

Abstract
Atopic sensitization and allergic diseases are increasing in modernized countries. These diseases affect millions of individuals, but the mechanisms behind their development are not fully understood. One hypothesis relates to early life respiratory viral infections driving the development of atopic disease including asthma. This review presents the current state of the field, focusing on epidemiologic data supporting a role for early life respiratory viruses in the development of specific IgE, both against aeroallergens and the respiratory virus. Our own work using the Sendai mouse model is then summarized to provide a potential mechanistic explanation for how a respiratory viral infection could drive development of atopic sensitization and disease. We then discuss the components of this mechanistic pathway that have and have not been validated in humans. Finally, we discuss areas ripe for research, as well as potential and current therapeutics that might disrupt the link between respiratory viral infections in early life and atopic sensitization/disease.
Keywords: Allergy, asthma, RSV, Sendai virus, respiratory viral infection
Graphical abstract
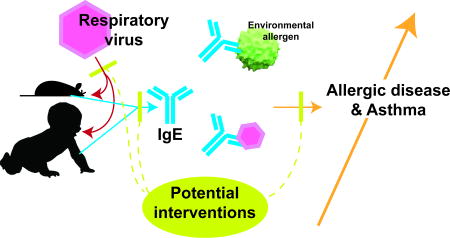
Human and mouse studies suggest early life respiratory viral infections with RSV (in human) and Sendai virus (in mouse) drive development of asthma and allergic disease through production of IgE against aeroallergens and viruses. Therapeutic implications of blocking the translation of a viral infection into atopic disease are also discussed.
Introduction
The prevalence of allergic diseases including asthma, allergic rhinitis and food allergy is increasing in developed nations, suggesting we are in the midst of a public health epidemic. Worldwide, nearly 300 million people suffer from asthma today, with 400 million to be affected by 2025 [1]. Allergic rhinitis affects up to 400 million individuals worldwide while an additional 250 million individuals are affected by food allergy [1]. Given health and economic implications, it is important that mechanism(s) driving increased prevalence of allergic diseases be determined. While the role of genetics, epigenetics, microbiome, environment and infections have been explored, no clear explanation for the rapid increase in disease prevalence has been identified.
Common to all allergic disease is generation of specific immunoglobulin E (sIgE) when exposed to an innocuous antigen – i.e., allergic sensitization. Once sIgE has loaded onto mast cells or basophils, subsequent exposure to the antigen leads to cross-linking of the high-affinity IgE receptor (FcεRI) and degranulation of the cells, driving allergic disease symptomatology. Therefore, preventing allergic sensitization should avert progression to disease and provide an excellent primary prevention strategy against allergic disease.
Given the fairly ubiquitous nature of respiratory viral infections in early life and the fact that allergic sensitization tends to develop early in life, it is tempting to hypothesize that early life respiratory viral infections underlie the increase in allergic disease. In fact, observational studies have associated allergic sensitization by one year of age with respiratory viral infections in infancy [2–3]. The Paramyxoviridae (respiratory syncytial virus (RSV) and parainfluenza viruses 1 and 3) and Piconaviridae (human rhinovirus (RV)) are the respiratory virus families most commonly associated with allergic sensitization [4–6]. This review presents an overview of literature linking early infection with respiratory viruses and development of atopic sensitization and allergic disease. We provide information on potential pathways that could be targeted and identify key areas of research still needed for full understanding of the role early life respiratory viral infections play in atopic sensitization.
Epidemiologic associations between respiratory viral infections and atopic sensitization
There are reasons to associate respiratory viral infections with atopy/allergic disease. While underappreciated, the immune response against many viral infections (not just respiratory viral infections) includes production of specific IgE (sIgE) against viral pathogens [7–16]. The significance of this anti-viral IgE is not fully understood; although, in our animal model it appears critical for translation of respiratory viral infection into atopic disease (discussed in more detail below). Human studies have demonstrated a correlation between the titer of antiviral IgE and severity of RSV symptoms such as wheezing in infants, and recurrent wheezing and development of asthma in older children [10–11, 14–16]. In addition to the presence of antiviral sIgE, respiratory viral infections have an added similarity with allergic disease—symptoms of an upper respiratory tract viral infection parallel those of allergic rhinitis, and viral infections clearly exacerbate asthma [17].
In 1979, Frick and co-workers suggested an association between respiratory viral infections and atopic disease development [18]. Using a birth cohort, they found 11 of 13 children developed atopic sensitization by 4 years of age, and all of these children had had an upper respiratory viral infection within the 1–2 months preceding the development of atopy [18].
