig 1). One case was positive for ACTB1 and GLI1 gene rearrangements by FISH (case 6). An additional case that was positive for GLI1 gene rearrangement by FISH but negative for ACTB1 was tested for MALAT1 gene abnormalities (case 2). FISH showed in this case a balanced break-apart signal (Supplem Fig 1) in keeping with a MALAT1-GLI1 fusion.
Table 1.
Clinicopathologic and Molecular Findings of GLI1-Fusion Positive Mesenchymal Tumors
| Case # | Age/Sex | Site | IHC | Fusion | FU |
|---|---|---|---|---|---|
| 1 | 20/M | Thigh | S100 (+) | ACTB-GLI1* | Lost FU |
| 2 | 16/M | C2 spine w spinal canal extension | S100 (+) | MALAT1-GLIΔ | Recentcase |
| 3 | 30/F | Foot (1.5 cm) | S100 (+) CD56 (+) | ACTB-GLI1* | LR* Inguinal LN met 21 mo AWD |
| 4 | 34/F | Submandibular ST/neck | S100 (+) | PTCH1-GLI1* | LR, Mets to LN & lung AWD 80 mo |
| 5 | 79/F | RP | All IHC tested neg | ACTB-GLI1* | Inguinal LN mets |
| 6 | 38/F | Chest wall (skeletal muscle) | CK (F+) | ACTB-GLI1Δ | Recent case |
F, female; M, male; RP, retroperitoneum; ST, soft tissue; FU; follow-up;
targeted RNA sequencing;
FISH; LN, lymph nodes; met, metastasis; mo, months, AWD, alive with disease; positive; neg, negative; (+), F+, focally positive.
Figure 4. ACTB1-GLI1 and novel PTCH1-GLI1 gene fusion structure and molecular correlates.
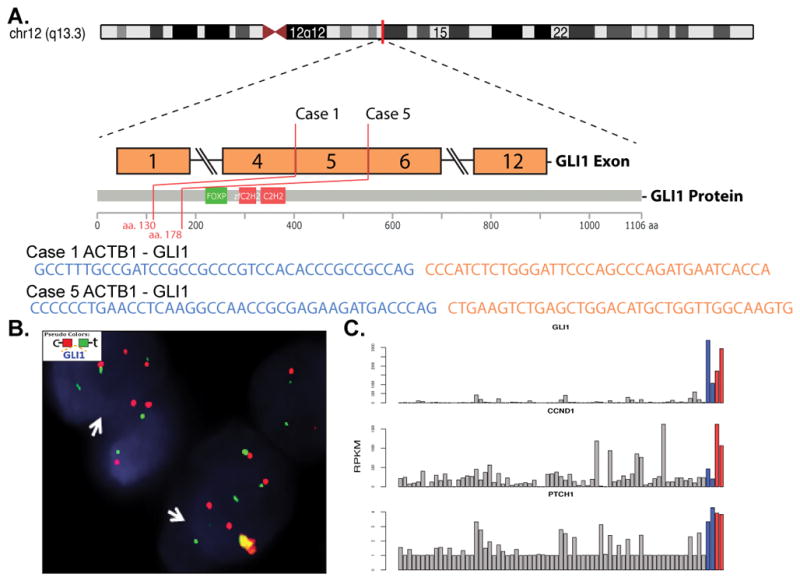
A. Diagrammatic representation of GLI1 (12q13.3) genomic location and exonic transcript composition in 2 positive cases for ACTB1-GLI1 fusion and one for PTCH1-GLI1 by targeted RNA sequencing B. FISH studies showing GLI1 break-apart (red, centromeric; green telomeric); C. Bar charts depicting mRNA expression from the RNAseq dataset in 2 ACTB1-GLI1 and 1 PTCH1-GLI1 fusion positive cases (red bars, cases 1 &5; orange bar, case 4), compared to 2 unrelated soft tissue tumors with 12q13-15 regional amplification (which includes GLI1 gene; blue bars) and other soft tissue tumors (gray). Upper and lower panel shows significant GLI1 and PTCH1 mRNA upregulation, respectively, in all 5 tumors with GLI1 gene abnormalities, while the middle panel shows CCND1 overexpression only in the ACTB1-GLI1 and PTCH1-GLI1 fusion positive tumors.
Overexpression of GLI1 mRNA was detected in all 4 tumors tested by targeted RNA sequencing, irrespective of the ACTB1 or PTCH1 fusion partner (Fig. 4C, upper panel, red and orange bars), at similarly high levels when compared to 2 unrelated tumors showing 12q13-15 amplifications that span GLI1 locus (blue bars). GLI1 overexpression was also present in the S100 protein negative tumor (case 5), tested on the same platform.
