

Abstract
Safety is prerequisite for preventive medicine, but non-toxic agents are generally ineffective as clinical chemoprevention. Here we propose a strategy overcoming this challenge by delivering molecular-targeted agent specifically to the effector cell type to achieve sufficient potency, while circumventing toxicity in the context of cancer chemoprevention. Hepatic myofibroblasts drive progressive fibrosis that results in cirrhosis and liver cancer. In a rat model of cirrhosis-driven liver cancer, a small molecule epidermal growth factor receptor inhibitor, erlotinib, was delivered specifically to myofibroblasts by a versatile nanoparticle-based system, targeting platelet-derived growth factor receptor-beta uniquely expressed on their surface in the liver. With systemic administration of erlotinib, tumor burden was reduced to 31%, which was further improved to 21% by myofibroblast-targeted delivery even with reduced erlotinib dose (7.3-fold reduction with equivalent erlotinib dose) and less hepatocyte damage. These findings demonstrate a strategy, cell type-specific kinase inhibition, for more effective and safer precision cancer chemoprevention.
Keywords: Cancer chemoprevention, liver cancer, kinase inhibitor, epidermal growth factor, nanoparticle
Graphical Abstract
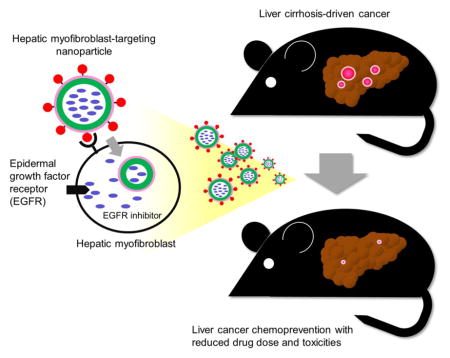
Nanoparticle-based system for cell type-specific delivery of selective kinase inhibitor was developed. The system enabled specific inhibition of epidermal growth factor signaling in hepatic myofibroblasts, the major driver of liver cancer development, and elicited cancer chemoprevention effect with reduced drug dose and toxicities.
Background
Recent development of specific molecular pathway modulators, especially kinase inhibitors, has revolutionized anti-cancer treatment by enabling personalized administration of highly selective inhibitors targeting patient-specific somatic molecular aberrations in tumors.1 In contrast, although preventing cancer development is deemed as theoretically the most impactful strategy to improve patient prognosis, development of cancer chemoprevention therapy has lagged far behind with almost no substantial progress translated to clinic over the past decades.2 One of the major hurdles is the higher requirement for drug safety as preventive medicine prescribed to asymptomatic, still cancer-free individuals. For this reason, less/non-toxic natural product-derived and/or nutritional substances such as vitamins have been preferably examined for chemoprevention purpose. However, these agents are generally not potent enough to elicit clinically meaningful efficacy.3 On the other hand, more potent selective molecular-targeted agents such as kinase inhibitors and therapeutic antibodies are associated with critical toxicities, which are generally acceptable only in the context of therapy for advanced cancer patients with limited life expectancy and no other therapeutic options. This dilemma may be solved by delivery of molecular-targeted agents pinpointing target organ/cells.
Liver cancer is the second leading cause of cancer death worldwide, which develops in cirrhosis, the terminal stage of progressive fibrosis caused by viral hepatitis or metabolic disorders.4 Hepatic myofibroblasts, originating mainly from transdifferentiation of hepatic stellate cells, drive liver fibrogenesis and generate cancer initiation-supporting tissue microenvironment.5 Our previous study suggested that pharmacological inhibition of epidermal growth factor receptor (EGFR) in myofibroblasts may serve as liver cancer chemoprevention,6 based on which a proof-of-concept clinical trial was initiated (ClinicalTrials.gov Identifier: NCT02273362). However, the known toxicities of the drug likely hamper its clinical deployment.7 To overcome the challenge of cancer chemoprevention development, in this study, we examined whether target cell type-specific delivery could serve as repurposing of existing potent kinase inhibitor for cancer chemoprevention using liver cancer as an example of malignancy developing in chronically diseased organs.