The early respiratory viral infection
A common viral infection in early childhood is RSV, a respiratory virus to which a relatively poor immune response is generated, allowing for repeat infections throughout life [19]. In fact, up to 80–90% of children have been infected at least once with RSV during the first 2 years of life [20]. While most of these infections are associated with symptoms of an upper respiratory infection that resolves in several days, a small percentage (0.5–2%) of infants require hospitalization for management—especially those infected in the 2 to 6 month age group [20]. It is these infants hospitalized with RSV that were shown in 1992 to have a higher incidence of skin prick test (a diagnostic test for the presence of allergic sensitization) positivity at 5.5 years of age (42.5% versus 15.0%, p<0.05) compared to control subjects who would have been infected with RSV, but not hospitalized [21].
Three years later in the landmark study by Sigurs et al., the authors found nearly 32% of infants hospitalized with RSV developed atopic sensitization by 3 years of age compared to only 9% of subjects who were not hospitalized (p=0.002) [22]. In a follow up study, RSV was shown to be a significant risk factor for the development of allergic sensitization as 34% of RSV hospitalized subjects demonstrated atopic sensitivity to inhalant allergens compared to only 15% of controls at age 7.5 years (p=0.013) [23].
The association of antiviral IgE with respiratory viral infections
As mentioned, antiviral IgE is produced as part of the antiviral immune response. In 1981, Welliver et al. demonstrated that the titer of anti-RSV IgE correlated with increasing severity of RSV symptoms (wheezing and bronchiolitis) [11]. Other studies have similarly reported presence of anti-RSV IgE in nasal secretions [11, 24–25] and in peripheral blood [9, 26–27]. Smith-Norowitz et al. showed that asthmatic patients (N=30, mean age 14 years) had greater anti-RSV IgE antibody titers compared to non-asthmatics (p = 0.003) [15]. Total serum IgE also correlated with anti-RSV IgE titer in asthmatics (p = 0.047) but not in non-asthmatics (p = 0.13) [15]. Although, it is important to note that one group was unable to detect sIgE against recombinant RSV F or G proteins, suggesting that anti-RSV sIgE is directed against other RSV proteins [28].
It is not just the production of antiviral sIgE that associates with respiratory viral infections early in life. In a cohort featuring subjects with confirmed RSV infection at less than one year of age, 14/42 of the RSV hospitalized subjects demonstrated elevated sIgE by 1 year of age, while only 2/84 of the control group developed sIgE by 1 year [29]. The sIgE tended to be directed against foods, with all 14 of the RSV hospitalized subjects who had elevated sIgE having food sIgE (2 also had aeroallergen sIgE) [29]. This finding contrasts somewhat with the Sigurs study, in which there was an equal distribution of food and aeroallergen sIgE positive subjects. One explanation for this difference might be that Sigurs et al. evaluated sIgE at later ages (3 and 7 years) rather than at 1 year [22–23].
Atopic sensitization in early life and environmental exposures
The Urban Environment and Childhood Asthma (URECA) study examined environmental exposures and their impact on asthma development. In this birth cohort study, 32.5% of children were sensitized to at least one allergen by one year of age [30–31]. Given that sensitization was evaluated at one year of age, the study was not designed to establish a causal relationship between early viral respiratory infection and sensitization. However, the majority of sensitized individuals were sensitized against food allergens (22% had sIgE against milk, 20% against egg and 13% against peanut) [30–31]. Similarly, the Childhood Origins of Asthma (COAST) study demonstrated a 25% prevalence of food sensitization at one year of age in children that had been infected with RV [32]. Thus, there are several studies that show an association between development of atopic sensitization—both to aeroallergens and foods—and previous history of a respiratory viral infection. See Table 1 for summary of aeroallergen and/or food allergen sensitization in the respective birth cohorts.
Table 1.
Prevalence of aeroallergen and food sensitization in featured birth cohorts.
| Study | Year | Cohort (n) |
Incidence of aeroallergen sensitization |
Incidence of food sensitization |
|---|---|---|---|---|
| McGowan et al.30 | 2015 | 516 | Not assessed | 55.4% at any age (milk 46.7%, egg 31.0%, peanut 20.9%) |
| Wood et al.31 | 2011 | 560 | 4% to cockroach by age 12 months | 25% by age 12 months |
| Rubner et al.32 | 2017 | 217 | 13% by age 12 months | 32% by age 12 months (milk 22%, egg 20%, peanut 12%) |
Additional studies with RV have demonstrated increased rates of atopic sensitization following infection [33–35]. In a cohort of 111 patients aged 3 to 23 months with wheeze, atopic sensitization was noted in 23% of all patients; further, RV infection was positively associated with age and other atopic factors including blood eosinophil count, eczema, cough duration and parental allergic rhinitis [33]. In the same cohort, illness severity (p < 0.05) and atopy (p = 0.001) associated with RV species A and C compared to counterparts with non-RV induced wheezing [34]. Similarly, a larger cohort featuring asthmatic patients aged 10–35 years of age showed that RV infected patients had higher rates of allergic sensitization (p=0.04) compared to RV negative controls [36].
