

Abstract
Linear ubiquitylation, in which ubiquitin units are covalently linked through N- and C-terminal amino acids, is a unique cellular signaling mechanism. This process is controlled by a single E3 ubiquitin ligase, the linear ubiquitin chain assembly complex (LUBAC), which is composed of three proteins – HOIL-1L, HOIP and SHARPIN. LUBAC is involved in the activation of the canonical NF-κB pathway and has been linked to NF-κB dependent malignancies In this work, we present HOIP-based stapled alpha-helical peptides designed to inhibit LUBAC through the disruption of the HOIL-1L-HOIP interaction and loss of the functional complex. We find our HOIP peptides to be active LUBAC ubiquitylation inhibitors in vitro, though through interaction with HOIP rather than HOIL. Active peptides were shown to have inhibitory effects on cell viability, reduced NF-κB activity and decreased production of NF-κB related gene products. This work further demonstrates the potential of LUBAC as a therapeutic target and of the use of stapled peptides as inhibitors of protein-protein interactions.
Keywords: Linear ubiquitylation, NF-κB, stapled peptides, protein-protein interactions
1. Introduction
Protein ubiquitylation is a post-translational modification that provides a versatile range in cellular signaling owing to the many ways in which monoubiquitin or polyubiquitin chains can be attached to a protein substrate.1 One of the more recently discovered ubiquitylation modes is linear ubiquitylation, in which each individual unit is covalently connected head-to-tail using the N-terminal amine of Met1, rather than through an internal lysine side chain.2 Currently, there is only one E3 ubiquitin ligase, the linear ubiquitin chain assembly complex (LUBAC), that is known to generate these linear chains.
LUBAC is composed of three different subunits - HOIP, HOIL-1L and SHARPIN (Figure 1A).3,4 HOIP contains the catalytic domains responsible for linear ubiquitylation in its ring-between-ring (RBR) region, while HOIL-1L and SHARPIN function as accessory subunits necessary for complete LUBAC activation. HOIP and HOIL-1L interact with one another via their ubiquitin-associated (UBA) and ubiquitin-like (UBL) domains, respectively, and this interaction has been shown to be necessary for LUBAC function.3,5 The UBA-UBL interaction is mediated by two helices (α8 and α9) located in the UBA region of HOIP and separated by a proline (P619) residue which enforces a bend between the two helices (Figure 1B). A number of amino acid residues in this region (Q613, L617, F620 and R623 – indicated in red) have been identified by alanine mutation studies as being key for binding between the bent HOIP helices and the HOIL-1L-UBL.6
Figure 1.

(A) Illustration of LUBAC structural domains. The HOIP-UBA/HOIL-UBL interaction is indicated. (B) Crystal structure of the HOIP-UBA/HOIL-UBL interaction (PDB: 4DBG). UBA – beige, UBL – green. Red indicates key residues for binding, blue indicates P619. (B) Circular dichroism spectra in H2O of peptides presented in this work (50 μM peptide concentration).
LUBAC can be activated by a wide variety of stimuli2 and constitutive LUBAC activity is associated with several kinds of human malignancies, including ovarian, pancreatic, colon, and lung carcinomas7–10 as well as inflammation processes.11,12 Of particular interest in cancer, LUBAC has been shown to be involved in the canonical NF-κB signaling pathway via linear ubiquitylation of NEMO (NF-κB essential modulator)5,13 following simulation by TNFα and recruitment to the TNF-R signalosome.14 Ubiquitylated NEMO is then able to recruit and activate the IκB Kinase (IKK) complex, resulting in phosphorylation and degradation of the NF-κB inhibitor, IκBα.10,14 This cascade of events allows p65, a component of the NF-κB transcription factor, to induce the expression of several proto-oncogenes, such as cyclin D1,15,16 allowing for the maintenance of malignant phenotypes in cancer cells. Direct inactivation of LUBAC through HOIP knockdown has been shown to sensitize cells to genotoxic stress through decreased NF-kB activity,8,9 making inhibition of LUBAC a promising therapeutic target for the treatment of diseases with overactive NF-κB. Chemical inhibition of LUBAC has recently been demonstrated, with a high-throughput screen identifying the fungal metabolite gliotoxin as an inhibitor of LUBAC through direct binding to the catalytic domain of HOIP resulting in decreased NF-κB activation.17
We previously demonstrated that a stapled α-helical peptide based on the HOIP-UBA helical contacts (HOIP-N, 2) sensitizes activated B-cell (ABC) diffuse large B-cell lymphoma (DLBCL) cells to genotoxic stress through interaction with LUBAC resulting in reduced NF-κB activation.18 Stapled α-helical peptides are a class of synthetic macrocycles in which the secondary α-helix structure is stabilized by the introduction of a hydrophobic bridge or “staple”.19 Hydrocarbon stapling has been shown to reduce susceptibility of peptides to proteolytic degradation and enhance their cell permeability,19 and has been successfully used to generate peptides capable of disrupting protein-protein interactions important for the treatment of numerous diseases, including cancer.20,21
In the current work, we explore a library of stapled α-helical peptides based on the α8 and α9 helices of HOIP-UBA to optimize their ability to downregulate LUBAC activity. We expand our knowledge further into the structure-activity relationships of the original HOIP-N peptide and investigate their mechanism of LUBAC inhibition. We show that peptide inhibition can be optimized by the choice of staple position and demonstrate that the entire macrohelix is, in fact, unnecessary for LUBAC inhibition. Finally, we tested a selection of active peptides for their ability to inhibit LUBAC and examined the resulting effects against a panel of carcinoma cells.
2. Results and discussion
2.1 Design and synthesis of stapled HOIP peptides
Our original design generated two peptides (HOIP WT, 1 and HOIP-N, 2) based on a 22-amino acid region of the HOIP-UBA (residues 606-627) shown to interact with the HOIL-1L-UBL domain.6 We sought to improve the performance of our existing stapled peptide by exploring what specific structural modifications within this sequence were necessary for the effective inhibition of LUBAC. To that end, we designed and synthesized an additional fourteen peptides (3-16, Table 1) based upon this dual helix region of HOIP-UBA shown to be responsible for interaction with HOIL-UBL. We systematically introduced small changes to the HOIP peptide parent sequence18 exploring three different staple positions, overall peptide flexibility, and minimum helix size while maintaining those residues previously shown to be important for binding (Q613, L617, F620 and R623 – indicated in red. Figure 1B). We then evaluated the effect that these modifications had on peptide shape in solution via circular dichroism (CD) spectroscopy.