Tumors associated with GLI1-related fusions have metastatic potential
Clinical follow-up was available for 3 patients, ranging 21–80 months. One patient was lost to follow-up, while the remaining two being recent cases. Two patients developed local recurrence before developing distant spread. Three patients developed metastatic disease to the lymph nodes and one additionally to the lung (Table 1) and are alive with disease after a mean follow-up of 50 months.
DISCUSSION
Pericytic neoplasms with recurrent t(7;12) translocations were first reported by Dahlen at al.1 as likely benign lesions occurring in young adults; tumors appear to often arise in the tongue, and occasionally the stomach and soft tissue. Morphologically the tumors were composed of monomorphic ovoid to spindle cells arranged in solid sheets around a conspicuous, arborizing vasculature. Immunohistochemically, all 5 tumors showed a variable degree of SMA reactivity (3 focal, 2 diffuse), while none of the 4 cases tested showed S100 protein or cytokeratin positivity.1 Based on the perivascular growth and actin reactivity the tumors were suggested to have a pericytic phenotype. Although some of the cases showed a prominent subendothelial protrusion into vascular lumina reminiscent of myopericytic neoplasms, they were believed to be a distinct entity from the wide spectrum of common perivascular myoid tumors, such as glomus tumors/myopericytomas, despite their shared immunoreactivity for SMA and ultrastructural features of modified smooth muscle cells1. Since the original description of 5 cases, 2 additional pericytomas harboring ACTB-GLI1 fusion positive have been reported, one in the bone (tallus)3 and one in the stomach4. All 7 cases reported thus far pursued a benign course, lacking local recurrences or metastases after long follow-up.
An alternative recurrent GLI1 fusion to MALAT1 gene has been recently reported in 2 distinct pathologic entities, including: gastric plexiform fibromyxoma7 and gastroblastoma8. MALAT1 (Metastasis associated lung adenocarcinoma transcript 1) is a long non-coding RNA, which is highly expressed in the nucleus of most cells and expected to provide a strong promoter that drives GLI1 overexpression9. Plexiform fibromyxoma is a rare gastric tumor occurring in young patients composed of multinodular growth of bland myofibroblastic-type spindle cells associated with myxoid stroma and a rich capillary network10,11. The tumors show expression of SMA, while being negative for S100 protein, CD117, DOG1, CD34 and EMA. A MALAT1-GLI1 fusion has been described recently in a subset (15–20%) of plexiform fibromyxoma, which showed nuclear and cytoplasmic GLI1 immunoreactivity, while tumors lacking the fusion were negative7. Additional functional studies performed revealed that the truncated GLI1 protein was overexpressed and retained the capacity to transcriptionally activate its downstream target genes7.
An identical MALAT1-GLI1 fusion was recently reported in 4 cases of gastroblastoma8. Gastroblastoma was first described by Miettinen and colleagues in 2009 as a rare biphasic neoplasm of young adults with a nested multinodular growth and potential for locoregional or distant spread11. The epithelial component of these lesions is composed of nests and rosette-like structures with eosinophilic material; these tumors contain immunoreactivity for cytokeratin and CD56. The spindle cells show bland cytologic features, with reticular or short fascicular growth patterns. One of the three tumors tested showed patchy SMA expression, and the only case reportedly tested for S100 protein was negative. As expected from the GLI1 mRNA up-regulation, MALAT1-GLI1 fusion positive gastroblastomas showed strong nuclear and cytoplasmic patterns of GLI1 immunoexpression8. Due to the biphasic phenotype and clinical presentation in young adults, the main differential diagnosis included biphasic synovial sarcoma. Despite identical MALAT1-GLI1 fusions (including similar fusion transcript structure), gastric plexiform fibromyxoma and gastroblastoma are morphologically and immunohistochemically distinct pathologic entities. It is quite intriguing that both tumors occur exclusively in the stomach. To complicate matters further, rare examples of gastric pericytomas with ACTB-GLI1 fusion have also been described1, including a recent example in the pyloric wall of a 9 year-old girl which lacked the typical SMA reactivity and instead showed CD56 and focal EMA positivity4. The latter tumor had a solid and cystic gross appearance and microscopically was composed of sheets of bland ovoid to spindle cells associated with a plexiform vasculature. The tumor appeared negative for cytokeratin and S100 protein and ultrastructurally lacked evidence of epithelial or smooth muscle lineage. Some of the morphologic features were quite similar to our cohort and the significance of its non-specific immunoprofile remains to be determined.