Methods
Synthesis of myofibroblast-targeting nanoparticles
Fluorescent dye-attached myofibroblast-targeting nanoparticles were synthesized as follows. Briefly, 50 mg Platelet-derived growth factor receptor β (PDGFRB)-targeting peptide (PPB) (GenScript USA) was dissolved in 2 mL dimethylformamide (DMF) together with 45 mg N-γ-maleimidobutyryl-oxysuccinimide ester (GMBS), and stirred for 6 hr at room temperature. The solvent was removed using high vacuum, and the formation of PPB-GMBS was confirmed by liquid chromatography–mass spectrometry (LC-MS). Subsequently, 10 mg mesoporous thiol (SH)-functionalized silica nanoparticles (200 nm in diameter) (Sigma Aldrich) and 17 mg PPB-GMBS were mixed in 0.5 mL DMF, stirred for 1 hr at room temperature. Approximately 68% of SH groups on the surface of nanoparticle were bound by PPB-GMBS as quantified by Ellman’s assay (Thermo Scientific) performed in 1 mg/mL nanoparticle suspension. Then, 25 mg fluorescein isothiocyanate (FITC)-maleimide (Sigma Aldrich) was added and stirred for additional 1 hr. Subsequently, polyethylene glycol (PEG) maleimide (NOF America) was added to cover unbound SH groups on the surface of the nanoparticles, and stirred for 24 hr at room temperature. Upon completion of the reaction, the nanoparticles (PPB-NP-FITC) were washed 3 times with methanol to remove unreacted PPB, FITC dye, and PEG polymer, and confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy. Non-myofibroblast-targeting nanoparticles were similarly synthesized without adding PPB-GMBS (NP-FITC).
Erlotinib-loaded myofibroblast-targeting nanoparticles (PPB-NP-erlotinib) were synthesized as follows. Ten mg of erlotinib (LC Laboratories) in 300 μL acetonitrile/methanol (1:1) was added to 10 mg PPB-attached pegylated nanoparticles and shaken for 24 hr. Drug-loaded nanoparticles were then centrifuged at 14,000 g for 5 min to remove supernatant, washed 5 times with 500 μL acetonitrile/methanol (1:1), and vacuum dried. Estimated amount of loaded erlotinib based on the concentration difference in the original solution and the supernatant after loading was 70% of the nanoparticle weight, which was higher than alternative formulation, where erlotinib was loaded before PPB attachment and pegylation (40%). Non-myofibroblast-targeting nanoparticles were similarly synthesized without adding PPB-GMBS (NP-erlotinib). The erlotinib-loaded nanoparticles was incubated at 37 °C for 30 min in 500 μL methanol, centrifuged at 14,000 g for 5 min to remove supernatant, and no degradation of erlotinib was confirmed by LC-MS. Morphology and size of nanoparticles (1 mg) in rat plasma (200 μL) were characterized for 18 days by transmission electron microscopy (TEM) in a drop of a sonicated aqueous solution of nanoparticles on a carbon-coated copper grid. The grids were negatively stained with an aqueous solution of 0.5% uranyl acetate, and analyzed on TEM microscope (HT7000, Hitachi). In vitro release of erlotinib from the nanoparticles was assessed in 2 mL rat plasma for 18 days. At each pre-determined time point, the solution was centrifuged at 10,000 g for 10 min, and 1 mL of supernatant was removed and replaced with 1 mL fresh plasma. Erlotinib concentration in the supernatant was determined by absorbance at 658 nm.
Cellular internalization of nanoparticles
Human myofibroblast cell lines, TWNT-4 and LX-2,8,9 and hepatoma cell line, Hep G2, (American Type Culture Collection) were cultured in DMEM with 10% fetal bovine serum (FBS) supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin (Mediatech CellGro). Cells were maintained at 37 °C in a humidified incubator with 5% CO2. The dye-attached nanoparticles (PPB-NP-FITC and NP-FITC) (100 μL at 1 mg/mL) were added to the cell culture and incubated at 37 °C for 24 hr. Cells were then washed with phosphate buffered saline (PBS) 3 times, and association and internalization of the nanoparticles in the cells were evaluated by fluorescence microscope (Leica DM6000) and confocal microscope (Zeiss LSM880 Airyscan) (excitation at 488 nm for green channel).
In vivo assessment of nanoparticle biodistribution and chemopreventive effect
For the assessment of nanoparticle biodistribution, male C57BL/6 mice (Charles River) were intraperitoneally injected carbon tetrachloride (0.1 mg/kg) twice per week for 2 weeks to activate hepatic stellate cells/myofibroblasts. Subsequently, PPB-NP-FITC or NP-FITC nanoparticles were injected via tail vein (n=2 for each condition), and mice were sacrificed 3 to 4 hr later. Each organ was immediately obtained and fluorescence imaging was performed by using IVIS small animal imaging system (Xenogen).