Table 1.
Summary of synthesized HOIP-based peptides
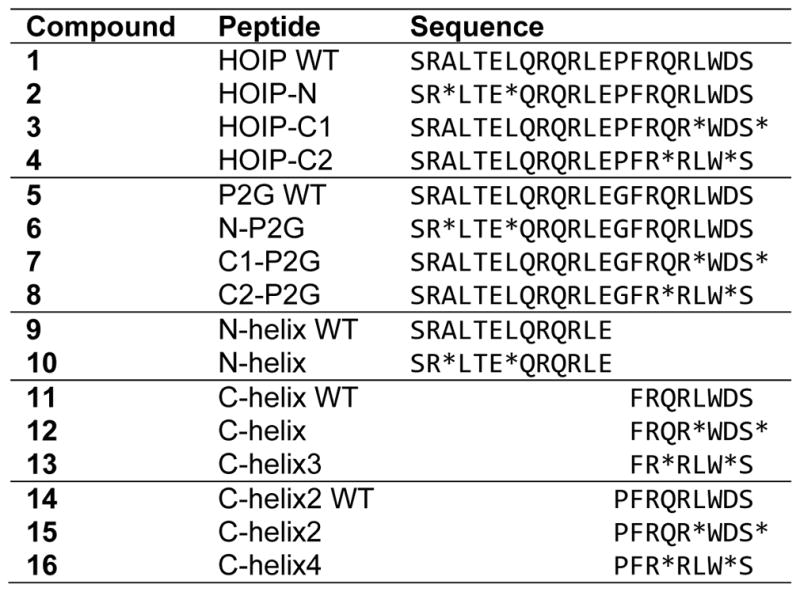
|
Denotes position of the cross-linking amino acid.
For comparison to our previously published peptides (unstapled HOIP WT (1) and N-terminal stapled HOIP-N (2)),18 we synthesized two additional peptides to study the effect of placing the hydrocarbon staple on the shorter C-terminal α9 helix. This generated the full-length peptides HOIP-C1 (3) and HOIP-C2 (4), which contained terminal and internal C-helix staples, respectively. Stapling of either helix in the full-length peptides only slightly increased their α-helical character when compared to unstapled HOIP-WT (1), as shown by CD spectroscopy (Figure 1C, “Original sequence”). This was not unexpected due to the presence of the central proline residue (P619) which would prevent complete, end-to-end helix nucleation in the full peptide. To explore the importance of this kinked structure, we substituted the rigid P619 with a more flexible glycine residue to generate a proline-to-glycine (“P2G”) series of compounds (5 – 8, “P2G sequence”). Compared to P2G WT (5), which remained unstructured in solution, the P2G modification was found to dramatically increase the overall helicity of the two C-stapled peptides, but had only minimal effect on the N-stapled P2G compound (6) (Figure 1C, “P2G sequence”). This latter effect was surprising, as we had expected to see greater overall helicity with 6 achieved by substitution of the proline residue.
The effect on peptide helicity from hydrocarbon crosslinking at these three staple positions became more apparent when we considered the two halves of the full peptide sequence as individual helices. We used P619 to define where the two helices met and individually synthesized both N- and C-terminal helices in WT and stapled forms. We found that stapling of the α8 helix as in N-helix (10) increased the α-helical character compared to WT (9) (Figure 1C, “Short N-helix”). Similarly, internal (C2) stapling of the short C-helix region was found to increase helicity as shown by C-helix3 (13). Despite the dramatic increase in helicity due to the terminal (C1) C-helix stapled observed for the full-length C1-P2G peptide (7), only a minimal effect was seen for the shorter C-helix peptide with the same staple position (12). We also found that inclusion of an N-terminal proline in peptides 14-16 did not have any further effect on helicity in solution, when compared to related compounds lacking proline (11-13) (Figure 1C, “Short C-helix”, solid vs. dashed lines).
2.2 Stapled HOIP peptides inhibit LUBAC ubiquitylation activity in vitro
We next evaluated whether our stapled peptides could inhibit linear ubiquitylation activity of LUBAC using a single peptide concentration (40 μM) in an in vitro auto-ubiquitylation assay. Since active, trimeric LUBAC is difficult to generate in sufficient quantity, we instead used a truncated sub-complex of LUBAC known as “Petit-LUBAC” that has been shown to be easily expressed and exhibits robust linear polyubiquitylation activity (Supplemental Figure 1A & 1B).17 Most important for this study, Petit-LUBAC contains residues 1-191 of human HOIL-1L and residues 474-1072 of human HOIP, encompassing their respective UBL (residues 37-128) and UBA (residues 480-636) domains in addition to the catalytic regions of HOIP, which would allow us to look at peptide activity in the presence of our targeted interaction.
We found that all stapled proline-containing full-length peptides (2-4) resulted in a discrete inhibition of linear ubiquitylation when compared to a vehicle-treated reaction (Figure 2A). Qualitatively, HOIP-C2 (4) was most effective at shutting down Petit-LUBAC activity as shown by a near complete absence of polyubiquitin bands, while the remaining peptides exhibited moderate decreases in ubiquitin laddering of the order HOIP-C1 (3) > HOIP-N (2). A similar effect was observed in the P2G family, as stapled C2-P2G (8) also exhibited the strongest effect with complete inactivation of Petit-LUBAC, while N-P2G (6) displayed only modest activity, and P2G WT (5) and C1-P2G (7) were inactive. These results suggest that staple position in this system has a significant effect on potency as the internal C-helix (C2) staple was most effective in both cases.
Figure 2.