Advances in treatments of respiratory viral infections
It stands to reason that if respiratory viral infections can drive atopic sensitization and allergic disease, then preventing or treating these infections should prevent or limit the development of atopy. In fact, there have been several studies that have demonstrated a reduction in atopy with treatment or prevention of RSV infection. Children (under 2 years of age) who were infected with RSV and treated with the antiviral ribavirin (n=40) had a rate of sensitization to at least one positive environmental specific IgE of only 26% compared to 75% for children who had RSV infection but were not treated with the antiviral (p<0.01) [37]. Treatment with Palivizumab, a monoclonal antibody against the RSV F protein that prevents RSV from binding and infecting cells (essentially preventing RSV infection), in the first 6 months of life led to a significant reduction by 3 years of age in amount of total IgE and dust mite specific IgE in treated (n=349) compared to untreated subjects (n=95) after adjustment for baseline factors (p=0.031) [38].
These human studies support the hypothesis that atopy is acquired early in life and associates with the immune response to respiratory viral infections. Understanding potential mechanisms that could translate a viral infection into atopic sensitization, thus, becomes important, as these studies will identify potential therapeutic targets to prevent atopic development.
Mechanisms connecting respiratory viral infections and atopic sensitization
We have utilized the mouse Paramyxovirus, parainfluenza virus type 1 (Sendai virus; SeV) to explore potential mechanisms involved in translation of a viral infection into atopic sensitization and disease. SeV, a negative single stranded RNA virus, is a natural rodent pathogen, and in mice leads to an infection that is limited to the airways with a subsequent antiviral inflammatory response occurring in the peribronchial and bronchiolar tissues—similar to the pathogenesis of RSV in humans. Mice that have been inoculated with SeV lose up to 20% of their body weight over 9 days, and then recover with the development of airway hyperreactivity (AHR) and mucous cell metaplasia (MCM) by 21 days post inoculation (PI) SeV [39]. Thus, this model mimics a very severe respiratory viral infection.
Using the SeV model, we demonstrated that development of AHR and MCM required expression of FcεRI on lung conventional dendritic cells (cDC) [39]. In order to induce expression of FcεRI on lung cDC, a specific subset of neutrophils (PMN) expressing CD49d needed to be recruited to the airways. These PMN express the receptor for cysteinyl leukotrienes, CysLTR1, and in vitro cysteinyl leukotrienes impair PMN apoptosis [40]. In vivo, blockade of CysLTR1 led to reduced accumulation of CD49d+ PMN in the airways (and prevented development of AHR and MCM) [40]. CD49d+ PMN in the airways then bound to airway cDC through CD11b/CD18 (the binding partner on the cDC remains unknown). This cognate interaction then led to induction of FcεRI on the cDC [41]. Furthermore, during the antiviral immune response IgE against SeV was produced, and crosslinking cDC FcεRI led to cDC production of CCL28, a chemokine that was required for recruitment of IL-13 producing, GATA-3+ CD4+ lymphocytes to the lung; IL-13 then drove development of airway hyperreactivity (AHR) and mucous cell metaplasia (MCM) [42]. In summary, we documented a mechanistic pathway (Figure 1) in the mouse beginning with a viral infection in the respiratory epithelium to the development of AHR and MCM (i.e., the mouse equivalent of human asthma).
Figure 1. Mechanistic pathway translating respiratory viral infection into atopic disease.

Sendai virus infects murine airway ciliated epithelial cells, leading to accumulation of CD49d expressing neutrophils (PMN) in a CysLTR1 dependent fashion. Through CD11b and an unknown ligand, these cells interact with lung conventional dendritic cells (cDC) driving expression of the high-affinity IgE receptor (FcεRI) on cDC. At the same time, anti-SeV IgE (Antiviral IgE) is made; crosslinking of FcεRI on the cDC leads to production of the chemokine CCL28, which recruits IL-13 producing lymphocytes to the airways. IL-13 drives development of mucous cell metaplasia and airway hyperreactivity. As shown on the right side of the figure, intranasal exposure to an innocuous environmental antigen (shown as peanut or pollen) during the antiviral immune response in mice results in IgE production against that antigen. See text for more details.
Interestingly, while the SeV model has been used to study development of chronic post-viral obstructive lung disease, we also found evidence of underlying allergic sensitization. While the mice made sIgE against SeV, there was also a large increase in total IgE [43]. In fact, total IgE remained elevated in mice long after the viral infection had resolved, while the SeV sIgE rapidly disappeared from circulation. This led us to explore whether exposure to an innocuous antigen during the respiratory viral infection could lead to production of sIgE against the antigen. We were able to demonstrate that i.n. exposure to ovalbumin at day 8 PI SeV led to production of ovalbumin sIgE at levels similar to that seen with the traditional i.p. ovalbumin and alum sensitization protocol [35]. A subsequent exposure to ovalbumin i.n. led to worsening AHR and MCM in the mice that had received SeV [43]. Thus, the SeV model provides a mechanistic pathway that not only leads to development of post-viral AHR and MCM, but also to atopic sensitization to innocuous environmental allergens (in this case the model allergen ovalbumin) and allergic disease to the innocuous allergen.