For the assessment of chemopreventive effect, male Wistar rats (Charles River) received weekly intraperitoneal injections of low-dose diethylnitrosamine (100 mg/kg) to induce liver fibrogenesis and resulting development of neoplastic nodules as previously reported6. Each rat was randomly assigned to either of the following four groups and received 6 weeks of treatment (n=8 in each group): (a) vehicle control: intraperitoneal injection of PBS 5 times per week; (b) erlotinib: intraperitoneal injection of erlotinib (2 mg/kg) 5 times per week (weekly erlotinib dose: 10 mg/kg) instead of intravenous injection, which is known to result in rapid elimination from the body;6 (c) PPB-NP: tail vein injection of PPB-NP nanoparticles (8.9 mg/kg) twice per week; (d) PPB-NP-erlotinib: tail vein injection of PPB-NP-erlotinib nanoparticles (12.9 mg/kg, containing 4 mg erlotinib) twice per week (weekly erlotinib dose: 8 mg/kg). Rats were sacrificed after a one-week washout period to eliminate acute effects of diethylnitrosamine, and each organ and plasma were fixed in 10% formalin and embedded in paraffin blocks or snap-frozen and stored at −80°C until use.
Animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” of the National Academy of Sciences. All animals were maintained and treated following the protocol approved by Institutional Animal Care and Use Committee (IACUC) at Icahn School of Medicine at Mount Sinai (protocol number: LA12-00338).
Western blotting
Cellular protein expression in TWNT-4, LX-2, and Hep G2 cell lines was assessed by Western blotting following the standard protocol. Briefly, cells were lysed using RIPA buffer with protease and protein phosphatase inhibitor mixture (Roche), subjected to SDS-PAGE (7.5%), and separated proteins were transferred to PVDF membrane. The membranes were blocked with TBST with 5% skimmed milk, and incubated with rabbit polyclonal antibody for either PDGFRB (Santa Cruz, sc-432), EGFR (Cell Signaling, 2232L), phospho-EGFR (p-EGFR) (Tyr1068, Cell Signaling, 2234L), GAPDH (Abcam, ab9385), and actin (Abcam, ab8227). After washings, the blots were incubated with HRP-conjugated goat anti-rabbit antibody (Dako), and developed using ECL solution (ThermoFisher) according to the manufacturer’s instruction. Intensity of each band was quantified by using ImageJ software (https://imagej.nih.gov/ij).
Immunostaining
For immunofluorescence, the tissues were cut into 5 μm-thick sections, fixed in acetone, incubated with the primary antibody against glial fibrillary acidic protein (GFAP) (Dako, M076029-2, 1:100 dilution) and with secondary antibodies. Nucleus were counterstained with DAPI (Life Technologies, D1306) and examined with fluorescence microscope (Leica). For immunohistochemistry, formalin-fixed paraffin-embedded tissue blocks were cut into 5 μm-thick sections, deparaffinized in xylene, and dehydrated in ethanol. Antigen retrieval was performed by steaming for 20 min in 10 mM citrate buffer (pH 6.0) for glutathione S-transferase pi 1 (GSTP1) or in 1 mM EDTA (pH 8.5) for glypican-3 (GPC3) and p-EGFR staining. Endogenous peroxidase activity was blocked with peroxidase inhibition for 10 min using EnVision kit (Dako). The slides were incubated with primary antibodies against PDGFRB (Abcam, ab32570), GSTP1 (Sigma-Aldrich, SAB3500265, 1:100 dilution), GPC3 (Abcam, ab129381, 1:200 dilution), and p-EGFR (Cell Signaling, #3777, 1:50 dilution) at 4 °C overnight, then with secondary HRP-labeled polymer anti-rabbit antibody (Dako, K4011) for GSTP1 and p-EGFR and secondary anti-mouse antibody (Abcam, ab170190) for GPC3 at room temperature for 30 min. Peroxidase activity was visualized using 3,3′-diaminobenzidine tetrahydrochloride (Dako) as chromagen (1.5 min incubation). Sections were counterstained with hematoxylin for nuclei. GSTP1- and GPC3-positive foci were counted based on size threshold of 100 μm (cluster of approximately 30 cells) and 10 μm (cluster of approximately 3 cells), respectively, in 40× images of 4 to 5 randomly chosen visual fields using ImageJ software ver. 1.48 (https://imagej.nih.gov/ij). Collagen was visualized by Sirius red staining using Direct Red 80 (Sigma-Aldrich) following manufacturer’s instruction, and amount of fibrous tissue was quantified as collagen proportionate area (CPA).