Inhibitory effects of HOIP-based stapled peptides on Petit-LUBAC activity. (A) Representative blots against linear ubiquitin of autoubiquitylation assays of Petit-LUBAC preincubated with 40 μM of the specified HOIP based peptides. Labels are compound numbers. (B) Immunoblots against total ubiquitin of dose response experiments for each specified peptide against petit-LUBAC. (C) Dose response graphs were generated by plotting % of LUBAC inhibition (normalizing each densitometric value of free ubiquitin with the one corresponding to 60 μM peptide) against [Peptide] (μM), and used to estimate an IC50 for each tested peptide.
We then turned our attention to evaluating inhibitory properties of the single N- and C-helix peptides. Of these shorter peptides, the highly-structured stapled N-helix (10) was unexpectedly inactive against Petit-LUBAC. In fact, most partial peptides were marginally inhibitory except for C-helix3 (13) and C-helix4 (16), both of which also contained the more strongly helix-promoting, internal C2 staple position. We likewise found that addition of the leading proline (C-helix4 (16)) slightly weakened inhibition of Petit-LUBAC. We also confirmed that the observed decreases in autoubiquitylation were due to peptide interaction with Petit-LUBAC rather than through binding to another component of the reaction mixture (E1, E2, or ubiquitin). Using HOIP-N and C-helix3 as representative peptides, we found that they did not affect ubiquitylation using a different E3 (HDM2 RING) in the place of Petit-LUBAC (Supplemental Figure 1C).
With the results of the Petit-LUBAC screen in hand, we next investigated the dose response behavior of some of the more active peptides. Of the four C2 stapled peptides that were shown to cause a near complete shutdown of ubiquitylation activity, we looked at the ability of three – C2-P2G (8), C-helix3 (13) and C-helix4 (16) – to inhibit the enzymatic activity of Petit-LUBAC up to a top peptide concentration of 60 μM (Figure 2B). We found that C-helix3 exhibited the lowest IC50 by far, while C2-P2G and C-helix4 were equipotent, albeit weaker by a factor of 5 (Figure 2C, and Table 2). These results suggest that the well-structured C-terminal helix of HOIP alone, without an adjacent N-terminal peptide or proline residue, is the minimal structural element necessary to block LUBAC activity.
Table 2.
Summary of peptide activity, in vitro inhibition and binding data
| Compound | Peptide | Petit-LUBAC IC50 (μM) | HOIP-UBA | HOIL-UBL | ||
|---|---|---|---|---|---|---|
| Kd (μM) | Chi2 | Kd (μM) | Chi2 | |||
| 1 | HOIP WT | n.b. | 8.64 | 0.108 | ||
| 2 | HOIP-N | 9.37 | 1.95 | 7.65 | 0.632 | |
| 3 | HOIP-C1 | 8.65 | 1.61 | n.b | ||
| 4 | HOIP-C2 | 6.77 | 8.01 | n.b | ||
|
| ||||||
| 5 | P2G WT | 81.4 | 1.14 | 97.3 | 1.24 | |
| 6 | N-P2G | 4.28 | 2.42 | n.d. | ||
| 7 | C1-P2G | n.b | n.d. | |||
| 8 | C2-P2G | 23.83 ± 3.5 | 6.38 | 12.1 | ** | |
|
| ||||||
| 9 | N-helix WT | n.b. | n.b | |||
| 10 | N-helix | n.b | n.b | |||
|
| ||||||
| 11 | C-helix WT | n.b. | n.b | |||
| 12 | C-helix | n.b | n.d. | |||
| 13 | C-helix3 | 3.74 ± 0.88 | n.b. | ** | ||
|
| ||||||
| 14 | C-helix2 WT | n.b. | n.b. | |||
| 15 | C-helix2 | n.b | 41.4 | 0.555 | ||
| 16 | C-helix4 | 21.68 ± 4.25 | 4.67 | 13.2 | n.b | |
n.b. = no binding at the highest tested concentration. n.d. = not determined.
unable to determine due to inconsistent behavior at highest tested concentration.
2.3 Stapled HOIP peptides unexpectedly bind to HOIP-UBA over HOIL-UBL
Given that our peptides displayed a range of activities against Petit-LUBAC, we wished to confirm that they were indeed causing inhibition through binding to HOIL-UBL and subsequent displacement HOIP-UBA. Using a native gel shift assay and fluorescently-labelled HOIP WT (1), we demonstrated that fluorescence intensity due to complexed peptide was indeed present at a higher shifted molecular weight upon inclusion of Petit-LUBAC as would be expected if the peptide was bound to the enzyme complex (Figure 3A, lane 8). As a control experiment, we looked for a similar shift with both HOIL-UBL and HOIP-UBA alone. Unexpectedly, no shift was observed with HOIL-UBL while a shift similar to the one observed with Petit-LUBAC was observed with HOIP-UBA (Figure 3A, lanes 6 and 10, respectively). We also found that the intensity of this shifted HOIP-UBA band could be decreased upon addition of unlabeled, acetylated peptide (Supplemental Figure 2), offering further confirmation that the observed shift in Petit-LUBAC was due to peptide binding to HOIP rather than HOIL.
Figure 3.

HOIP based peptides bind to HOIP-UBA over HOIL-UBL. (A) Representative gels from fluorescent hrCNE. Bands representing FITC labeled HOIP WT peptide in absence (lanes 1–4), or presence (lanes 6, 8, and 10) of specified recombinant proteins. Lanes 5, 7, and 9 were loaded with each of the recombinant proteins, but without FITC-peptide. (B) Representative blot against total ubiquitin of the HOIL-UBL and HOIP-UBA competition assays. Petit-LUBAC and the indicated peptides were incubated in presence or absence of 90 μg/mL of HOIL-UBL or HOIP-UBA.
To further investigate this counterintuitive result, we performed a competition experiment with Petit-LUBAC in which we added a three-fold excess of either HOIL-UBL or HOIP-UBA to act as competing bait for peptide binding. We found that while addition of either domain to Petit-LUBAC itself had no effect on reducing its activity (Figure 3B, lanes 1–3), the addition of HOIP-UBA but not HOIL-UBL rescued previously inhibited Petit-LUBAC from peptide treatment with both HOIP-N (2) and C-helix3 (13) (Figure 3B, lanes 4–9), again indicating that HOIP peptides preferentially bind to the HOIP-UBA over HOIL-UBL.
