

Abstract
Diagnosis of clinical toxoplasmosis remains a challenge, thus limiting the availability of human clinical samples. Though murine models are an approximation of human response, their definitive infection status and tissue availability makes them critical to the diagnostic development process. Hydrogel mesh nanoparticles were used to concentrate antigen to detectable levels for mass spectrometry. Seven T. gondii isolates were used to develop a panel of potential peptide sequences for detection by parallel reaction monitoring (PRM) mass spectrometry. Nanoparticles were incubated with decreasing concentrations of tachyzoite lysate to explore the limits of detection of PRM. Mice whose toxoplasmosis infection status was confirmed by quantitative real-time PCR had urine tested by PRM after hydrogel mesh concentration for known T. gondii peptides. Peptides from GRA1, GRA12, ROP4, ROP5, SAG1, and SAG2A proteins were detected by PRM after nanoparticle concentration of urine, confirming detection of T. gondii antigen in the urine of an infected mouse.
Keywords: Nanoparticles, Toxoplasma gondii, Multiple Reactions Monitoring
Graphical Abstract
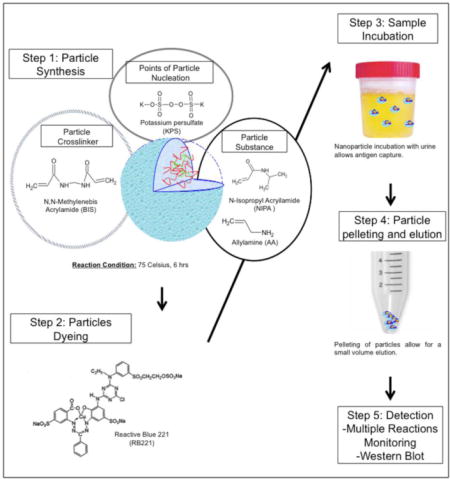
Background
T. gondii infection is a common, relatively asymptomatic infection. However, in those individuals with a compromised immune system or infected congenitally, severe consequences including cerebral or ocular involvement and death can occur. [1] Current diagnostic options for both toxoplasmic encephalitis (TE) and congenital toxoplasmosis are limited.
The difficulty of human diagnosis of toxoplasmosis increases the need for murine models in which disease status has been confirmed. Animal models provide a verifiable means of exploring the type of antigens shed systemically at different stages in the infection. Furthermore, strain variation in T. gondii is critical as to the virulence of the parasite, thus impacting its clinical manifestations. [2] Though it has been well established in murine models that type I strains are virulent and type II and III strains are avirulent; the relationship between infected strain and impact on human disease presentation remains a topic of discussion. [3] Several groups have successfully established murine infection models for RH and ME49 strains of T. gondii. There has only been one group that has compared these two infection models by conventional PCR in different organs.[4]
Utilization of biological fluids, such as urine, is an attractive approach for diagnostic development because of the non-invasive nature of sample collection. Furthermore, mass spectrometry specificity allows for confidence in detection of specific pathogens in any biological fluid. Detection of microorganisms in human urine is not unprecedented. In mice, Wang et al were able to detect Schistosoma mansoni in the urine based on biomarkers and a changed metabolic profile using H NMR spectroscopy. [5] Zhou et al showed a metabolic shift in the serum of mice infected with T. gondii using Liquid chromatography–mass spectrometry (LC-MS) analysis. [6] Ferreira et al (2011) demonstrated the possibility of detecting bacteria and yeast in human urine samples, if the concentration was greater than 105 colony-forming units. [7] However, no group has previously demonstrated detection of T. gondii antigens potentially shed by the parasite in the urine of infected animals.
One barrier to mass spectrometry’s analysis of large volumes of biological fluids is low relative abundance of antigen. Hydrogel mesh nanoparticles have increased the limits of detection of immunoblot by semi-specifically concentrating antigens in such fluids. [8–10] Thus the application of these nanoparticles in mass spectrometry seemed a logical progression. Hydrogel nanoparticles function by exclusion of abundant large proteins. Hydrogel nanoparticles also semi-specifically bind antigens, thus allowing the concentration of antigens of interest. [11,12] Semi-specific binding is mediated by industrial dye moieties. [11,12]
Here we demonstrate the success of the hydrogel mesh nanoparticles in their capture of T. gondii antigens. We describe the development of a T. gondii specific parallel reaction monitoring (PRM) system and its limit of detection for various T. gondii antigens. Furthermore we document the variability in brain, cardiac tissue, and blood of T. gondii in mice infected with ME49 and RH strains through detection by qPCR and histology. Finally, we assess the possibility of antigen detection from urine from these infected mice using hydrogel mesh nanoparticles to concentrate antigen for detection by PRM mass spectrometry.
Methods
Culture of T. gondii isolates strain
RH isolates were maintained by successive passage in monolayers of cells line LLC-MK2 (Macaca mulatta, monkey, rhesus, ATCC/CCL-7) in growth media consisting of RPMI 1640 (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), pyruvate (1 mM; Gibco), non-essential amino acid mixture 1:100; (Gibco), gentamicin (45 µg·mL−1; Gibco), and penicillin (100 U/mL; Gibco), incubated at 37°C and 5% CO2. Growth media was changed every four days and cultures were passed to a new monolayer of cells every ten days. In order to obtain total tachyzoite lysate, tachyzoites obtained from cell culture were sonicated (Sonicator 3000, Misonix). Four sonication cycles of 30 seconds at 4 Hz were done with one minute rest between each cycle. Then, the obtained product was centrifuged at 3000 g for 15 minutes. Finally, the protein concentration was measured using Bradford method.