Structural and statistical data analysis
The 3D modeling of docking between PPB and extracellular PDGF-binding domain of PDGFRB protein was visualized using PyMol software (www.pymol.org) based on crystal structure of PDGF-PDGFRB complex.10 Continuous and categorical data were tested by using Wilcoxon rank-sum test and Fisher’s exact test, respectively. Multiple hypothesis testing was corrected by using Bonferroni correction as needed. Two-tailed p-value <0.05 was regarded as statistically significant. All statistical analyses were performed using R statistical language (www.r-project.org).
Results
Synthesis and characterization of myofibroblast-targeting nanoparticles
Platelet-derived growth factor receptor β (PDGFRB) is uniquely expressed on the surface of fibrogenic myofibroblasts in the liver,5 which can be specifically bound by a short cyclic PDGFRB-binding peptide (PPB) for delivery of directly-conjugated macromolecules such as interferon-γ protein.11 We first developed a versatile system for small molecule delivery to myofibroblasts by utilizing biodegradable mesoporous silica nanoparticles (200 nm in diameter) with an array of PPB peptides attached on their surface via N-γ-maleimidobutyryl-oxysuccinimide ester (GMBS) linker and coated by polyethylene glycol (PEG) polymer to control drug release and reduce non-specific uptake by the reticuloendothelial system (PPB-NP) (Figure 1A). In vitro degradation of the particles in rat plasma started at day 7, but average size of non-degraded particles was maintained between 150 and 200 nm as assessed by transmission electron microscopy (TEM) (Figure 1B, C). A small molecule EGFR kinase inhibitor, erlotinib, loaded in the nanoparticles (PPB-NP-erlotinib), was stably released in rat plasma for 12 days maintaining consistent drug concentration until all loaded erlotinib was released (Figure 1D, E). Given the short half-life of erlotinib (36 hr) and reversibility of its EGFR inhibition,12 the inhibitory activity is assumed to be proportional to the concentration. Thus, myofibroblast-targeting, erlotinib-loaded nanoparticles were successfully formulated.
Figure 1.

Myofibroblast-targeting nanoparticles. (A) Schematic presentation of erlotinib-loaded nanoparticles that target PDGFRB-expressing myofibroblasts (PPB-NP-erlotinib). Erlotinib is released after cellular internalization and inhibits intracellular kinase domain of EGFR protein. In the 3D structural modeling, PPB fits the extracellular domain of PDGFRB with close proximity to the site of PDGF ligand binding. (B) Morphology of the nanoparticles in rat plasma over time by TEM imaging. Scale bar indicates 200 nm. (C) Size of nanoparticles in rat plasma over time measured on TEM images (median of 59 nanoparticles at each time point). Boxes represent 75th and 25th percentile, horizontal line is median, and whiskers mark lowest and highest values. Outliers outside 1.5× of inter-quartile range are shown as open circles. (D) Concentration of erlotinib released from the nanoparticles over time in vitro in rat plasma. (E) Cumulative amount (%) of erlotinib released from the nanoparticles over time in vitro in rat plasma.
PDGFRB: platelet-derived growth factor receptor-β, PPB: PDGFRB-binding peptide, NP: nanoparticle, GMBS: N--maleimidobutyryl-oxysuccinimide ester, PEG: polyethylene glycol, EGFR: epidermal growth factor receptor, TEM: transmission electron microscopy
In vitro selective targeting of myofibroblasts
Next, selective uptake of the PDGFRB-targeting nanoparticles conjugated with fluorescein isothiocyanate (FITC) dye (PPB-NP-FITC) was used to assess cellular uptake in PDGFRB-expressing human myofibroblast cell lines, TWNT-4 and LX-2, and hepatoma cell line, Hep G2, which lacks PDGFRB expression. As expected, after 24 hr of incubation, the nanoparticles showed distinctly higher association with TWNT-4 and LX-2 cells compared to Hep G2 cells when PPB is attached (PPB-NP-FITC) as confirmed by fluorescence imaging (Figure 2A). Internalization of PPB-NP-FITC in TWNT-4 cells was further confirmed in the images of 0.7 μm-thick sections obtained by confocal microscopy (Figure 2B, C). p-EGFR suppression by 6 hr treatment with PPB-NP-erlotinib was observed in TWNT-4 and LX-2 in a dose-dependent manner, but not in Hep G2 cells (Figure 2D). Collectively, these results support in vitro selective uptake of the PPB-conjugated nanoparticles and EGFR inhibition in PDGFRB-expressing myofibroblast cells.