We attempted to quantify this unexpected result using surface plasmon resonance (SPR) spectroscopy to directly measure binding to each protein domain of interest. Full-length proline-containing peptides in general were found to favor binding to HOIP-UBA with affinities ranging from 7–10 μM (Table 2), with the exception of HOIP-N (2) which also bound to HOIL-UBL with a similar affinity (7.6 μM). These affinities against HOIP correlated with the order of peptide inhibition previously seen against Petit-LUBAC where HOIP-C2 > HOIP-C1 > HOIP-N. Given that both C-stapled peptides showed no interaction with HOIL-UBL and were the strongest inhibitors, it is plausible that the C-stapled peptides more specifically bind to a single HOIP region that is then responsible for the observed inhibition while the effectiveness of the N-stapled peptide is reduced through a lack of selectivity as shown by binding to both proteins equipotently. Full peptides containing the proline-to-glycine modification also appeared to favor HOIP over HOIL with the strongest binding to HOIP corresponding to the two most active peptides of this series, C2-P2G (8) and the weaker N-P2G (6). All unstapled peptides exhibited little to no binding towards either protein, indicative of the high energetic penalty necessary to structure these peptides for binding in solution.
In the case of the single helix peptides, weak-to-no binding to either protein domain was seen for most of the inactive peptides such as N-helix (10) and both internal C1 peptides, C-helix (12) and C-helix2 (15). C-helix4 (16) was found to bind tightly to HOIP-UBA (Kd = 4.7 μM) while binding of C-helix3 (13) proved difficult to measure accurately at the higher end of concentrations used. We found in many cases that our measured binding constants became tighter at lower peptide concentrations, which could indicate a tendency for some peptides to aggregate or self-associate at higher concentrations, especially for the more flexible (P2G) or shorter single-helix peptides such as 8 and 13, respectively. Self-association would also not be unexpected, since we have now shown that these HOIP-based peptides bind to HOIP itself.
Comparison of affinities from the short vs. long stapled peptides was especially interesting as it showed that nearly all HOIP-binding affinity displayed by the C2-stapled peptides originates from the short, stapled C-helix exemplified by C-helix4 (16). Similarly, given that the structured N-helix peptide (10) did not bind to either protein but full-length peptides containing this stapled helix did, it again appears that the C-terminal helix is the more critical of the two in terms of peptide affinity as well as peptide inhibition.
While the determination of binding constants by SPR addressed the question of which protein these peptides bind to, it does not address the question of where on each protein that binding was taking place. We found that the ability of peptides to bind to a given protein (HOIL or HOIP) did not directly correlate in all cases to that peptide’s degree of LUBAC inhibition. For example, both N-P2G (6) and C-helix4 (16) exhibited the tightest binding constants to HOIP (Table 2, 4.3 vs 4.7 μM, respectively) but significantly different responses to Petit-LUBAC (Figure 2A). Further structural studies to elucidate exactly how and where each peptide binds, how hydrocarbon stapling influences the binding location, and how this mode of binding affects enzymatic activity are necessary to fully understand this mechanism of inhibition.
We were specifically intrigued by the results of C-helix3 (13), which, despite exhibiting the strongest inhibitory activity against Petit-LUBAC, did not have any measurable direct binding to either HOIP or HOIL. Consequently, we wanted to determine whether this peptide was in fact binding to any component in Petit-LUBAC by using Petit-LUBAC itself. Native gels confirmed that the C-helix3 (13) peptide binds weakly to the HOIP-UBA, and in a lesser extent to HOIL-UBL (Supplemental Figure 3A, lanes 4 and 5). However, when 13 was exposed to Petit-LUBAC, a higher shifted band than previously observed, appeared. Western blot assays revealed that this peptide shift correlates with the position where HOIL and HOIP co-migrated on native electrophoresis (Supplemental Figure 3B). Together these observations suggest that the C-helix3 peptide preferentially binds to the HOIL-HOIP complex rather than to its individual components, raising the possibility that this peptide inhibits LUBAC activity without disrupting the protein complex.
2.4 Active HOIP peptides affect cancer cell viability through the NF-κB pathway
LUBAC activity has been related to the malignant phenotype of some kinds of cancer.8,9,18,22 Having determined which HOIP-peptides were most active against Petit-LUBAC, we then assessed their cellular effects including cell viability, mechanism of cell death (if present), and any effect on aspects of the downstream LUBAC pathway, including NF-κB. We tested a selection of active and inactive HOIP-peptides against a range of carcinomas with known sensitivities to HOIP knockdown, including fibrosarcoma, pancreatic, colon, and cervical carcinomas.9,22 We observed reductions in cell viability ranging from 20–50% in all tested cell lines with the exception of HT1080 fibrosarcoma cells (Figure 4A). In most cases, the most effective peptides tested were C1-P2G (7), C-helix3 (13), and C-helix4 (16) while HOIP WT (1) and N-helix (10) were the least effective, correlating with the Petit-LUBAC results presented earlier. These observations are in agreement with other published work which has shown that HOIP knockdown sensitizes cells to genotoxic induced death.9 Also, our observations on HT1080 cells are in agreement with Niu, et al.,22 who showed that HOIP knockdown by itself was insufficient to induce cell death in this cell line. Moreover, our results also showed that some cells respond better to peptide treatment than others, suggesting that LUBAC activity may have specific functions in different types of cancer.
Figure 4.

HOIP-based peptides decrease viability of cancer cells through cell-type specific mechanisms. (A) Cell viability as measured after 72 h of peptide treatment. Data were normalized to the untreated control. Each point represents mean ± SEM. (B) After 24 h treatments with 25 or 50 μM of each specified peptides caspase-3/7 activity was determined. Each bar represents mean ± SEM. *p≤0.05 vs. 0 μM. (C) Lysates of HT1080, PANC1, and HeLa cells treated for 24 h with 50 μM of each of the specified peptides were analyzed by immunoblot of phosphorylated and total IκBα, cyclin D1, and GAPDH used as loading control.