Similar findings have been demonstrated in the work of Dakhama et al. who infected newborn mice with RSV and found wild-type (WT) mice developed anti-RSV IgE following neonatal infection [44]. Upon reinfection 5 weeks later, the WT mice had increased AHR and mucus production, which was not observed following RSV reinfection in mice deficient in IL-4 and IL-13 (Il4−/−/Il13−/−); further, Il4−/−/Il13−/− mice lacked anti-RSV IgE. Upon reinfection, mice genetically deficient in FcεRI had significantly attenuated AHR and mucus production, further highlighting IgE and this receptor’s role in enhancing a TH2 phenotype in an anti-viral immune response [44]. In a separate study lending further support to viral mechanisms of atopic disease, increased IL-4 and IL-13 were noted in BALB/c mice sensitized to ovalbumin and inoculated with RV-1B compared to mice inoculated with ultraviolet light inactivated RV-1B and ovalbumin [45]. The authors of this manuscript also noted viral infection enhanced AHR and Muc5AC and Muc5B protein abundance compared to controls [45].
Studies have demonstrated that components of this pathway are intact in humans. In one of our studies, we found that atopic subjects have a greater frequency of CD49d+ PMN in their blood (p=0.0007) and baseline nasal lavage (p=0.0045) samples [46]. Nasal challenge with an antigen to which the subjects had atopic sensitization led to significant increases in the frequency and number of CD49d+ PMN in the nasal lavage. Interestingly, challenge with an antigen to which the subject was not sensitized did not lead to an increase in this PMN subset [46]. Finally, we recently have shown that human CD49d+ PMN (but not CD49d− PMN) from nasal lavage express CysLTR1 [40].
The expression of FcεRI in conventional dendritic cells of mice and humans
Expression of FcεRI on cDC is critical in the development of atopic sensitization in our mouse model. In mice, cDC do not express FcεRI until the viral infection (and mouse pDC are not known to express FcεRI under any conditions); however, in adult humans cDC and pDC are known to express FcεRI. Therefore, we hypothesized that in children there would be a correlation between subject’s age (as a surrogate for respiratory viral infections) and expression of cDC FcεRI. Using a cohort of 27 patients (aged 12–188 months), we found expression of FcεRI receptor on cDC, but noted that there was no correlation with age [47]. In fact, human cDC appear to express FcεRI from birth—demonstrating a major difference between the human and mouse.
Although baseline expression of FcεRI may be dissimilar between mice and human, the effect of a viral infection on the levels of cDC FcεRI expression appears to be similar. Examining 67 children (mean age 7.3 ± 0.4 age) who had an acute asthma exacerbation, with 80% being due to a respiratory viral infection (95% of which were RV), FcεRI expression on cDC (and pDC), as well as total IgE was found to be significantly elevated during the acute exacerbation when compared to convalescence (p<0.05) [48]. Another cohort featuring allergic asthmatic children (N = 14) showed increased surface expression of the FcεRI receptor on plasmacytoid DCs (p = 0.009) and myeloid DCs (p=0.04) compared to nonallergic non-asthmatic children (N = 14) [49]. Cross-linking of the FcεRI receptor (in PBMC) led to significantly impaired RV-induced IFN-α responses in allergic asthmatics compared to nonallergic non-asthmatics (p = 0.002) or allergic non-asthmatics (p=0.004) [49]. Thus, the viral signals that drive IgE and FcεRI on cDC may be similar between humans and mice.
Humans make antiviral sIgE, including against RV, the most common virus to be implicated in causing asthma exacerbations [7]. Therefore, treatment with anti-IgE should prevent viral induced asthma exacerbations, if they are due to antiviral sIgE. The Inner City Asthma Consortium (ICAC) examined the effect of anti-IgE therapy on asthma exacerbations in a cohort of 419 inner city children (ages 6–20 years) with persistent allergic asthma greater than one year [50]. Treatment with anti-IgE significantly reduced the frequency of asthma exacerbations—both during the spring/fall allergy seasons and during the fall/winter respiratory viral season [50]. This provides indirect support for the hypothesis that antiviral sIgE may play a significant role in atopic disease.