Figure 2.

Cellular internalization of myofibroblast-targeting nanoparticles. (A) Upper panels show expression of PGDFRB and GAPDH proteins by Western blotting, and lower panels show fluorescence microscope images in human myofibroblast cell lines, TWNT-4 and LX-2, and hepatoma cell line, Hep G2, incubated with FITC-labeled nanoparticles with (PPB-NP-FITC) or without (NP-FITC) PPB for 24 hr. Scale bar indicates 400 μm. (B) Confocal microscope image of internalized nanoparticles in 0.7 μm-thick slice in TWNT-4 cells incubated with PPB-NP-FITC for 24 hr. Scale bar indicates 40 μm. (C) Fluorescence intensity inside TWNT-4 cells treated with PPB-NP-FITC or NP-FITC (randomly chosen 7 cells for each group). Boxes represent 75th and 25th percentile, horizontal line is the median, and whiskers mark lowest and highest values. (D) Western blotting to quantify p-EGFR suppression by PPB-NP-erlotinib at two doses (0.125 mg and 0.0078 mg). Numbers indicate ratios of p-EGFR to total EGFR protein normalized to untreated controls.
PDGFRB: platelet-derived growth factor receptor-β, PPB: GAPDH: glyceraldehyde-3-phosphate dehydrogenase, PDGFRB-binding peptide, NP: nanoparticle, FITC: fluorescein isothiocyanate, EGFR: epidermal growth factor receptor.
In vivo biodistribution of myofibroblast-targeting nanoparticles
Subsequently, anticipated myofibroblast-specific delivery of the PDGFRB-targeting nanoparticles was evaluated in C57BL/6J mice with fibrogenic myofibroblast activation induced by intraperitoneal injection of carbon tetrachloride, a well-established stimuli to activate and proliferate myofibroblasts in the liver and lung.5,13 The fluorescent dye-conjugated nanoparticles with (PPB-NP-FITC) and without PPB (NP-FITC) were injected via tail vein in the mice following the myofibroblast induction. Without the PDGFRB-targeting PPB, 4% and 12% of nanoparticles (NP-FITC) quantified in the imaged organs were entrapped by spleen and kidneys, which were reduced to 3% and 9% with the use of PPB-NP-FITC, respectively (Figure 3A, B). Accumulation of FITC signal in the liver was high even with NP-FITC (78%), and slightly increased by the use of PPB-NP-FITC. FITC signals in the lungs were increased from 5% to 14% by attachment of PPB, where resident myofibroblasts were indeed activated and proliferated in peri-capillary and fibrotic area by carbon tetrachloride (Figure 3B, C). Because lung myofibroblasts are not activated in liver cirrhosis patients, relative non-specific accumulation to lungs is expected to remain minor. Microscopically, the fluorescence signals from PPB-NP-FITC were detected in hepatic sinusoids (vascular channels that connect arterial/portal blood flows to hepatic vein),5 which were co-localized with peri-sinusoidal cells expressing a hepatic myofibroblast marker, glial fibrillary acidic protein (GFAP) (Figure 3D, E). Taken together, in vivo enrichment of the PPB-conjugated nanoparticles in the organs with activated myofibroblasts was confirmed.
Figure 3.

In vivo biodistribution of myofibroblast-targeting nanoparticles. (A) Fluorescence signals from each organ isolated from carbon tetrachloride-treated mice after tail vein injection of FITC-labeled nanoparticles with (PPB-NP-FITC) or without (NP-FITC) PPB by IVIS imaging. (B) Quantified fluorescence signals from each organ (n=2 per group). Error bars indicate standard error of mean. (C) Immunohistochemistry of PDGFRB in lung tissues from mice intraperitoneally injected with carbon tetrachloride or PBS (control). Scale bar indicates 100 μm. (D) Immunofluorescence images of liver tissues of PPB-NP-FITC-injected mice. Scale bar indicates 50 μm. (E) Immunofluorescence images of three representative peri-sinusoidal cells in liver tissues of PPB-NP-FITC-injected mice.
PPB: platelet-derived growth factor receptor-β-binding peptide, NP: nanoparticle, FITC: fluorescein isothiocyanate, PDGFRB: platelet-derived growth factor receptor-β receptor, glial fibrillary acidic protein (GFAP).