In order to have a deeper understating of the cell type differences observed by treatment with some of the peptides, we analyzed their cell permeability by confocal microscopy. Our results indicate that the WT (1), as well as the HOIP-N (2) showed low cell permeability, while C-helix3 (13) showed higher levels of peptide internalization in all tested cell lines (Supplemental Figure 4, 5 and 6). Thus, the differences observed in cell viability because of C-helix3 (13) treatment are not due to the ability of each cell to internalize the peptide. We then analyzed the expression levels of the LUBAC components in these cell lines, and we found that while HT1080 cells expressed lower amounts of HOIP and HOIL, HeLa cells have higher expression levels of these proteins (Supplemental Figure 7). Taken together, these observations suggest that the impact on cell viability due to peptide treatment on the tested cell lines is due to its intrinsic dependence on LUBAC activity and not on the permeability properties of the compounds or the cells.
To characterize the observed reduction in viability, we evaluated caspase-3/7 activation, a hallmark of apoptosis in all tested cell lines (Figure 4B). After treatment with 50 μM peptide, we found an increase in caspase activity by C-helix3 (13) in PANC1 and C1-P2G (7) in HCT116 cells. Interestingly in HeLa cells, where we had observed the greatest reduction in viability, showed no significant change in caspase-3/7 activity or cytotoxicity (Supplemental Figure 8) indicating that cell death was not contributing to the decreased viability.
We also sought markers further downstream the LUBAC pathway which includes NF-κB (Figure 4C). In both PANC1 and HeLa cells, we found decreased levels of p-IκB and increased IκB, which could be related to LUBAC inhibition. The lack of alterations in cyclin D1 and cleaved caspase-3 in PANC1 cells suggest that other LUBAC related pathways may be affected by peptide treatment.14,23 Surprisingly, in HeLa cells, we also observed a slight reduction in cyclin D1 expression, a known NF-κB gene target,16 which suggests that the reduced viability may be due to cell cycle arrest, instead of induction of cell death, as no changes in cytotoxicity were induced in this cell line due to peptide treatment.
2.5 Active HOIP peptides exhibit inhibitory effects on the NF-κB pathway following TNFα stimulation
We next looked whether C-helix3 (13) could prevent NF-κB activation following stimulation with TNFα in the cell lines shown to be most responsive to peptide treatment. After 24 hours of peptide treatment, PANC1 and HeLa cells were with stimulated with 20 ng/mL TNFα, and the resulting NF-κB activity and secreted IL-8 were measured at 10, 60, and 240 minutes. We found that C-helix3 (13) partially reduces NF-κB activation after 60 and 240 minutes post-stimulation in both cell lines (Figure 5A), while IL-8 secretion decreased after 10 and 60 minutes of stimulation (Figure 5B). There was no difference in secreted IL-8 by 240 minutes compared to the untreated control, likely due to accumulation of IL-8 over the longer measured time. We evaluated the levels of phosphorylated IκBα by western blot (Figure 5C), and observed that, in DMSO-treated cells, IκBα levels peaked after 60 minutes of TNFα stimulation. Peak levels of IκBα are not detected until 240 minutes post-stimulation in cells treated with C-helix3 (13). Taken together, these observations suggest that the blockade of LUBAC only delays NF-κB activation, and cells likely find an alternative pathway to activate this transcription factor as has been shown previously.24
Figure 5.

C-helix3 (13) peptide delayed TNF-α NF-κB activation and IL-8 secretion. After 24h of treatment with α9 (50 μM) HeLa and PANC1 cells were stimulated with 20 ng/mL of TNF-α: (A) Cell lysates were analyzed for NF-κB activity by DNA binding ELISA. (B) Media was removed and analyzed for secreted IL-8 by specific ELISA. (C) Cell lysates were analyzed for p-IκB and GAPDH (used as loading control) expression by Western blot. Bars on A and B represent mean ± SEM, *p ≤ 0.05 vs. DMSO at the same time of TNF-α stimulation.
3. Conclusions
Specific inhibition of LUBAC, through direct inhibition of its catalytic domain or inhibition of other functional aspects, remains an attractive therapeutic target for inflammatory and oncologic diseases due to its position upstream of the NF-κB pathway. In this work, we have developed and optimized a small family of HOIP-based peptides designed to disrupt the HOIL-UBL to HOIP-UBA interaction. Though our compounds were unexpectedly found to bind to HOIP over HOIL, we showed that the α9 helix of this interacting region is most important for their LUBAC inhibition. Compounds that rigidified this area such as HOIP-C2 (4), C2-P2G (8), C-helix3 (13) and C-helix4 (16) were most effective at reducing LUBAC activity both in vitro and in cell-based studies. Surprisingly, C-helix3 (13), which contains only the α9 helix itself, was found to be the most potent peptide, and preliminary evidence suggests that LUBAC inhibition with these peptides occurs due to peptide binding but not through complex disruption as we had originally intended.
Irrespective of their mechanism of inhibition, we have shown that our active peptides reduced cellular LUBAC activity and cell viability to different extents in a panel of cancer cell lines. Interestingly, there appear to be cell-type specific effects with respect to LUBAC inhibition which are dependent on the intracellular levels of each LUBAC component. Further studies will be needed to understand the molecular basis for this cell-specific difference in susceptibility. We are currently exploring ways to improve the potency of these peptides through better structural understanding of this inhibition towards the possibility of using them in combination with other genotoxic compounds to improve their therapeutic value.