Selection, preparation, and analysis of T. gondii Isolates for LC MS/MS
Isolates were selected from Biodefense and Emerging Infections Research Resources Repository (BEI Resources) to ensure coverage of the three major circulating T. gondii strains (I, II, and III). [13] One isolate of a type IV strain (MAS) and one isolate of type V strain (CAST) were selected for their clinical importance. Isolates were thawed and lysed in 1% Rapigest (Waters, Millford, MA) in 50mM ammonium bicarbonate with 10mM TCEP for 10 minutes at 100°C. Isolates were then alkylated with 50mM iodoacetamide for 15 minutes in the dark at room temperature, then diluted 10-fold in 50mM ammonium bicarbonate to lower the Rapigest concentration to 0.1%. Samples were then digested with trypsin overnight at 37°C. Digestion was halted by adding trifluoroacetic acid to a final concentration of 0.1%. Samples were desalted with C-18 spin columns (ThermoFisher Scientific, Waltham, MA, USA), dried by vacuum centrifugation, and then reconstituted in 0.1% formic acid in water.
LC-MS/MS experiments were performed on an Orbitrap Fusion (ThermoFisher) equipped with a nanospray EASY-nLC 1200 HPLC system (ThermoFisher). Peptides were separated using a reversed-phase PepMap RSLC 75 µm i.d. × 15 cm long with 2 µm, C18 resin LC column (ThermoFisher). The mobile phase consisted of 0.1 % aqueous formic acid (mobile phase A) and 0.1 % formic acid in 80% acetonitrile (mobile phase B). After sample injection, the peptides were eluted by using a linear gradient from 5% to 50 % B over 90 min and ramping to 100% B for an additional 2 min. The flow rate was set at 300 nL/min. The Orbitrap Fusion was operated in a data-dependent mode in which one full MS scan (60,000 resolving power) from 300 Da to 1500 Da using quadrupole isolation, was followed by MS/MS scans in which the most abundant molecular ions were dynamically selected by Top Speed, and fragmented by collision-induced dissociation (CID) using a normalized collision energy of 35%. “Peptide Monoisotopic Precursor Selection” and “Dynamic Exclusion” (8 sec duration), were enabled, as was the charge state dependency so that only peptide precursors with charge states from +2 to +4 were selected and fragmented by CID.
Tandem mass spectra were searched against the database ToxoDB: The Toxoplasma Genomics Resource using Proteome Discover v 2.1 with SEQUEST using tryptic cleavage constraints. Mass tolerance for precursor ions was 5 ppm, and mass tolerance for fragment ions was 0.5 Da. Data were analyzed with oxidation (+15.9949 Da) on methionine as a variable post translation modification, and carbamidomethyl cysteine (+57.0215) as a fixed modification. A 1% false discovery rate (FDR) was used as a cut-off value for reporting peptide spectrum matches (PSM) from the database. A BLAST search was conducted for the sequences of interest in the National Institute of Health’s U.S. National Library of Medicine’s National Center for Biotechnology Information to ensure that the sequences were specific to T. gondii.
Mouse infection model
Two-month-old female Swiss mice were infected with either 103–104 RH tachyzoites IP or orally with 12–15 ME49 cysts from the brain of a previously infected mouse. ME49 infected mice were immunosuppressed with dexamethasone (100 mg/kg) 2 months after infection.
Management of T. gondii isolates
Laboratorio de Inmunología Experimental Instituto Nacional de Pediatría, México and Instituto de Ciencias Biológicas, Universidad Federal de Minas Gerais, Brazil, were kind enough to share their RH and ME49 strains, respectively. The RH strain was then injected intraperitoneally (I.P.) into adolescent Swiss mice, ME49 strain mice were infected orally. One month (ME49) or four days (RH) post infection these mice were sacrificed and the tachyzoites were obtained as per Dubey et al [14]. Resulting RH+ tachyzoites were plated with LLC-MK2 cells, and incubated at 37°C and 5% CO2. ME49 strain is maintained in vivo. All the animals used were managed in accordance to there protocols approved by the Animal Use Committee of Universidad Cayetano Heredia (Approval No. 64220)
Mouse specimen collection
Blood specimens were taken from the tail of the mouse. Urine specimens were obtained after the administration of furosemide (1 mg/kg). Mice were sacrificed with xylacine and ketamine. Terminal cardiac bleeds and organ excisions were then preformed. RH mice were scarified four days post-infection. ME49 infected mice were scarified at day 49 post-infection, day 4-post immunosuppression.
DNA extraction of Tissues
After fixation, 25 mg of tissue were cut into small pieces and rehydratated. Then, they were transferred to a Lysing Matrix H 2 mL tube (MP Biomedicals) and treated with 300 μL of guanidine hydrochloride 6M (Sigma-Aldrich, USA). After this, tissues were treated with an agitation cycle (6 m/s – 40″) in a FastPrep-24™ 5G machine to ensure tissue disaggregation before the exposition to Proteinase K. After this process, DNA from all samples was extracted using High Pure PCR Template Preparation Kit (Roche Diagnostic Corporation, USA) following the manufacturer’s instructions.
qPCR
Quantitative Real Time Polymerase Chain Reaction was completed as previously described using by Wahab et al.[15]Briefly, primers for REP 529 5′-GCTCCTCCAGCCGTCTTG (forward) and 5′- TCCTCACCCTCGCCTTCAT (reverse) using probe FAM - AGGAGAGATATCAGGACTGTA - 3′MGB, or primers for BBZ were 5′- GCATTGCCCGTCCAAACT- 3′ (forward) and 5′-AGACTGTACGGAATGGAGACGAA-3′ (reverse) using probe FAM -AGGAGAGATATCAGGACTGTA - 3′MGB were used on a light cycler (Applied Biosystems). The denaturing stages were: two minutes at 50°C, followed by 10 minutes at 95°C and then 15 seconds per cycle at 95°C. The annealing stage was one minute at 58°C and the elongation stage was one minute at 72°C. Forty cycles were used.
Nanoparticle synthesis
Nanoparticles were synthesized as previously described[11,12,16,17]. Briefly, nine grams N-Isopropylacrylamide (NIPA- Sigma 415324) and 0.280g N,N′ methylenbisacrylamide (BIS- Sigma M7279) were dissolved in 250millileters (mL) of MilliQ water and filtered using a nitrocellulose membrane (0.45um, Millipore) into a 1000mL round bottom flask. Nitrogen was used to purge the system for a half hour. Allylamine (676ul) was added and the Nitrogen purging was continued for another 15 minutes. The solution was then heated to 70–80°C and 0.1g of potassium persulfate (KPS, Sigma- 379824) was added in MilliQ water. The solution was incubated with heating, stirring, and condensing for six hours. After six hours the solution was stirred overnight. Nanoparticles were then washed 4–5 times with deionized water at 19000g for 40–50 minutes/wash.