In vivo liver cancer chemopreventive effect of myofibroblast-targeted EGFR inhibition
Finally, in vivo liver cancer chemopreventive effect of PPB-NP-erlotinib was assessed in an established rat model of cirrhosis-driven liver cancer induced by weekly injection of low-dose diethylnitrosamine, which recapitulates global transcriptome dysregulation in human fibrotic/cirrhotic livers at risk of cancer development.6,14 The rats developed bridging liver fibrosis and cirrhotic nodules followed by emergence of diffuse clusters of epithelial cells positive for glutathione S-transferase pi 1 (GSTP1) protein expression, a widely used marker of neoplastic nodules in the liver3 (Figure 4A). Consistent with our previous observation, systemic daily administration of the EGFR inhibitor, erlotinib, showed a trend of reduced number of the GSTP1-positive foci to 31% compared to vehicle-treated controls (p=0.16) (Figure 4B). Animals treated with biweekly injection of the myofibroblast-targeting nanoparticles (PPB-NP-erlotinib) resulted in 10% more reduction of neoplastic foci, which reached statistical significance (i.e., 21% compared to controls, p=0.01) with 17% less total cumulative dose of erlotinib (19.9 mg for systemic erlotinib vs. 16.4 mg for PPB-NP-erlotinib per animal). With equivalent dose of erlotinib with the systemic treatment, PPB-NP-erlotinib could reduce the tumor burden to 4% (7.3-fold reduction, Fisher’s exact test p<0.001) if the tumor reduction is assumed to be proportional to cumulative dose of erlotinib. In addition, a clinically used early HCC marker, glypican 3 (GPC3), was also examined.15 Similarly, PPB-NP-erlotinib resulted in more significant reduction of GPC3-positive foci compared to systemic erlotinib treatment (Figure 4C, D).
Figure 4.

In vivo chemopreventive effect of myofibroblast-targeted EGFR inhibition. (A) Immunohistochemistry of GSTP1 protein. Scale bar indicates 200 μm. (B) Number of GSTP1-positive foci per visual field normalized to respective controls according to the treatment groups (n=8 per group). Wilcoxon rank-sum test p-values (Bonferroni-corrected) are shown. Longer and shorter horizontal bars indicate mean and standard error of mean, respectively. (C) Immunohistochemistry of GPC3 protein. Closed arrow head indicates GPC3-positive focus. Scale bar indicates 200 μm. (D) Number of GPC3-positive foci per visual field normalized to respective controls according to the treatment groups (n=8 per group). (E) Immunohistochemistry of p-EGFR protein. Open arrow head indicates p-EGFR-positive cell. Closed arrow head indicates neoplastic foci with hyper-eosinophilic cytoplasm and GSTP1 positivity in serial tissue section as shown in panel A. Scale bar indicates 100 μm. (F) Number of p-EGFR-positive cells normalized to respective controls according to the treatment groups (n=8 per group). (G) Sirius red staining to visualize and quantify fibrotic tissue. Scale bar indicates 200 μm. (H) Collagen proportionate area (CPA) per visual field normalized to respective controls according to the treatment groups (n=8 per group).
*Vehicle control for systemic erlotinib administration (i.e., intraperitoneal injection). **Vehicle control for myofibroblast-targeting nanoparticles (i.e., PPB-NP). PPB: platelet-derived growth factor receptor-β-binding peptide, NP: nanoparticle.
In the vehicle-treated animals, phospho-EGFR (p-EGFR) protein, indicative of EGFR pathway activation, was present in elongated fibroblast-like cells around the portal veins consistent with our previous observation of p-EGFR positivity in hepatic myofibroblasts6 (Figure 4E). The p-EGFR positivity was restricted mainly to the perivascular cells, and we did not observe any positively stained cell in the neoplastic nodules in the vehicle-treated animals, suggesting that the neoplastic cells are not direct target effector cells of EGFR inhibition (Figure 4E). PPB-NP-erlotinib more significantly reduced the p-EGFR positivity compared to systemic erlotinib treatment (Figure 4E, F). Hepatic fibrosis quantified by collagen proportionate area (CPA) was significantly reduced in both the systemic and myofibroblast-targeted treatments at similar level (Figure 4G, H). Even with adjustment of erlotinib dose, improvement of fibrosis reduction was modest and not statistically significant, suggesting that EGFR signaling in hepatic myofibroblasts is more associated with carcinogenesis than fibrogenesis.