4. Experimental section
4.1 Chemistry procedures
4.1.1 Peptide synthesis and purification
All peptides were synthesized using fluorenylmethyloxycarbonyl (Fmoc)-based solid phase peptide chemistry on Rink amide AM resin (100–200 mesh, Novabiochem) at 30 μM peptide per reaction using a Tetras Peptide Synthesizer (Advanced ChemTech, Louisville, KY). In brief, the synthesis protocol consists of removal of the Fmoc-protecting group with 25% piperidine in N-methyl-2-pyrrolidone (NMP), washing with NMP, and subsequent amino acid coupling using 10 equiv. amino acid (1 mL, 0.3 M), HCTU (0.99 mL, 0.3 M), and diisopropylethylamine (DIPEA, 2.0 mL, 0.3 M) in NMP for 60 minutes before draining and washing. Crosslinking residues were coupled twice using 5 equiv. amino acids (0.5 mL, 0.3 M), HCTU (0.49 mL, 0.3 M), and DIPEA (1.0 mL, 0.3 M) for 120 minutes each to ensure reaction completion. All peptide sequences include an N-terminal β-alanine residue as a flexible linker between the helical peptide and select capping groups. The short N-helix peptide (10) also includes an additional tryptophan residue (W) prior to the β-alanine to have a spectroscopic handle for peptide quantification. Stapled peptides underwent a further ring-closing metathesis reaction on the resin-bound N-terminal Fmoc-protected peptide to complete the macrocyclization. After washing with NMP and 1,2-dichloroethane (DCE), the resin was exposed to three 6-hour cycles of bis(tricyclohexylphosphine)benzylidine ruthenium (IV) dichloride (Grubbs’ First generation catalyst, 3 mL, 0.6 mM) at room temperature in DCE. Peptides were primarily N-terminal acetylaed for use in biochemical and cell-based assays. The acetylation reaction consisted of deprotection of the Fmoc-group as described, followed by reaction with neat acetic anhydride (1 mL) and DIPEA (2.0 mL, 0.3 M in NMP) for 1 hour. Other experiments made use of N-terminal fluoresceinated peptides. To this end, Fmoc-deprotected peptides were exposed to fluorescein isothiocyanate (FITC, 2.8 mL, 25 mM) and DIPEA (0.2 mL, 0.3 M) in DMF for 12 hours. Final capped peptides were resin-cleaved and fully deprotected by exposure to a cleavage solution containing 95% TFA, 2.5% water, and 2.5% triisopropylsilane (TIPS) for 2.5–3 hours. Cleaved peptides were precipitated into ice-cold 1:1 methyl-tert-butyl ether (MTBE)/hexanes, re-suspended in water and lyophilized to yield crude dry peptides. The lyophilized peptides were purified by C-18 reverse phase HPLC on an Agilent 1200 HPLC system (Santa Clara, CA).
4.1.2 Circular dichroism spectroscopy
Acetylated compounds were dissolved in water to concentrations of 50 μM. Final compound concentration were determined by measuring sample absorbance at 280 nm (tryptophan, ε = 5560 cm−1 M−1) using a NanoDrop2000 spectrophotometer (ThermoScientific, Wilmington, DE). Spectra were obtained on an Aviv Circular Dichroism Spectrometer, Model 420 (Aviv Biomedical, Inc, Lakewood, New Jersey) at 25°C. The spectra were collected using a 0.1 cm path length quartz cuvette (Hellma Analytics, Germany) with the following measurement parameters: wavelength, 240–190 nm; step resolution, 0.50 nm; averaging time, 5.0 sec per step. Spectra were processed using Aviv CDS Program software and converted to mean residue molar ellipticity using the cuvette path length (0.1 cm), the measured concentration, and the number of amino acids in the peptide (cross-linking amino acids and β-alanine cap were included as amino acids in this count).
4.1.3 Surface plasmon resonance (SPR)
Interactions between peptides and GST-HOIP-UBA or His6-HOIL-UBL were analyzed by surface plasmon resonance using a Biacore T200 system (GE Healthcare). GST-HOIP-UBA or His-HOIL-UBL (50 μg/mL) in Acetate 4.5 immobilization buffer (10 mM sodium acetate pH 4.5, GE Healthcare) were covalently immobilized on a CM5 chip via amine coupling to a final response of 500 RU in 25 mM Hepes pH 7.3 at 25°C. Peptide samples were prepared in degassed, filtered HBS-P+ (diluted from 10x concentrated stock solution containing 0.1 M HEPES, 1.5 M NaCl and 0.5% v/v Surfactant P20, GE Healthcare) buffer. Single cycle kinetic experiments were carried out using five injections (30 μL/min) of increasing concentration of peptides (0.1–10 μM or 1–100 μM) passed over the sensor chip for 150 s association followed by a 420 s dissociation. Following buffer and reference subtraction, kinetic constants and binding affinities were determined utilizing the Biacore T200 evaluation software (GE Healthcare).
4.2 Cell lines and maintenance
HT-1080, PANC1, HeLa, and HCT116 cell lines were purchased from American Type Culture Collection (ATCC), PEA1 were from Sigma-Aldrich. All cell lines were adherent cells and their source is as follows: HT-1080, fibrosarcoma; PANC-1, pancreatic cancer; HeLa, cervical cancer; PEA1, ovarian cancer; HCT116, colon cancer. HT-1080, PANC-1, and HeLa cells were maintained in Dulbecco’s minimum essential medium (DMEM) with Glutamax-I, 4.5 g/L D-glucose, 10% fetal bovine serum (FBS; Gemini Bio-Products), 5% penicillin/streptomycin, and without sodium pyruvate. PEA1 cell lines were maintained in RPMI 1640 with L-glutamine media with 10% FBS, and 5% penicillin/streptomycin. Finally, HCT116 cell lines were maintained in McCoy’s 5A media with 10% FBS and 5% penicillin/streptomycin. All cell lines were maintained at 37°C in a 5.0% CO2 atmosphere.
4.3 Preparation of purified proteins
Plasmids for expression of Petit-LUBAC, GST-HOIP-UBA, and His6-HOIL-UBL were a kind donation of Dr. Kazuhiro Iwai, Department of Molecular and Cellular Physiology, Graduate School of Medicine, Kyoto University. Plasmids were cloned on Rosetta (DE3) (EMD Millipore Corporation), expressed and purified as previously described.6,17,25
4.4 In vitro autoubiquitylation assays
All reagents used for In vitro ubiquitylation assays were from Enzo Life Sciences unless specified. All reactions were performed as previously described17 with slight modifications. Briefly, 10 μg/mL E1 (BML-UW9410-0050), 20 μg/mL Ubc-H5c E2 (BML-UW9070-0100), 30 μg/mL Petit-LUBAC E3, and 50 μg/mL ubiquitin (BML-UW8795-0005) were incubated in 1X ubiquitylation buffer (BML-KW9885-0001) supplemented with an activating solution containing 5 μM ATP, and 5 μM MgCl2 (BML-EW9805-0100) and with or without 40 μM of specified peptides. Reactions were incubated at 37 °C for 4h, and Western blots for anti Linear-ubiquitin (MABS199) (EMD Millipore corporation) or ubiquitin (U5379-1VL) (Sigma-Aldrich Co. LCC) were performed.