Nanoparticle dyeing
3.96g of sodium bicarbonate was dissolved in 240mL of MilliQ water. 1.8g of Reactive Blue 221d dye was then added. Dye solution was filtered using a nitrocellulose membrane (0.45µm, Millipore). Dye solution and washed nanoparticles were combined for overnight incubation with stirring. Nanoparticles were then washed 4–5 times with deionized water until the supernatant appears clear at 19000g for 40–50 minutes/wash.
Particle incubation with urine
Urine was then centrifuged at 3500g for 10 minutes for the removal of cellular debris. 200µl of Nanoparticles were added to the remaining sample. Urine and particles were incubated for 30 minutes at room temperature with rotation. Following incubation, particles were spun down at 13000g for 10 minutes. Supernatant was removed and remaining particles were resuspended with one milliliter of MilliQ water. After resuspension, particles were centrifuged at 13000g for 10 minutes. The wash was then removed and the samples were eluted from the particles with 1% Rapigest (Waters, Milford, MA) in 50mM ammonium bicarbonate with 10mM TCEP for mass spectrometry experiments, or in 2X Lamealli’s Sample Buffer (Biorad) for gel electrophoresis and western blotting for 10 minutes at 100°C.
Electrophoresis and Western blotting
Eluates were loaded into a 4–20% Tris-glycine gel (Life Technologies) and run at 200V for 1 hour. After gel separation, proteins were transferred to PVDF membranes (Biorad) for 1.5 hours at 35V. Membranes were then blocked in 5% dry milk in PBS-Tween (0.1%). Post blocking, membranes were incubated for 1 hour in either monoclonal mouse Tg17-43 anti-GRA1 or polycolonal mouse anti-M2AP, or overnight in monoclonal mouse anti-SAG1 (ThermoFisher). Membranes were washed and incubated for 1 hour with either anti-mouse of anti-rabbit HRP. Super signal Dura western (ThermoFisher) was used for chemiluminescent detection.
Eluate processing for mass spectrometry analysis
Nanoparticle eluates were alkylated with 50mM iodoacetamide for 15 minutes in the dark at room temperature, then diluted 10-fold in 50mM ammonium bicarbonate to lower the Rapigest concentration to 0.1%. Samples were then digested with trypsin overnight at 37°C. Digestion was halted by adding trifluoroacetic acid to a final concentration of 0.1%. Samples were desalted with C-18 spin columns (ThermoFisher), dried by vacuum centrifugation, and then reconstituted in 0.1% formic acid in water.
PRM mass spectrometry analysis of eluates
Digested eluate samples were analyzed on an Orbitrap Fusion mass spectrometer (ThermoFisher) equipped with a nanospray EASY-nLC 1200 HPLC system (ThermoFisher). Peptides were separated using a reversed-phase PepMap RSLC 75 μm i.d. × 15 cm long with 2 μm, C18 resin LC column (ThermoFisher). The mobile phase consisted of 0.1 % aqueous formic acid (mobile phase A) and 0.1% formic acid in 80% acetonitrile (mobile phase B). After sample injection, the peptides were eluted by using a linear gradient from 5% to 50% B over 15 min and ramping to 100% B for an additional 2 minutes. The flow rate was set at 300nL/min. The Orbitrap Fusion was operated using parallel reaction monitoring where only the precursor m/z values of the peptides of interest were permitted to pass through to be fragmented by HCD. Fragment ions were detected in the Orbitrap with resolution at 60,000. Data were analyzed with Skyline v3.6 (University of Washington, MacCoss Lab) to determine the presence or absence of proteins/peptides of interest.
Results
Selection of peptides for Parallel Reaction Monitoring
The limited availability of antibodies for the diverse antigens associated with T. gondii significantly limits antigen identification by western blot. Additionally, biological processing of antigens may result in their cleavage of epitopes preventing their detection by antibodies. Thus we utilized seven isolates (see table 1) of T. gondii for LC-MS/MS experiments. The seven strains were chosen to allow for coverage of the most common T. gondii isolates and clinical manifestations. The seven isolates covered all three major T. gondii strains and two less common haplotypes. They were isolated from both infected humans and animals. LC-MS/MS experiments revealed a total of 1,054 non-redundant proteins (see supplemental table 1). From these 1,054 proteins, 34 peptides, representing 12 proteins (Table 2) were chosen for further monitoring by PRM. Proteins were selected based on their biological significance (Figure 1), and peptides were chosen based on their protein ion fragmentation pattern compatibility with PRM and biological relevance. Figure 1 is a basic schematic of the invasion of a tachyzoite into a host cell for reference in the biological relevance of the proteins selected for further monitoring by PRM.
Table 1. Seven Isolates sequenced by LC-MS/MS.
Seven reference strains from BEI resources were tryptically digested, alkylated, acidified, and sequenced by LC-MS/MS.
| Isolate | Stain/Haplotype | Source |
|---|---|---|
| NR-44106 | I/III | Toxoplasma gondii (T. gondii), strain EGS was isolated in 1998 from amniotic fluid of a human patient with congenital toxoplasmosis |
| NR-223 | I | Toxoplasma gondii, strain RH-88 was derived from a clone of the RH strain. The RH strain was isolated in 1939 from a six year old boy with a lethal case of encephalitis in Cincinnati, Ohio |
| NR-225 | II | Toxoplasma gondii (T. gondii), ME49 (originally isolated from a sheep in California and available as BEI Resources NR-362) was passed singly through a cat to obtain a line that was capable of producing oocysts, and then further cloned by limiting dilution to produce the B7 clone (also referred to as P and PLK strain). |
| NR-362 | II | Toxoplasma gondii (T. gondii), ME49 was isolated from sheep muscle. |
| NR-20730 | III | Toxoplasma gondii (T. gondii), strain VEG was isolated from the blood of an AIDS patient with toxoplasmic encephalitis in California in 1989. |
| NR-20731 | IV | Toxoplasma gondii (T. gondii), strain MAS was isolated from a human with severe congenital toxoplasmosis in France in 1991 |
| NR-20734 | V | Toxoplasma gondii (T. gondii), strain CAST was isolated from an AIDS patient with toxoplasmic encephalitis in California, 1988 |
Table 2. Peptide Sequences identified by PRM by isolate.