The chemokine CCL28 is downstream of antiviral sIgE and was found to translate a respiratory viral infection into atopic disease in our mouse model as mentioned above. Using purified peripheral blood cDC (N=28) from atopic and nonatopic human subjects we found that crosslinking IgE led to increased CCL28 production regardless of atopic status, again suggesting possible common mechanisms for development of atopic disease in mice and human [42]. In a prospective cohort featuring 206 children with severe RSV infection diagnosed at 12 months of age or less, there was an increased risk of asthma when mRNA for the chemokine CCL5 (RANTES) could be isolated from nasal epithelial cells during the acute RSV infection [51]. CCL5 plays an important role in the mouse model primarily through recruitment of macrophages that are needed to clear cellular debris during the antiviral immune response; however, it is not critical for dendritic cell driven post-viral atopic disease [52].
Role of the CysLTR1 receptor and development post-viral airway disease
As mentioned above, our mouse model is dependent upon the CysLTR1 and cysteinyl leukotrienes for the accumulation of CD49d+ PMN. There are also other studies demonstrating a role of cysteinyl leukotrienes in the anti-viral immune response. Viruses such as RSV and SeV have been shown to acutely increase the presence of cysteinyl leukotrienes in bronchoalveolar lavage (BAL) fluid in mice [40]. In one animal study, montelukast, a CysLTR1 antagonist, was administered daily for 7 days starting 1 day prior to RSV inoculation and significant reductions in lymphocytes and AHR were noted in both neonatal and adult mice treated with montelukast by day 7 [53]. This protective effect of reduced AHR, decreased goblet cell metaplasia and reduce airway eosinophilia was also observed in neonatal mice re-infected with RSV 5 weeks later [53].
A cohort study with patients aged 3 to 36 months (N=130) randomized to receive montelukast (5 mg) or placebo for 28 days after being diagnosed with acute RSV, showed that subjects treated with montelukast had significantly increased symptom-free days (p = 0.015), reduced cough (p = 0.04) and delayed presence of exacerbations (p < 0.05) compared to placebo [54]. However, 5 years later, a follow up study by Bisgaard et al. failed to identify a protective effect of montelukast in a larger cohort (N=979) of infants aged 3 to 24 months with acute RSV infection [55]. Whether early life blockade of CysLTR1 before a viral infection would work as a preventive strategy to prevent development of allergies and asthma waits to be determined.
Future directions
Although our mechanistic work and the human epidemiologic data might suggest a causative role for respiratory viral infection in the development of some forms of atopic sensitization, no study was able to demonstrate a clear causal relationship between early life viral infection and atopic sensitization. Clearly this is an area of needed research. Another area that is not well defined includes the role of genetics in this process. It is well known that development of atopy has a strong genetic component—how or whether genetics play a role in these antiviral responses remains unclear.
One glimpse into the role of genetics is provided by a study from Forten et al. in which the authors analyzed single nucleotide polymorphisms (SNPs) in RSV infected infants (N = 78) and compared data to 1045 controls; revealed a high risk haplotype was across IL-13, CNS-1 and IL-4 (p < 0.0001) and such haplotypes were associated with increased IL-13 production and earlier age of infection (< 6 months) [56]. The mechanistic link between these SNPs and atopy and susceptibility to infection awaits further evaluation. A similar area that is not well understood is the role of the microbiome in translating viral infections into atopic disease. In fact, it is not unreasonable to assume that for a respiratory viral infection to drive development of atopic sensitization, the individual would have to have a viral infection in the appropriate genetic and microbiome environment. These issues are actively being investigated.
Finally, there is no clear understanding why IgE would be part of the antiviral immune response. While treatment with anti-IgE does not appear to have any adverse effects in terms of respiratory viral infections, it is still quite intriguing that viral sIgE is made. Future studies will hopefully be able to answer the question as to why this IgE is produced and what (if any) role it plays in the antiviral immune response.
Conclusion
Research and epidemiologic studies increasingly show that atopic diseases are on the rise. Although the mechanisms driving this public health crisis are multifactorial, evidence does suggest that early life respiratory viral infections may play a role. In this review, we discussed the epidemiologic data suggesting an association between RSV and RV infection in the first year of life, and subsequent atopic sensitization (development of sIgE against innocuous antigens). Although the human data cannot demonstrate causation, they are provocative and provide rationale for a study directly looking at the timing of early life respiratory viral infections and atopic development.
Using the SeV mouse model, we have delineated a pathway that translates viral infection into both atopic sensitization and allergic disease. Some, but not all, components of this pathway appear to be intact in humans. Whether these pathways truly drive development of atopy in humans awaits further studies, but the mouse studies provide a potential framework upon which future therapies may be focused.
In conclusion, infection with respiratory RNA viruses appears to drive an immune response that has similarities with atopic disease in that both can lead to production of sIgE. Further, clinically significant infection with these viruses appears to correlate with increased atopic sensitization and allergic disease in humans. These findings may help to explain the rise of atopic disease prevalence in developed nations and will possibly delineate potential therapeutic targets for prevention of atopic development.