4.5 High Resolution Clear Native Electrophoresis (hrCNE)
hr-CNE-2 were modified from its original version to visualize peptide-protein interactions.26 Briefly, 5% acrylamide resolving gels were prepared by mixing of 3.1 mL of a 3X Native Binding Buffer (NBB) (150 mM bis-Tris (Sigma-Aldrich, Co.); pH=7.0), 4.2 mL of 30% acrylamide (Bio-Rad Laboratories), 12.0 mL of 50% glycerol, 100 μL ammonium persulfate (APS) (Sigma-Aldrich Co.) (10%), and 10 μL of TEMED (Bio-Rad Laboratories). After polymerization, the stacking gel was prepared and poured over the resolving gel. 3.2% acrylamide stacking gel was prepared by mixing 4.0 mL of 3X NBB, 1.3 mL of 30% acrylamide, 6.7 mL of water, 150 μL of 10% APS, and 15 μL of TEMED. Gels were placed on a Mini-PROTEAN cell (Bio-Rad Laboratories), upper chamber was filled with cathode buffer (50 mM Tricine, 7.5 mM Imidazole, 0.05% Deoxycholic acid, 0.05 Triton X-100, pH=7.0), and the tank was filled with anode buffer (50 mM Bis-Tris, pH=7.0). Gels inside the electrophoresis apparatus were left overnight at 4 °C.
Binding reactions were performed by the mixing 1X ubiquitylation buffer, 50 μg/mL of each recombinant protein, and 2.5 μM of each FITC labeled peptides, and if the case 20 μM of unlabeled peptide. Reactions were incubated for 1h at 37°C, and then added with orange loading dye (LI-COR Inc.), loaded on an hr-CNE gels, and resolved at 80 mV during 4–5h at 4 °C. Fluorescent peptide bands were visualized using a Typhoon FLA 7000 imager (GE Healthcare Life Sciences).
4.6 Cell Viability Assay
5×104 cells/well were seeded in an opaque white 96-well plate (Costar), then treated for 72 hours with stapled peptides at concentrations of 3, 6, 12, 25, and 50 μM; or DMSO as vehicle control. After treatment, viability was measured by CellTiter-Glo® (Promega, WI, USA). Luminescence was recorded on an Infinite M1000 Pro plate reader (Tecan Trading AG). Cell viability was normalized to that of DMSO as 100%.
4.7 Cell Cytotoxicity & Apoptosis Assay
1.5×104 cells/well were seeded in an opaque white 96-well plate (Costar). Cells were treated as for cell viability, except that for this experiment treatment with peptides was for 24 hours. Then cytotoxicity and caspase-3/7 activity were measured by ApoTox-Glo™ Triplex Assay Kit (Promega WI, USA). Fluorescence at 485EX/520EM, as well as Luminescence were recorded on an Infinite M1000 Pro plate reader (Tecan Trading AG). Both cytotoxicity and caspase-3/7 activity were normalized against DMSO as 100%.
4.8 Western Blots
1.0×106 cells/well were seeded on a 6-well plate (Corning). Cells were treated with 50 μM of peptide for 24 hours. Cells were harvested and lysed with IP lysis buffer (Thermo Scientific) supplemented with Halt protease and phosphatase inhibitor (Thermo Scientific). Protein concentration was quantified by BCA (Thermo Scientific). Equivalent volumes to 30 μg protein of each sample were loaded in NOVEX 10% TBE gels (Invitrogen). After electrophoresis proteins were transferred to PVDF membranes (Millipore). Membranes were blocked and blotted with the specific p-IκBα (sc-371) (Santa Cruz Biotech), p-IkBα (5A5), cyclin D1 (92G2), cleaved caspasee3 (5A1E) (Cell Signaling Technology), GST (10000-0-AP) (Proteintech), HOIL-1L (0AAB15606) (Aviva Systems Biology), HOIP (16289-1-AP) (Proteintech) and GAPDH (631402) (Biolegend) primary antibodies. Proteins were visualized using IRDye rabbit (926-32213) or mouse (926-68051) secondary antibodies in a Li-Cor Odyssey 9120 Imaging System (LI-COR inc.).
4.9 Peptide Cell Permeability
Cell permeability was addressed using FITC labeled peptides. Briefly, 3×105 cells were treated with 20 μM of the WT (1), HOIP-N (2) or C-Helix3 (13) peptide on complete media for 2 hours. Then, cells were fixed with a mix of 30% Formaldehyde-10% Methanol. Nuclei Was stained with Hoechst 33342. Cells were observed and microphotographed under an LSM 710 confocal microscope (Zeiss).
4.10 TNF-α stimulation, NF-κB p65 DNA binding, and IL-8 ELISAs
1×106 cells of HeLa, and PANC1 cell were seeded on a 12-well plate and treated for 24 h with C-helix3 (13) at 50 μM in a final volume of 500 μL. After peptide treatment, cell media was exchanged by new one containing 20ng/mL of TNF-α. After 0, 10, 60, and 240 min of TNF-α stimulation supernatants were aspirated and stored at −80°C until analyzed. Cells were harvested and lysed with IP lysis buffer supplemented with Halt Protease and Phosphatase inhibitor (Thermo Scientific). Protein concentration was measured by BCA (Thermo Scientific).
For NF-κB DNA binding ELISA. Each sample was diluted in lysis buffer to a final concentration of 2 μg/μL of whole-cell extract. 20 μL/well of each diluted lysate was used to determine NF-κB p65 activity, measured using a TransAM NF-κB p65 Transcription Factor ELISA Kit (Active Motif), and following manufacturer’s protocol.