Peptides identified in BEI reference isolates (see table 1) by LC-MS/MS chosen for PRM analysis of RH tachyzoite lysate and urine from mice infected with this same lysate.
| BEI Reference Isolate
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parent Protein | Peptide Sequence | NR44106 | NR223 | NR225 | NR362 | NR20730 | NR20371 | NR20374 | 1e6 Tachyzoite Lysate | RH+ Murine urine |
| GRA1 | VERPTGNPDLLK | x | x | x | x | x | x | |||
| VIDDVQQLEK | x | x | x | x | x | x | ||||
| GRA3 | KVEEELSLLR | x | x | x | x | x | x | x | ||
| AVILSLATSAAIGGR | x | x | x | |||||||
| GRA7 | REPEPLETEPDEQEEVHFR | x | x | x | x | x | x | x | x | |
| NSDFFDGQAPVDSLRPTNAGVDSK | x | x | x | x | x | x | x | |||
| SDAEVTDDNIYEEHTDRK | x | x | x | |||||||
| GVGSDAVTDDHIYEENTDRK | x | |||||||||
| GRA12 | AATVAAGNELFK | x | x | x | x | x | x | x | ||
| TEFIEGIITK | x | x | x | x | x | x | ||||
| ASETGSGLAASFLNTVEVR | x | x | x | x | x | x | x | x | ||
| RON8 | FPDGTEATLR | x | ||||||||
| SIIDLVGASDGTQPLVLK | x | x | ||||||||
| ATVIPVIETFGPDGELLK | x | x | x | x | x | x | ||||
| LLDLASPVTIVGDLTR | x | x | x | x | ||||||
| ROP4 | VVVPAEYDEYTPPEGR | x | x | x | x | x | x | x | x | |
| LLALQAIETPEYR | x | x | x | x | x | x | x | x | x | |
| ROP5 | VPASSTTTSASEGIFR | x | x | x | x | x | ||||
| LHEATFAAAR | x | x | x | x | ||||||
| TLDFLYVLR | x | x | x | |||||||
| TNDLASGTPHVAR | x | x | ||||||||
| ROP7 | VVAENAQGFSPPEVR | x | x | x | x | x | x | x | x | |
| HQALAIGLFGK | x | x | x | x | x | x | ||||
| FVEELYELPTEDRPLADAAR | x | x | x | x | x | x | ||||
| VWPERPQPVFTEGDPPDLETNSLYYR | x | x | x | x | x | x | x | |||
| MIC10 | KQEAVIQELK | x | x | |||||||
| RIDEQQANYEQR | x | x | ||||||||
| M2AP | NSFSGEDSSEIDEKEVSLPIK | x | x | |||||||
| SAG1 | TALTEPPTLAYSPNR | x | x | x | x | x | x | |||
| STAAVILTPTENHFTLK | x | x | x | x | x | x | x | x | x | |
| LSENPWQGNASSDNGATLTINK | x | x | x | x | ||||||
| AVTLSSLIPEAEDSWWTGDSASLDTAGIK | x | x | x | x | x | x | x | |||
| SAG2A | LTISPSGEGDVFYK | x | x | x | x | x | x | x | ||
| KLTTVLPGAVLTAK | x | x | x | x | x | |||||
Figure 1. Necessary proteins for tachyzoite invasion of host cell.
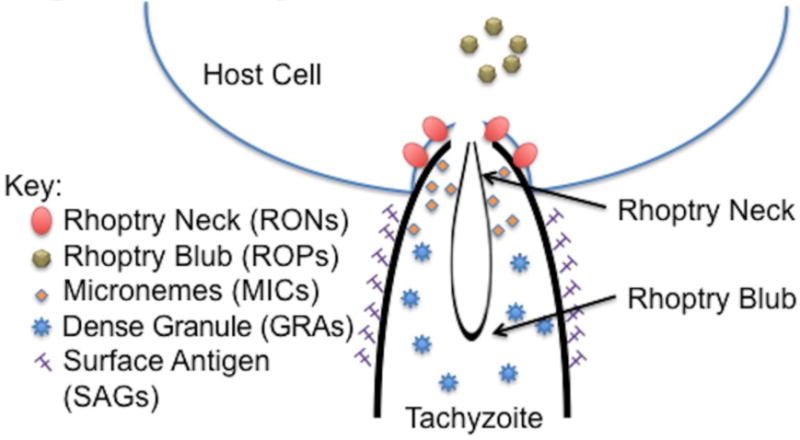
Microneme (MIC) proteins aggregate near the apical end of the tachyzoite. [18–20] MIC and Rhoptry Neck (RON) proteins then work to attach to the host cell membrane. [18,21,22] Attachment is followed by aggregation of Rhoptry Blub (ROP) proteins near the host’s membrane, and later secretion of dense granules (GRA). [22–25] SAGs coat the surface of the tachyzoite.
Sensitivity of detection of RH tachyzoites by Parallel Reaction Monitoring
The analysis of the seven isolates allowed for the establishment of a protein profile commonly seen in various isolates of T. gondii. We then wanted to confirm that our in-house live culture’s protein profile was similar to the seven BEI isolates as well as explore the limits of detection of PRM after hydrogel nanoparticle concentration. Mass spectrometry analysis has previously been limited due to a lack of ability to detect low concentrations of protein. To establish the limits of detection of the utilization of hydrogel nanoparticles in combination with parallel reaction monitoring, 106 to 101 RH strain tachyzoites were spiked into one milliliter of human urine, and the urine samples then incubated with 200µL of nanoparticles.