We next assessed whether the use of myofibroblast-targeted erlotinib delivery modifies manifestation of drug toxicities. In rodents, adverse effects of erlotinib frequently seen in human such as hepatotoxicity is present, whereas others, e.g., skin rash and diarrhea, are not observed as reported in our previous study.6 Therefore, hepatocyte damage as well as body/organ weight change were examined to assess adverse effects. Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling (TUNEL) staining showed hepatocyte apoptosis distributed throughout the fibrotic/cirrhotic livers of vehicle-treated rats, indicating hepatic damage (Figure 5A). Whereas systemic erlotinib did not significantly reduced the number of TUNEL-positive area, PPB-NP-erlotinib-treated rat livers showed significant reduction of the area, indicating a less cytotoxic condition in the animals treated with the myofibroblast-targeted erlotinib (p=0.04) (Figure 5B). Although not statistically significant, there were trends of better body weight restoration as well as lower weight of target organs with myofibroblasts, i.e., liver and lung, in the rats treated with PPB-NP-erlotinib compared to systemic erlotinib (Figure 5C–E). Weight of heart and kidney was almost unchanged, whereas spleen weight was reduced in drug-treated rats, reflecting improved splenomegaly due to cirrhosis-associated portal hypertension16 (Figure 5F–H). The improvement of spleen weight was less obvious in PPB-NP-erlotinib-treated rats compared to systemic erlotinib, which may be due to splenic entrapment of nanoparticles. These results collectively indicate that the myofibroblast-targeted EGFR inhibition can enhance chemopreventive effect with reduced dose of the drug, thereby enabling more potent cancer chemoprevention therapy with reduced toxicity.
Figure 5.

Change in hepatocyte totoxicity and body weight after systemic or myofibroblast-targeted delivery of erlotinib. (A) Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling (TUNEL) staining. Scale bar indicates 100 μm. (B) TUNEL-positive area per visual field normalized to respective controls according to the treatment groups (n=8 per group). (C) Body weight of the rats at the time of sacrifice normalized to respective controls according to the treatment groups (n=8 per group). Weight of livers (D), lungs (E), hearts (F), kidneys (G), and spleens (H) from the rats normalized to respective controls according to the treatment groups (n=8 per group). Wilcoxon rank-sum test p-values (Bonferroni-corrected) are shown. Longer and shorter horizontal bars indicate mean and standard error of mean, respectively.
*Vehicle control for systemic erlotinib administration (i.e., intraperitoneal injection). **Vehicle control for myofibroblast-targeting nanoparticles (i.e., PPB-NP). PPB: platelet-derived growth factor receptor-β-binding peptide, NP: nanoparticle.
Discussion
Our study demonstrated a proof of concept of safer, repurposed use of selective kinase inhibitor for cancer chemoprevention by means of cell type-enriched drug delivery. This new strategy will open the path toward more flexible cancer chemoprevention development, leveraging the breadth of rapidly expanding inventory of selective molecular-targeted agents with less concern about drug toxicities. The use of hollow silica nanoparticles can be flexibly applied for delivery of not only small molecules, but also genetic reagents such as short-hairpin RNA and guide RNA for CRISPR/Cas9-based genome editing.17,18 The spherical shape of our nanoparticles can also provide an advantage to hepatic accumulation compared to cylindrical and discoidal shapes.17 The targeting moiety-independent accumulation of a subset of nanoparticles to the spleen and liver is assumed to be due to uptake by organ-resident macrophages, as typically observed for nanoparticles of this size (150 to 200 nm).17 This could in fact further enhance the liver cancer chemopreventive effect of our nanoparticle-based strategy given a recent study reporting contribution of EGFR activation in hepatic macrophages to hepatocarcinogenesis.19 On the other hand, nanoparticle uptake may lead to killing of macrophages and result in unfavorable immunocompromised state, which should be clarified in future studies and may be overcome by ongoing efforts of technical development.17
Whereas this study represents a conceptual advancement in the development of cancer chemoprevention therapy, repeated intravenous nanoparticle injection employed in this study is not ideal as a widely applicable cancer chemoprevention therapy in clinic. Enhanced stability of nanoparticles by chemical and/or biological engineering is expected to enable less invasive (e.g., oral administration) and more convenient (e.g., longer treatment interval) clinical application. Nevertheless, nanoparticle injection could be justifiable in the setting of clinical trials to prove on-target chemopreventive effect in human, which will justify further clinical optimization and development of drug design and delivery system. In summary, our strategy, cell type-specific kinase inhibition, will facilitate cancer chemoprevention development and contribute to substantial improvement of the still dismal cancer mortality.