The present cohort showed a predilection for soft tissue location and had a wide age range at diagnosis, although most patients were younger than 40 years of age. Microscopically all tumors were composed of monomorphic round to epithelioid cells arranged with a distinctive nested growth pattern, separated by a delicate stroma and capillary network. Some of the tumors showed features reminiscent of pericytoma or glomus tumor, while others resembled myoepithelial neoplasms, due to cords, sieve-like or reticular growth patterns within myxoid stroma. Immunohistochemically, all cases except two cases showed strong and diffuse S100 protein expression. One of the S100 protein negative cases showed diffuse reactivity for CD56 and focal Cytokeratin reactivity, as single cells and small nests. The second S100 protein negative neoplasm showed a vimentin-only immunoprofile. Based on these findings, one tumor (case#3) was initially diagnosed as an epithelioid schwannoma due to its monomorphic epithelioid appearance and strong S100 protein immunoreactivity, and subsequently reclassified as an S100 positive malignant neoplasm after the patient developed metastasis to the groin lymph nodes. Three other cases were interpreted as myoepithelial carcinomas, due to the presence of a nested appearance or tubular structure formation, within variably myxoid stroma, and immunoreactivity for either S100 protein or focal cytokeratin. One tumor was classified as an unusual pericytic tumor, despite the strong S100 protein positivity, based on its morphologic appearance reminiscent of a cellular glomus and the presence of the ACTB1-GLI1 gene fusion. However, none of the tumors showed immunoexpression of actin. Three patients developed lymph node metastases, while one patient additionally developed distant spread to the lung. All patients with available follow-up are still alive with disease, after a mean follow-up of 50 months. This pattern of lymph node metastasis would be highly unusual for a conventional pericytic neoplasm. Case 6 (a chest wall deep soft tissue tumor) was the only case that showed focal Cytokeratin positivity (as rare single cells and clusters) and diffuse reactivity for CD56, while being negative for S100 protein and SMA. The tumor showed scattered tubular structures within the predominant solid epithelioid areas, but lacked a spindle cell component. Based on the presence of GLI1 gene rearrangement and above immunoprofile, this case suggests a phenotypic overlap with the recently described MALAT1-GLI1 positive gastroblastoma. Further larger studies are needed to establish whether entities such as gastroblastoma are indeed organ-specific and/or might be associated with other GLI1 gene partners than MALAT1. At present it remains possible these entities fall under the same rubric as the group of tumors presented herein.
GLI expression in adult tissues is restricted to the fallopian tube, myometrium and testis. GLI functions as an effector of the sonic hedgehog (SHH) pathway, inducing up- or down- regulation of multiple downstream targets. GLI activation has been identified in various neoplasms as a result of mutations in the PTCH (patched) and SMOH (smoothened) genes, encoding components of the SHH pathway. Furthermore, GLI1 gene amplification has been detected in various tumor types, such as glioblastoma, B-cell lymphoma and sarcomas12. The targeted RNA sequencing showed that the known downstream targets of GLI1, such as PTCH1 was upregulated in all 4 cases with GLI1 gene abnormalities, while CCND1 was highly upregulated only in the GLI1-fusion positive tumors, while not in the tumors with regional 12q13-15 amplifications, which included GLI1 (Fig. 4). Remarkably, the targeted RNA sequencing in case 4 (a recurrent/metastatic head and neck tumor), showed the presence of a novel PTCH1-GLI1 fusion which was associated with overexpression of both GLI1 and PTCH1 mRNA. Particularly noteworthy is the oncogenic activation of two members of SHH pathway through chromosomal translocation. To our knowledge, this is the first example of gene fusion involving PTCH1 in neoplasia.
Our cohort highlights yet another translocation-associated, S100 protein positive/SOX10 negative tumor; this is similar to the immunophenotype of PAX3-MAML3 positive biphenotypic sinonasal sarcoma, NTRK1-fusion positive lipofibromatosis-like neural tumor, TFG-RET-positive in infantile fibrosarcoma-like tumor, etc13–15. The presence of strong S100 positivity in the absence of SOX10 – in the context of a monomorphic spindle or epithelioid histology – should include a fusion-positive neoplasm, outside the more common neuroectodermal/schwannian line of differentiation.