Figure 2 displays the relationship between the number of urine-spiked tachyzoites and the area under the curve for the specified peptide ion fragmentation peaks by PRM for two Surface Antigen 1 (SAG1) peptides (TALTEPPTLAYSPNR and STAAVILTPTENHFTLK) and one Dense Granule Protein 1 (GRA1) peptide (VERPTGNPDLLK). These were the only 3 peptides that could be detected down to the 100 tachyzoite level. Less sensitive, but still detectable in the sample containing 106 tachyzoites were three peptides from GRA12 and GRA7, two peptides from rhoptry bulb proteins (ROP) 4 and GRA3, and one peptide each from GRA1, ROP5, ROP7, SAG2A, and micronemal protein 10 (MIC10) (Table 2).
Figure 2. Relationship between the area under the fragmentation peak and the tachyzoite count.
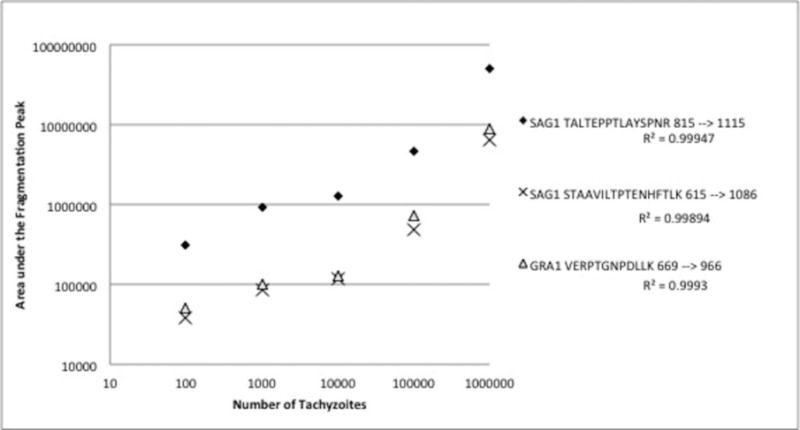
Decreasing counts of tachyzoites were spiked into urine prior to hydrogel nanoparticle incubation. After incubation with, and elution from the nanoparticles PRM analysis revealed a linear relationship between the number of tachyzoites and the area under the fragmentation peak for SAG1 peptides (TALTEPPTLAYSPNR and STAAVILTPTENHFTLK) and one GRA1 peptide (VERPTGNPDLLK).
Sensitivity of detection of tachyzoites by western blot significantly improved with nanoparticles
An identical protocol as described for the PRM spike-in studies was used for the preparation of samples for western blotting (with the exception of the elution) to compare the sensitivities of PRM results with western blotting. Figure 3A and 3B demonstrate the concentration capacity of the nanoparticles. Without the nanoparticles it is only possible to detect 105 tachyzoites of GRA1 and 104 tachyzoites of SAG1. In contrast, after nanoparticle concentration, SAG1 was detectable in the lysate with as few as 10 tachyzoites (Figure 3A). One thousand tachyzoites were required for detection of GRA1 (Figure 3B).
Figure 3. Nanoparticles bind target antigens from whole tachyzoite lysate.
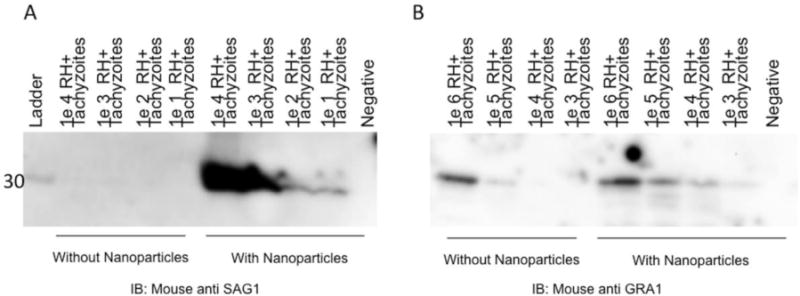
Cultured RH+ tachyzoites were lysed and spiked into 1mL of human urine with and without nanoparticles. Following nanoparticle incubation, nanoparticles were eluted and gel electrophoresis and immunoblotting for SAG1 (Figure 3A) and GRA1 (Figure 3B) followed.
Establishment of successful murine infection model with RH and ME49 infected mice by qPCR of blood and tissues
After characterization of the RH strain tachyzoites by PRM and western blot, mice were infected. The virulence of the RH strain is well established. Additionally, mice were infected with the ME49 strain, which is well established for its chronic infection capacity. Tissues from mice infected with each of these strains were analyzed by qPCR (Table 3). In the ME49 infected mice 12 of 12 brain and 10 of 12 (83%) heart tissues of mice were positive for T. gondii. However, none of these mice had parasite circulating in their blood. This is in contrast to all five of five RH infected mice, which had parasites in their blood. Similar to the ME49 infected mice, the RH infected mice had 3 of 4 (75%) heart and 2 of 2 brain tissue samples positive for parasite by qPCR.
Table 3. qPCR results from uninfected mice and mice infected with RH or ME49 strains of T. gondii.
Mice infected with T. gondii provided urine and blood specimens and were then euthanized prior to homogenization and DNA extraction of heart and brain specimens. qPCR was then completed on all specimens.
| Group | Blood | Heart | Brain |
|---|---|---|---|
| ME49 | 0 of 12 (0%) |
10 of 12 (83%) |
12 of 12 (100%) |
| RH+ | 5 of 5 (100%) |
3 of 4 (75%) |
2 of 2 (100%) |
| Uninfected | 0 of 7 (0%) |
0 of 7 (0%) |
0 of 7 (0%) |
| Uninfected and Immunosuppressed |
0 of 10 (0%) |
0 of 10 (0%) |
0 of 10 (0%) |
Detection of antigens in urine from ME49 and RH infected mice by PRM
Urine samples from 4 mice infected with ME49 and 4 mice infected with RH were collected and pooled by infection strain. Urine was incubated with nanoparticles and prepared for PRM analysis as described above. PRM analysis of the RH mouse urine revealed two peptides each from SAG1 (TALTEPPTLAYSPNR, STAAVILTPTENHFTLK), GRA1 (VERPTGNPDLLK, VIDDVQQLEK), and ROP4 (VVVPAEYDEYTPPEGR, LLALQAIETPEYR), and one peptide each from SAG2A (KLTTVLPGAVLTAK), and ROP5 (TLDFLYVLR). (Table 2 and Figure 4). While the majority of these peptides had previously been identified in the BEI tachyzoites lysate, the ROP5 peptide, though identified in NR-20734 and NR-223 reference isolates, was not identified in the lysate. None of the targeted proteins were found by PRM analysis in the chronic strain ME49 infected mouse urine.