Acknowledgments
Funding
This work is supported by Uehara Memorial Foundation (to S.N.), Japan Society for the Promotion of Science, Program for advancing strategic international networks to accelerate the circulation of talented researchers (to T.H.), and U.S. NIH/NIDDK (DK099558), European Commission (ERC-2014-AdG-671231), Irma T. Hirschl Trust, and U.S. Department of Defense (W81XWH-16-1-0363) (to Y.H.).
Advanced microscopy was performed by Tisch Cancer Institute (TCI) Microscopy Shared Resource Facility (MSRF).
Footnotes
Conflict of interest
The authors have declared that no conflict of interest exists.
Author contributions
M.D., S.N., T.H., and Y.H. designed, planned and performed experiments and analyzed the results.
A.V., A.V., A.P.K., N.G., H.H., C.B.B., N.F., A.O., H.H., M.E., N.B.A. M.R.dG., and A.L. supported experiment and data analysis.
R.S. performed 3D structural modeling analysis.
B.C.F., K.P., J.P. provided intellectual inputs for study conception, and critical reading of the manuscript.
M.D. and Y.H. conceptualized and supervised the project, analyzed results and wrote the manuscript.
B.C.F., and Y.H. are the members of the Precision Liver Cancer Prevention Consortium.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim RS, Goossens N, Hoshida Y. Use of big data in drug development for precision medicine. Expert Rev Precis Med Drug Dev. 2016;1:245–53. doi: 10.1080/23808993.2016.1174062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoshida Y, Fuchs BC, Tanabe KK. Prevention of hepatocellular carcinoma: potential targets, experimental models, and clinical challenges. Curr Cancer Drug Targets. 2012;12:1129–59. [PMC free article] [PubMed] [Google Scholar]
- 4.Goossens N, Nakagawa S, Hoshida Y. Molecular prognostic prediction in liver cirrhosis. World J Gastroenterol. 2015;21:10262–73. doi: 10.3748/wjg.v21.i36.10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017 doi: 10.1016/j.addr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs BC, Hoshida Y, Fujii T, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. 2014;59:1577–90. doi: 10.1002/hep.26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas MB, Chadha R, Glover K, et al. Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer. 2007;110:1059–67. doi: 10.1002/cncr.22886. [DOI] [PubMed] [Google Scholar]
- 8.Shibata N, Watanabe T, Okitsu T, et al. Establishment of an immortalized human hepatic stellate cell line to develop antifibrotic therapies. Cell Transplant. 2003;12:499–507. doi: 10.3727/000000003108747064. [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Hui AY, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–51. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shim AH, Liu H, Focia PJ, Chen X, Lin PC, He X. Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc Natl Acad Sci U S A. 2010;107:11307–12. doi: 10.1073/pnas.1000806107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bansal R, Prakash J, Post E, Beljaars L, Schuppan D, Poelstra K. Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice. Hepatology. 2011;54:586–96. doi: 10.1002/hep.24395. [DOI] [PubMed] [Google Scholar]
- 12.Akita RW, Sliwkowski MX. Preclinical studies with Erlotinib (Tarceva) Semin Oncol. 2003;30:15–24. [PubMed] [Google Scholar]
- 13.Paakko P, Ala-Kokko L, Ryhanen L. A light microscopic and biochemical study of carbon tetrachloride-induced pulmonary fibrosis in rats: the preventive effect of malotilate. Eur J Clin Invest. 1987;17:340–6. doi: 10.1111/j.1365-2362.1987.tb02198.x. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa S, Wei L, Song WM, et al. Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell. 2016;30:879–90. doi: 10.1016/j.ccell.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capurro M, Wanless IR, Sherman M, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89–97. doi: 10.1016/s0016-5085(03)00689-9. [DOI] [PubMed] [Google Scholar]
- 16.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–51. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–51. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ha JS, Lee JS, Jeong J, et al. Poly-sgRNA/siRNA ribonucleoprotein nanoparticles for targeted gene disruption. Journal of controlled release: official journal of the Controlled Release Society. 2017;250:27–35. doi: 10.1016/j.jconrel.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Lanaya H, Natarajan A, Komposch K, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nature cell biology. 2014;16:972–81. 1–7. doi: 10.1038/ncb3031. [DOI] [PMC free article] [PubMed] [Google Scholar]