Acknowledgments
NIH R01 HL087778 and R01 AI120655 and the Research Institute at Nationwide Children’s Hospital (all to MHG).
MHG has received funding through the National Institutes of Health, the Research Institute at Nationwide Children’s Hospital, Children’s Research Institute (of Children’s Hospital of Wisconsin), Polyphor Ltd. and Genentech. MHG is on the Board of Directors for the American Academy of Allergy, Asthma and Immunology, and the Asthma and Allergy Foundation of America.
Footnotes
Conflicts of Interest: LMM declares no commercial or financial conflict of interest.
References
- 1.Pawankar R. Allergic diseases and asthma: a global public health concern and a call to action. World Allergy Organ J. 2014;7(1):12. doi: 10.1186/1939-4551-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peebles RS., Jr Viral infections, atopy, and asthma: is there a causal relationship? J Allergy Clin Immunol. 2004;113(1 Suppl):S15–8. doi: 10.1016/j.jaci.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 3.Martinez FD. Viruses and atopic sensitization in the first years of life. Am J Respir Crit Care Med. 2000;162(3 Pt 2):S95–9. doi: 10.1164/ajrccm.162.supplement_2.ras-8. [DOI] [PubMed] [Google Scholar]
- 4.Sigurs N. A cohort of children hospitalised with acute RSV bronchiolitis: impact on later respiratory disease. Paediatr Respir Rev. 2002;3(3):177–83. doi: 10.1016/s1526-0542(02)00191-4. [DOI] [PubMed] [Google Scholar]
- 5.Sigurs N, Bjarnason R, Sigurbergsson F, et al. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161(5):1501–7. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- 6.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178(7):667–72. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith-Norowitz TA, Kusonruksa M, Wong D, et al. Long-term persistence of IgE anti-influenza A HIN1 virus antibodies in serum of children and adults following influenza A vaccination with subsequent H1N1 infection: a case study. Journal of Inflammation Research. 2012;5:111–116. doi: 10.2147/JIR.S34152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tam JS, Jackson WT, Hunter D, et al. Rhinovirus Specific IgE Can Be Detected in Human Sera. The Journal of allergy and clinical immunology. 2013;132(5):1241–1243. doi: 10.1016/j.jaci.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bui RH, Molinaro GA, Kettering JD, et al. Virus-specific IgE and IgG4 antibodies in serum of children infected with respiratory syncytial virus. J Pediatr. 1987;110(1):87–90. doi: 10.1016/s0022-3476(87)80295-0. [DOI] [PubMed] [Google Scholar]
- 10.Welliver RC, Sun M, Rinaldo D, et al. Predictive value of respiratory syncytial virus-specific IgE responses for recurrent wheezing following bronchiolitis. J Pediatr. 1986;109(4):776–80. doi: 10.1016/s0022-3476(86)80692-8. [DOI] [PubMed] [Google Scholar]
- 11.Welliver RC, Wong DT, Sun M, Middleton E, Jr, et al. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305(15):841–6. doi: 10.1056/NEJM198110083051501. [DOI] [PubMed] [Google Scholar]
- 12.Perelmutter L, Phipps P, Potvin L. Viral infections and IgE levels. Ann Allergy. 1978 Sep;41(3):158–9. [PubMed] [Google Scholar]
- 13.Nordbring F, Johansson SG, Espmark A. Raised serum levels of IgE in infectious mononucleosis. Scan J Infect Dis. 1972;4(2):119–24. doi: 10.3109/inf.1972.4.issue-2.10. [DOI] [PubMed] [Google Scholar]
- 14.Welliver RC, Duffy l. The relationship of RSV-specific immunoglobulin E antibody responses in infancy, recurrent wheezing, and pulmonary function at age 7–8 years. Pediatr Pulmonol. 1993;15(1):19–27. doi: 10.1002/ppul.1950150104. [DOI] [PubMed] [Google Scholar]
- 15.Smith-Norowitz TA, Mandal M, Joks R, et al. IgE anti-respiratory syncytial virus antibodies detected in serum of pediatric patients with asthma. Hum Immunol. 2015;76(7):519–24. doi: 10.1016/j.humimm.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Welliver RC. Respiratory syncytial virus and other respiratory viruses. Pediatr Infect Dis J. 2003;22(2 Suppl):S6–10. doi: 10.1097/01.inf.0000053880.92496.db. [DOI] [PubMed] [Google Scholar]
- 17.Steinke JW, Borish L. Immune responses in rhinovirus-induced asthma exacerbations. Curr Allergy Asthma Rep. 2016;16(11):78. doi: 10.1007/s11882-016-0661-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frick OL, German DF, Mills J. Development of allergy in children. I. Association with virus infections. J Allergy Clin Immunol. 1979;63(4):228–41. doi: 10.1016/0091-6749(79)90106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall CB, Simőes EA, Anderson LJ. Clinical and epidemiologic features of respiratory syncytial virus. Curr Top Microbiol Immunol. 2013;372:39–57. doi: 10.1007/978-3-642-38919-1_2. [DOI] [PubMed] [Google Scholar]