Figure 4. PRM for RH Strain Tachyzoites (Tz) and RH infected Murine Urine.
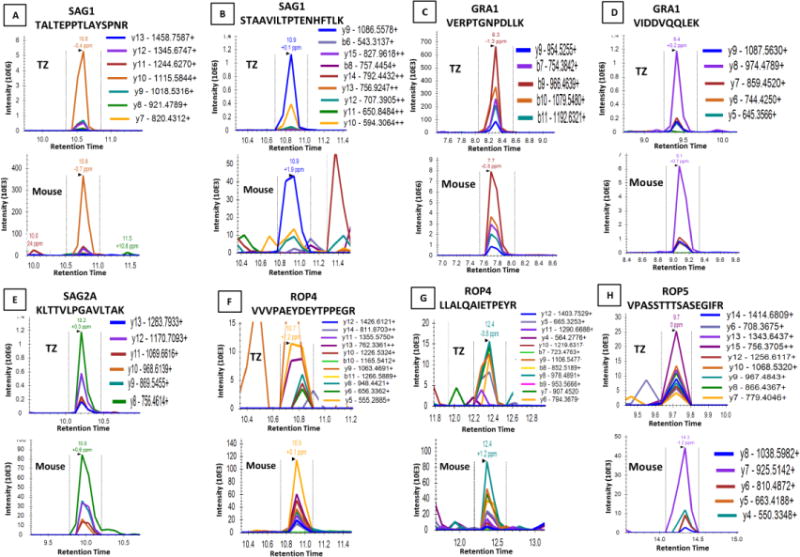
PRM spectra for the 106 RH strain tachyzoites after incubation with nanoparticles acting as a positive control, and the pooled urine collected from 4 RH strain infected mice after incubation with nanoparticles. The spectra shown are for the proteins detected in both the tachyzoite positive control (upper panels) and the pooled mouse urine sample (lower panels). The proteins detected are SAG1 (A) and (B), GRA1 (C) and (D), SAG2A (E), ROP4 (F) and (G), and ROP5 (H). Except for protein ROP5, the same peptides from each protein were detected in both samples. For ROP5, a different peptide was present for the tachyzoite sample and the mouse sample. The fragmentation ions detected for each peptide are depicted by the color scheme shown adjacent to each spectrum.
Discussion
Antigen detection by immunologic assay has been limited by the availability of highly specific antibodies. Mass spectrometry offers high specificity for antigen detection because of its capacity for exact peptide matches, but has previously been limited by poor limits of detection for low abundance targets. Through the joining of the nanoparticle concentration technique that semi-specifically increases the concentration of antigen found in biological fluids with the specificity of mass spectrometry, we have identified T. gondii parasitic antigens in the urine of infected mice.
The T. gondii genome is still being explored for differences between serotypes and strains. Thus, we selected a variety of isolates to ensure the greatest possible coverage and identification of possible protein targets for PRM. The finding that of the 1054 non-redundant proteins across all seven strains SAG and GRA proteins were some of the most abundant, given other protein related studies of T. gondii was unsurprising. [18,26]
Of the three families of proteins necessary for T. gondii invasion into new host cells- rhoptry blub proteins (ROPs), rhoptry neck proteins (RONs), and microneme (MICs)- ROPs and MICs were identified in the tachyzoite lysate (Figure 1). Initial parasite invasion is marked by the discharge of MICs for attachment to the host cell. Our group identified MIC10. [18–20] These same micronemal proteins work with the RONs to create a junction with the host cell’s plasma membrane. [18,21,22] Our group did not identify any proteins from the RON family. This is likely due to their large size, which would have been excluded by the nanoparticles. Finally, ROPs aggregate with the parasite vacuole membrane. [22–25] Our results identified ROP5 and ROP7. For GRA proteins, which are secreted after the invasion process, we identified GRA1, 3, 7, and 12. [26] The identification of these invasion sequence proteins increases our confidence that our method is both representative of the biological interaction between host and parasite, and supportive of the use of urine as an appropriate biofluid to observe such interactions.
Successful detection of decreasing concentrations of tachyzoite lysate with monoclonal anti-SAG1 and monoclonal anti-GRA1 illustrates the success of the nanoparticles ability to capture and concentrate the tachyzoites. The presence of SAG1 in urine could be predicted given that SAG1 coats the surface of the tachyzoite. [27] GRA1 and its dense granule compatriots (GRA2, 3, 5, 7, and 8) are secreted by the tachyzoite, after the parasite creates the parasitophorous vacuole. [26] We posit the varying levels of detection between the two antigens are a function of differing concentration of each antigen produced by the tachyzoites.
Terra et al preformed a series of comparison studies using conventional PCR between RH and ME49 looking at tissue specific infection. [4] They found that 50% of infected mice (infected with RH or ME49 strains) had heart infiltration by parasite components. [4] In contrast, in our study, cardiac infection was found in 83% of mice infected with the ME49 strain and 75% infected with the RH strain. This difference is likely a function of our use of qPCR in comparison to Terra et al use of conventional PCR. [4] Parasites in brain tissue were consistent across all mice in both groups, regardless of strain of infection, as was the case in our specimens as well. [4] Numerous other groups have used murine infection models to identify T. gondii with both conventional and qPCR. Dadimoghaddam et al and Daryani et al found mice intraperitoneally infected with RH T. gondii had varying levels of parasite in numerous organs depending on days post infection. [28,29] Notably, the high levels of parasite in the kidney offers one explanation as to why detection of antigen in urine is possible.