In summary, we report recurrent GLI1-related gene fusions in a group of malignant mesenchymal neoplasms involving soft tissue, and occasionally bone, with an often nested epithelioid phenotype and strong S100 immunoreactivity. Although some of the cases have a morphologic appearance reminiscent of either pericytoma or myoepithelioma, their immunoprofile and clinical behavior suggest a novel pathologic entity, which can be regarded, at least temporarily, under the descriptive terminology of ‘malignant epithelioid neoplasm with GLI1 fusions’. Further investigations are needed to confirm if this lesion represents a unique neoplastic entity, or perhaps an unusual clinical and immunophenotypic manifestation within the spectrum of previously described lesions with GLI1-related fusions. Our findings may also have potential therapeutic implications, as tumors with GLI1 oncogenic activations, and subsequent PTCH1 overexpression, might be sensitive to SHH pathway inhibitors16.
Supplementary Material
FISH showing break-apart signals for ACTB1 (A) and PTCH1 (B) (white arrows; red, centromeric; green, telomeric).
Acknowledgments
Supported in part by: P50 CA140146-01 (CRA); P30-CA008748 (CRA); Kristen Ann Carr Foundation (CRA); Cycle for Survival (CRA)
Footnotes
Conflicts of interest: none
References
- 1.Dahlen A, Fletcher CD, Mertens F, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12) Am J Pathol. 2004;164:1645–1653. doi: 10.1016/s0002-9440(10)63723-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dahlen A, Mertens F, Mandahl N, et al. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12) Biochem Biophys Res Commun. 2004;325:1318–1323. doi: 10.1016/j.bbrc.2004.10.172. [DOI] [PubMed] [Google Scholar]
- 3.Bridge JA, Sanders K, Huang D, et al. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum Pathol. 2012;43:1524–1529. doi: 10.1016/j.humpath.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castro E, Cortes-Santiago N, Ferguson LM, et al. Translocation t(7;12) as the sole chromosomal abnormality resulting in ACTB-GLI1 fusion in pediatric gastric pericytoma. Hum Pathol. 2016;53:137–141. doi: 10.1016/j.humpath.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Hung YP, Fletcher CDM. Myopericytomatosis: Clinicopathologic Analysis of 11 Cases With Molecular Identification of Recurrent PDGFRB Alterations in Myopericytomatosis and Myopericytoma. Am J Surg Pathol. 2017;41:1034–1044. doi: 10.1097/PAS.0000000000000862. [DOI] [PubMed] [Google Scholar]
- 6.Mosquera JM, Sboner A, Zhang L, et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer. 2013;52:1075–1087. doi: 10.1002/gcc.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spans L, Fletcher CD, Antonescu CR, et al. Recurrent MALAT1-GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J Pathol. 2016;239:335–343. doi: 10.1002/path.4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham RP, Nair AA, Davila JI, et al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol. 2017;30:1443–1452. doi: 10.1038/modpathol.2017.68. [DOI] [PubMed] [Google Scholar]
- 9.Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi Y, Shimizu S, Ishida T, et al. Plexiform angiomyxoid myofibroblastic tumor of the stomach. Am J Surg Pathol. 2007;31:724–728. doi: 10.1097/01.pas.0000213448.54643.2f. [DOI] [PubMed] [Google Scholar]
- 11.Miettinen M, Makhlouf HR, Sobin LH, et al. Plexiform fibromyxoma: a distinctive benign gastric antral neoplasm not to be confused with a myxoid GIST. Am J Surg Pathol. 2009;33:1624–1632. doi: 10.1097/PAS.0b013e3181ae666a. [DOI] [PubMed] [Google Scholar]
- 12.Stein U, Eder C, Karsten U, et al. GLI gene expression in bone and soft tissue sarcomas of adult patients correlates with tumor grade. Cancer Res. 1999;59:1890–1895. [PubMed] [Google Scholar]
- 13.Wang X, Bledsoe KL, Graham RP, et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet. 2014;46:666–668. doi: 10.1038/ng.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 Gene Fusions Define a Novel Subset of Locally Aggressive Lipofibromatosis-like Neural Tumors. Am J Surg Pathol. 2016;40:1407–1416. doi: 10.1097/PAS.0000000000000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flucke U, van Noesel MM, Wijnen M, et al. TFG-MET fusion in an infantile spindle cell sarcoma with neural features. Genes Chromosomes Cancer. 2017;56:663–667. doi: 10.1002/gcc.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rimkus TK, Carpenter RL, Qasem S, et al. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) 2016:8. doi: 10.3390/cancers8020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FISH showing break-apart signals for ACTB1 (A) and PTCH1 (B) (white arrows; red, centromeric; green, telomeric).