- 20.Psarras S, Papadopoulos NG, Johnston SL. Pathogenesis of respiratory syncytial virus bronchiolitis-related wheezing. Paediatr Respir Rev. 2004;5(Suppl A):S179–184. doi: 10.1016/s1526-0542(04)90034-6. [DOI] [PubMed] [Google Scholar]
- 21.Murray M, Webb MS, O'Callaghan C, Swarbrick AS, Milner AD. Respiratory status and allergy after bronchiolitis. Archives of disease in childhood. 1992;67(4):482–7. doi: 10.1136/adc.67.4.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sigurs N, Bjarnason R, Sigurbergsson F, et al. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics. 1995;95(4):500–5. [PubMed] [Google Scholar]
- 23.Sigurs N, Bjarnason R, Sigurbergsson F, et al. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161(5):1501–7. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- 24.Soto ME, Sly PD, Uren E, et al. Bronchodilator response during acute viral bronchiolitis in infancy. Pediatr Pulmonol. 1985;1(2):85–90. doi: 10.1002/ppul.1950010206. [DOI] [PubMed] [Google Scholar]
- 25.Russi JC, Delfraro A, Borthagaray MD, et al. Evaluation of immunoglobulin E-specific antibodies and viral antigens in nasopharyngeal secretions of children with respiratory syncytial virus infections. J Clin Microbiol. 1993;31(4):819–23. doi: 10.1128/jcm.31.4.819-823.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabatic S, Gagro A, Lokar-Kolbas R, et al. Increase in CD23+ B cells in infants with bronchiolitis is accompanied by appearance of IgE and IgG4 antibodies specific for respiratory syncytial virus. J Infect Dis. 1997;175(1):32–7. doi: 10.1093/infdis/175.1.32. [DOI] [PubMed] [Google Scholar]
- 27.Aberle JH, Aberle SW, Dworzak MN, et al. Reduced interferon-gamma expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am J Respir Crit Care Med. 1999;160(4):1263–8. doi: 10.1164/ajrccm.160.4.9812025. [DOI] [PubMed] [Google Scholar]
- 28.De Alarcon A, Walsh EE, Carper HT, et al. Detection of IgA and IgG but not IgE antibody to respiratory syncytial virus in nasal washes and sera from infants with wheezing. J Pediatr. 2001;138(3):311–7. doi: 10.1067/mpd.2001.111277. [DOI] [PubMed] [Google Scholar]
- 29.Schauer U, Hoffjan S, Bittscheidt J, et al. RSV bronchiolitis and risk of wheeze and allergic sensitization in the first year of life. Eur Resp J. 2002;20(5):1277–83. doi: 10.1183/09031936.02.00019902. [DOI] [PubMed] [Google Scholar]
- 30.McGowan EC, Bloomberg GR, Gergen PJ, et al. Influence of early-life exposures on food sensitization and food allergy in an inner-city birth cohort. J Allergy Clin Immunol. 2015;135(1):171–8. doi: 10.1016/j.jaci.2014.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wood RA, Bloomberg GR, Kattan M, et al. Relationships among environmental exposures, cord blood cytokine responses, allergy, and wheeze at 1 year of age in an inner-city birth cohort (Urban Environment and Childhood Asthma study) J Allergy Clin Immunol. 2011;127(4):913–9. e1–6. doi: 10.1016/j.jaci.2010.12.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubner FJ, Jackson DJ, Evans MD, et al. Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J Allergy Clin Immunol. 2017;139(2):501–7. doi: 10.1016/j.jaci.2016.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turunen R, Koistinen A, Vuorinen T, et al. The first wheezing episode: respiratory virus etiology, atopic characteristics and illness severity. Pediatr Allergy Immunol. 2014;25(8):796–803. doi: 10.1111/pai.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turunen R, Jartti T, Bochkov YA, et al. Rhinovirus species and clinical charactersitics in the first wheezing episode in children. J Med Virol. 2016;88(12):2059–68. doi: 10.1002/jmv.24587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kantor DB, Stenquist N, McDonald MC, et al. Rhinovirus and serum IgE are associated with acute asthma exacerbation severity in children. J Allergy Clin Immunol. 2016;138(5):1467–71.e9. doi: 10.1016/j.jaci.2016.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Öhrmalm L, Malinovschi A, Wong M, et al. Presence of rhinovirus in the respiratory tract of adolescents and young adults with asthma without symptoms of infection. Respir Med. 2016;115:1–6. doi: 10.1016/j.rmed.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen CH, Lin YT, Yang YH, et al. Ribavirin for respiratory syncytial virus bronchiolitis reduced the risk of asthma and allergen sensitization. Pediatr Allergy Immunol. 2008;19(2):166–72. doi: 10.1111/j.1399-3038.2007.00610.x. [DOI] [PubMed] [Google Scholar]