The detection of multiple antigens in the RH infected mouse urine, but not in the ME49 infected mouse urine, suggests that the chronically infected mice are not shedding antigen, or that the antigen is below the limits of detection. This finding is promising when looking forward to antigen detection in human urine of clinical toxoplasmosis cases. However, it has yet to be ascertained if the lack of antigen in the urine is a function of the infecting strain of T. gondii or the chronic infection status of the host. The relative strength of the signal for GRA1 peptides from the RH infected mice were significantly stronger than that of what was seen in the tachyzoites. This finding is in keeping with the predicted expression of GRA1 as it is most readily secreted immediately after the parasite enters new cells, which is likely the case in the RH infected mice four days post-infection. The lower diversity of detected peptides in the murine urine samples compared to the tachyzoite lysate is likely a function of the limits of detection of this assay.
Taken together these results offer a new method of detection for T. gondii. The successful capture of multiple antigens by the hydrogel nanoparticles allows for the adequate concentration of these antigens for mass spectrometry. The application of the standard discovery process of LC-MS/MS allowed for the identification of peptides for PRM, which could then be monitored in the tachyzoite lysate-spiked urine and murine urine samples. The linear response of the SAG1 and GRA1 peptides with PRM creates assurance that we have identified specific peptides, as well as determined the relative limits of detection of the PRM technique for antigen detection in T. gondii. The difference in peak magnitude between the tachyzoite lysate from culture and the RH infected murine urine sample elucidates the relationship between the behavior of the tachyzoites when they have infected the majority of possible host cells (as is expected in culture) and the behavior of these same tachyzoites as they continue to seek new host cells for infection in the mouse. Finally, the successful capture and identification of T. gondii antigens by PRM of GRA1, ROP4, ROP5, SAG1, and SAG2A in murine urine samples offers proof of concept that antigen detection in urine is possible. Finally, the successful capture and identification of T. gondii antigens by PRM of GRA1, ROP4, ROP5, SAG1, and SAG2A in murine urine samples offers proof of concept that antigen detection in urine is possible using an appropriated affinity concentration method (in this case affinity hydrogel nanoparticles) prior to mass spectrometry analysis. As analytical sensitivity improves, we envision that mass spectrometry will be used in the future for culture-free, antibody-free pathogen diagnosis and functional characterization in vivo.
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. Rafael Saavedra and Dr. Ricardo Vitor for their kind donations of the RH and ME49 strain tachyzoites respectively. Mouse monoclonal antibody Tg17-43 α-GRA1 was originally provided by Dr. Marie-France Cesbron Delauw. The following reagent were obtained through BEI Resources, NIAID, NIH: Toxoplasma gondii- Strain VEG, NR-20730; Strain MAS, NR-20731; Strain CAST, NR 20734; Strain EGS, NR-44106; Strain RH, NR-223; Strain ME-49, NR-225, NR 362.
Funding from INNOVATE PERU (137-PNICP-PIAP) allowed for the collaboration of Universidad Peruana Cayetano Heredia and Universidad Nacional Mayor de San Marcos as well as equipment, training and materials. Dr. Gilman’s 1D43TW010074-01 supported the training of many of the Peruvian authors. This study was partially funded by the following grants to Dr. Liotta: NIH NCI R33CA173359 and R33CA206937, NIAID R21AI099851 and R21AI117425, NIAMS 1R01AR068436. Drs. Liotta and Luchini are inventors on patents related to the nanoparticles. Ceres Nanosciences licensed the rights of these patents that are owned by George Mason University. Drs. Liotta and Luchini own shares of Ceres Nanosciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Desmonts G, Couvreur J. Congenital toxoplasmosis: a prospective study of 378 pregnancies. N Engl J Med. 1974;290(20):1110–1116. doi: 10.1056/NEJM197405162902003. [DOI] [PubMed] [Google Scholar]
- 2.Boothroyd JC, Grigg ME. Population biology of Toxoplasma gondii and its relevance to human infection: do different strains cause different disease? Curr Opin Microbiol. 2002;5(4):438–442. doi: 10.1016/s1369-5274(02)00349-1. [DOI] [PubMed] [Google Scholar]
- 3.Saeij JP, Boyle JP, Boothroyd JC. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 2005;21(10):476–481. doi: 10.1016/j.pt.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Terra Mrcia Andreia Barge Louo, Bello AR, Bastos OM, Amendoeira RR, Coelho Janice Mary Chicarino de Oliveira, Ferreira LF, et al. Detection of Toxoplasma gondii DNA by polymerase chain reaction in experimentally desiccated tissues. Memrias do Instituto Oswaldo Cruz. 2004;99(2):185–188. doi: 10.1590/s0074-02762004000200012. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Holmes E, Nicholson JK, Cloarec O, Chollet J, Tanner M, et al. Metabonomic investigations in mice infected with Schistosoma mansoni: an approach for biomarker identification. Proc Natl Acad Sci U S A. 2004;101(34):12676–12681. doi: 10.1073/pnas.0404878101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou C, Zhou D, Elsheikha HM, Zhao Y, Suo X, Zhu X. Metabolomic profiling of mice serum during toxoplasmosis progression using liquid chromatography-mass spectrometry. Scientific reports. 2016;6:19557. doi: 10.1038/srep19557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira L, Sánchez-Juanes F, Muñoz-Bellido JL, González-Buitrago JM. Rapid method for direct identification of bacteria in urine and blood culture samples by matrix-assisted laser desorption ionization time-of-flight mass spectrometry: intact cell vs. extraction method. Clinical Microbiology and Infection. 2011;17(7):1007–1012. doi: 10.1111/j.1469-0691.2010.03339.x. [DOI] [PubMed] [Google Scholar]