To determine secreted IL-8. 200 μL of supernatants from cell experiments after TNF-α stimulation was used for the quantitative measure IL-8 through sandwich ELISA. ELISA microplates were prepared and IL-8 levels quantified using a HUMAN IL-8 (CXCL8) Standard TMB ELISA Development Kit (PEPROTECH) according to the manufacturer’s protocol.
4.11 Quantification and Statistical Analysis
Unless otherwise specified, data are represented as the means of three, independent experiments (n=3) with error bars reflecting the standard error of the mean (SEM). P-values were determined with a one or two-way ANOVA test compared to vehicle treatment (DMSO): (* = ≤ 0.05). All statistical analyses were performed with GraphPad Prism 7.01.
Supplementary Material
Acknowledgments
We are grateful to Professor Kazuhiro Iwai (Kyoto University) for providing us with the Petit-LUBAC and related protein constructs and for enlightening discussions about LUBAC in general. We also wish to thank the staff of the Biophysics Resource Facility in the Structural Biophysics Laboratory (NCI-Frederick) for assistance with instrumentation, and the Protein Complex Characterization Section of the Cancer Research Technology Program (CCR/NCI/NIH) for assistance with our binding studies.
Funding
This research was supported by a CONACYT fellowship [grant 215132] to F.A.A. and the Intramural Research Program of the NIH and the National Cancer Institute.
Abbreviations
- LUBAC
linear ubiquitin chain assembly complex
- UBA
ubiquitin-associated domain
- UBL
ubiquitin-like domain
- RBR
ring-between-ring domain
References
- 1.Dove KK, Klevit RE. RING-Between-RING E3s ligases: Emerging themes amid the variations. J Mol Bio. 2017;429(22):3363–3375. doi: 10.1016/j.jmb.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rittinger K, Ikeda F. Linear ubiquitin chains: enzymes, mechanisms and biology. Open Biol. 2017 doi: 10.1098/rsob.170026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirisako T, Kamei K, Murata S, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25(20):4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ikeda F, Deribe YL, Skanland SS, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nat. 2011;471(7340):637–641. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tokunaga F, Sakata S-i, Saeki Y, et al. Involvement of linear polyubiquitylation of NEMO in NF-[kappa]B activation. Nat Cell Biol. 2009;11(2):123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 6.Yagi H, Ishimoto K, Hiromoto T, et al. A non-canonical UBA-UBL interaction forms the linear-ubiquitin-chain assembly complex. EMBO Rep. 2012;13(5):462–468. doi: 10.1038/embor.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jung J, Kim JM, Park B, et al. Newly identified tumor-associated role of human Sharpin. Mol Cell Biochem. 2010;340(1–2):161–167. doi: 10.1007/s11010-010-0413-x. [DOI] [PubMed] [Google Scholar]
- 8.Tomonaga M, Hashimoto N, Tokunaga F, et al. Activation of nuclear factor-kappa B by linear ubiquitin chain assembly complex contributes to lung metastasis of osteosarcoma cells. Int J Oncol. 2012;40(2):409–417. doi: 10.3892/ijo.2011.1209. [DOI] [PubMed] [Google Scholar]
- 9.MacKay C, Carroll E, Ibrahim AFM, et al. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res. 2014;74(8):2246–2257. doi: 10.1158/0008-5472.CAN-13-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tokunaga F. Linear ubiquitination-mediated NF-kappaB regulation and its related disorders. J Biochem. 2013;154(4):313–323. doi: 10.1093/jb/mvt079. [DOI] [PubMed] [Google Scholar]
- 11.Tokunaga F, Iwai K. LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes Infect. 2012;14(7):563–572. doi: 10.1016/j.micinf.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Gerlach B, Cordier SM, Schmukle AC, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nat. 2011;471(7340):591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 13.Iwai K, Tokunaga F. Linear polyubiquitination: a new regulator of NF-κB activation. EMBO Rep. 2009;10(7):706–713. doi: 10.1038/embor.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas TL, Emmerich CH, Gerlach B, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36(5):831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Chen ZD, Xu L, Tang KK, et al. NF-kappaB-dependent transcriptional upregulation of cyclin D1 exerts cytoprotection against hypoxic injury upon EGFR activation. Exp Cell Res. 2016;347(1):52–59. doi: 10.1016/j.yexcr.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 16.St Claire MC, Ragland DR, Bollinger L, Jahrling PB. Animal Models of Ebolavirus Infection. Comparative Medicine. 2017;67(3):253–262. [PMC free article] [PubMed] [Google Scholar]
- 17.Sakamoto H, Egashira S, Saito N, et al. Gliotoxin suppresses NF-kappaB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC) ACS Chem Biol. 2015;10(3):675–681. doi: 10.1021/cb500653y. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Schmitz R, Mitala J, et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov. 2014;4(4):480–493. doi: 10.1158/2159-8290.CD-13-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 20.Bernal F, Wade M, Godes M, et al. A Stapled p53 Helix Overcomes HDMX-Mediated Suppression of p53. Cancer Cell. 2010;18(5):411–422. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walensky LD, Bird GH. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J Med Chem. 2014;57(15):6275–6288. doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu J, Shi Y, Iwai K, Wu ZH. LUBAC regulates NF-kappaB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J. 2011;30(18):3741–3753. doi: 10.1038/emboj.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimizu Y, Taraborrelli L, Walczak H. Linear ubiquitination in immunity. Immunol Rev. 2015;266(1):190–207. doi: 10.1111/imr.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 25.Uekusa Y, Mimura S, Sasakawa H, et al. Backbone and side chain 1H, 13C, and 15N assignments of the ubiquitin-like domain of human HOIL-1L, an essential component of linear ubiquitin chain assembly complex. Biomol NMR Assign. 2012;6(2):177–180. doi: 10.1007/s12104-011-9350-1. [DOI] [PubMed] [Google Scholar]
- 26.Wittig I, Karas M, Schagger H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics. 2007;6(7):1215–1225. doi: 10.1074/mcp.M700076-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.