- 38.Yoshihara S, Kusuda S, Mochizuki H, et al. Effect of palivizumab prophylaxis on subsequent recurrent wheezing in preterm infants. Pediatrics. 2013;132(5):811–8. doi: 10.1542/peds.2013-0982. [DOI] [PubMed] [Google Scholar]
- 39.Grayson MH, Cheung D, Rohlfing MM, et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. 2007;204(11):2759–69. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheung DS, Sigua J, Simpson PM, et al. CysLTR1 and CD49d expressing neutrophils during viral infection contribute to post-viral atopic airway disease. J Allergy Clin Immunol. 2017 In press. [Google Scholar]
- 41.Cheung DS, Ehlenbach SJ, Kitchens RT, et al. Cutting edge: CD49d+ neutrophils induce FcepsilonRI expression on lung dendritic cells in a mouse model of postviral asthma. J Immunol. 2010;185(9):4983–7. doi: 10.4049/jimmunol.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan SH, Grayson MH. Cross-linking IgE augments human conventional dendritic cell production of CCL28. Journal Allergy and Clin Immunol. 2010;125(1):265. doi: 10.1016/j.jaci.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheung DS, Ehlenbach SJ, Kitchens T, Riley DA, Grayson MH. Development of atopy by severe paramyxoviral infection in a mouse model. Ann Allergy Asthma Immunol. 2010;105(6):437–43 e1. doi: 10.1016/j.anai.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dakhama A, Lee YM, Ohnishi H, et al. Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J Allergy Clin Immunol. 2009;123(1):138–145.e5. doi: 10.1016/j.jaci.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 45.Bartlett NW, Walton RP, Edwards MR, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14(2):199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sigua JA, Buelow B, Cheung DS, et al. CD49d-expressing neutrophils differentiate atopic from nonatopic individuals. J Allergy Clin Immunol. 2014;133(3):901–4 e5. doi: 10.1016/j.jaci.2013.09.035. [DOI] [PubMed] [Google Scholar]
- 47.Vasudev M, Cheung DS, Pincsak H, et al. Expression of high-affinity IgE receptor on human peripheral blood dendritic cells in children. PloS one. 2012;7(2):e32556. doi: 10.1371/journal.pone.0032556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subrata LS, Bizzintino J, Mamessier E, et al. Interactions between innate antiviral and atopic immunoinflammatory pathways precipitate and sustain asthma exacerbations in children. J Immunol. 2009;183(4):2793–800. doi: 10.4049/jimmunol.0900695. [DOI] [PubMed] [Google Scholar]
- 49.Durrani SR, Montville DJ, Pratt AS, et al. Innate immune responses to rhinovirus are reduced by the high-affinity receptor in allergic asthmatic children. J Allergy Clin Immunol. 2012;130(2):489–95. doi: 10.1016/j.jaci.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busse WW, Morgan WJ, Gergen PJ, et al. Randomized trial of Omalizumab (Anti-IgE) for asthma in inner-city children. N Eng J Med. 2011;364(11):1005–1015. doi: 10.1056/NEJMoa1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bacharier LB, Cohen R, Schweiger T, et al. Determinants of asthma after severe respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol. 2012;130(1):91–100.e3. doi: 10.1016/j.jaci.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyner JW, Uchida O, Kajiwara N, et al. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11(11):1180–7. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han J, Jia Y, Takeda K, et al. Montelukast during primary infection prevents airway hyperresponsiveness and inflammation after reinfection with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182(4):455–463. doi: 10.1164/rccm.200912-1811OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bisgaard H. A randomized trial of montelukast in respiratory syncytial virus postbronchiolitis. Am J Respir Crit Care Med. 2003;167(3):379–83. doi: 10.1164/rccm.200207-747OC. [DOI] [PubMed] [Google Scholar]
- 55.Bisgaard H, Flores-Nunez A, Goh A. Study of montelukast for the treatment of respiratory symptoms of post-respiratory syncytial virus bronchiolitis in children. Am J Respir Crit Care Med. 2008;178(8):854–60. doi: 10.1164/rccm.200706-910OC. [DOI] [PubMed] [Google Scholar]
- 56.Forton JT, Rowlands K, Rockett K, et al. Genetic association study for RSV bronchiolitis in infancy at the 5q31 cytokine cluster. Thorax. 2009;64(4):345–52. doi: 10.1136/thx.2008.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]