- 8.Castro-Sesquen YE, Gilman RH, Galdos-Cardenas G, Ferrufino L, Sánchez G, Ayala EV, et al. Use of a novel chagas urine nanoparticle test (Chunap) for diagnosis of congenital chagas disease. PLoS neglected tropical diseases. 2014;8(2) doi: 10.1371/journal.pntd.0003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro-Sesquen Y, Gilman RH, Bern C, Luchini A, Choi J, Reimer M, et al. Use of a Chagas urine nanoparticle test (Chunap) for prediction of parasitemia levels. T cruzi/HIV co-infected patients PLOS Neglected Tropical Diseases. 2016 doi: 10.1371/journal.pntd.0004407. Manuscript in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magni R, Espina BH, Shah K, Lepene B, Mayuga C, Douglas TA, et al. Application of Nanotrap technology for high sensitivity measurement of urinary outer surface protein A carboxyl-terminus domain in early stage Lyme borreliosis. Journal of translational medicine. 2015;13(1):346. doi: 10.1186/s12967-015-0701-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douglas TA, Tamburro D, Fredolini C, Espina BH, Lepene BS, Ilag L, et al. The use of hydrogel microparticles to sequester and concentrate bacterial antigens in a urine test for Lyme disease. Biomaterials. 2011;32(4):1157–1166. doi: 10.1016/j.biomaterials.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredolini C, Tamburro D, Gambara G, Lepene BS, Espina V, Petricoin EF, III, et al. Nanoparticle technology: amplifying the effective sensitivity of biomarker detection to create a urine test for hGH. Drug testing and analysis. 2009;1(9–10):447–454. doi: 10.1002/dta.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen KD, Camejo A, Melo MB, Cordeiro C, Julien L, Grotenbreg GM, et al. Toxoplasma gondii superinfection and virulence during secondary infection correlate with the exact ROP5/ROP18 allelic combination. MBio. 2015;6(2):2280. doi: 10.1128/mBio.02280-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubey JP. Refinement of pepsin digestion method for isolation of Toxoplasma gondii from infected tissues. Vet Parasitol. 1998;74(1):75–77. doi: 10.1016/s0304-4017(97)00135-0. [DOI] [PubMed] [Google Scholar]
- 15.Wahab T, Edvinsson B, Palm D, Lindh J. Comparison of the AF146527 and B1 repeated elements, two real-time PCR targets used for detection of Toxoplasma gondii. J Clin Microbiol. 2010;48(2):591–592. doi: 10.1128/JCM.01113-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luchini A, Fredolini C, Espina BH, Meani F, Reeder A, Rucker S, et al. Nanoparticle technology: addressing the fundamental roadblocks to protein biomarker discovery. Curr Mol Med. 2010 Mar;10(2):133–141. doi: 10.2174/156652410790963268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luchini A, Espina V, Liotta LA. Protein painting reveals solvent-excluded drug targets hidden within native protein–protein interfaces. Nature communications. 2014:5. doi: 10.1038/ncomms5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Hajj H, Demey E, Poncet J, Lebrun M, Wu B, Galéotti N, et al. The ROP2 family of Toxoplasma gondii rhoptry proteins: proteomic and genomic characterization and molecular modeling. Proteomics. 2006;6(21):5773–5784. doi: 10.1002/pmic.200600187. [DOI] [PubMed] [Google Scholar]
- 19.Dubremetz JF, Achbarou A, Bermudes D, Joiner KA. Kinetics and pattern of organelle exocytosis duringToxoplasma gondii/host-cell interaction. Parasitol Res. 1993;79(5):402–408. doi: 10.1007/BF00931830. [DOI] [PubMed] [Google Scholar]
- 20.Carruthers VB, Sibley LD. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur J Cell Biol. 1997;73(2):114–123. [PubMed] [Google Scholar]
- 21.Alexander DL, Mital J, Ward GE, Bradley P, Boothroyd JC. Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog. 2005;1(2):e17. doi: 10.1371/journal.ppat.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lebrun M, Michelin A, El Hajj H, Poncet J, Bradley PJ, Vial H, et al. The rhoptry neck protein RON4 relocalizes at the moving junction during Toxoplasma gondii invasion. Cell Microbiol. 2005;7(12):1823–1833. doi: 10.1111/j.1462-5822.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- 23.SAFFER LD, MERCEREAU-PUIJALON O, DUBREMETZ J, SCHWARTZMAN JD. Localization of a Toxoplasma gondii rhoptry protein by immunoelectron microscopy during and after host cell penetration. J Protozool. 1992;39(4):526–530. doi: 10.1111/j.1550-7408.1992.tb04844.x. [DOI] [PubMed] [Google Scholar]
- 24.Beckers CJ, Dubremetz JF, Mercereau-Puijalon O, Joiner KA. The Toxoplasma gondii rhoptry protein ROP 2 is inserted into the parasitophorous vacuole membrane, surrounding the intracellular parasite, and is exposed to the host cell cytoplasm. J Cell Biol. 1994;127(4):947–962. doi: 10.1083/jcb.127.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carey KL, Jongco AM, Kim K, Ward GE. The Toxoplasma gondii rhoptry protein ROP4 is secreted into the parasitophorous vacuole and becomes phosphorylated in infected cells. Eukaryotic cell. 2004;3(5):1320–1330. doi: 10.1128/EC.3.5.1320-1330.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou XW, Kafsack BF, Cole RN, Beckett P, Shen RF, Carruthers VB. The opportunistic pathogen Toxoplasma gondii deploys a diverse legion of invasion and survival proteins. J Biol Chem. 2005;280(40):34233–34244. doi: 10.1074/jbc.M504160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Yin H. Research progress on surface antigen 1 (SAG1) of Toxoplasma gondii. Parasit Vectors. 2014;7(180):24. doi: 10.1186/1756-3305-7-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dadimoghaddam Y, Daryani A, Sharif M, Ahmadpour E, Hossienikhah Z. Tissue tropism and parasite burden of Toxoplasma gondii RH strain in experimentally infected mice. Asian Pacific journal of tropical medicine. 2014;7(7):521–524. doi: 10.1016/S1995-7645(14)60087-0. [DOI] [PubMed] [Google Scholar]
- 29.Daryani A, Sharif M, Dadimoghaddam Y, Souteh MBH, Ahmadpour E, Khalilian A, et al. Determination of parasitic load in different tissues of murine toxoplasmosis after immunization by excretory–secretory antigens using Real time QPCR. Exp Parasitol. 2014;143:55–59. doi: 10.1016/j.exppara.2014.05.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.