

Abstract
Generation of the second messenger molecule cAMP mediates a variety of cellular responses which are essential for critical cellular processes. In response to elevated cAMP levels, cAMP dependent protein kinase (PKA) phosphorylates serine and threonine residues on a wide variety of target substrates. In order to enhance the precision and directionality of these signaling events, PKA is localized to discrete locations within the cell by A-kinase anchoring proteins (AKAPs). The interaction between PKA and AKAPs is mediated via an amphipathic α-helix derived from AKAPs which binds to a stable hydrophobic groove formed in the dimerization/docking (D/D) domain of PKA-R in an isoform-specific fashion. Although numerous AKAP disruptors have previously been identified that can inhibit either RI- or RII-selective AKAPs, no AKAP disruptors have been identified that have isoform specificity for RIα versus RIβ or RIIα versus RIIβ. As a strategy to identify isoform-specific AKAP inhibitors, a library of chemically stapled protein-protein interaction (PPI) disruptors was developed based on the RII-selective AKAP disruptor, STAD-2. An alanine was substituted at each position in the sequence, and from this library it was possible to delineate the importance of longer aliphatic residues in the formation of a region which complements the hydrophobic cleft formed by the D/D domain. Interestingly, lysine residues that were added to both terminal ends of the peptide sequence to facilitate water solubility appear to contribute to isoform specificity for RIIα over RIIβ while having only weak interaction with RI. This work supports current hypotheses on the mechanisms of AKAP binding and highlights the significance of particular residue positions that aid in distinguishing between the RII isoforms and may provide insight into future design of isoform-selective AKAP disruptors.
Keywords: constrained peptide, PKA, AKAP, isoform specificity, AKAP inhibitor, stapled peptide
Graphical abstract
1. Introduction
Reversible protein phosphorylation mediated by the action of protein kinases is a universal strategy involved in almost every cellular function. Notably, approximately 30% of all human proteins are covalently modified with phosphoryl moieties.1 cAMP-dependent protein kinase (PKA) was amongst the first to be discovered and provides a framework for the entire protein kinase family.2 The inactive holoenzyme tetramer is composed of a regulatory (R) subunit dimer, binding the second messenger cAMP, and two catalytic (C) subunits.3 The synthesis of cAMP results in activation of signaling pathways that are instrumental to a wide-ranging variety of cellular processes. Thereby, PKA is a key effector that coordinates the response to this second messenger to induce signaling responses within the cell. PKA is regulated through multiple mechanisms including isoform diversity of the R-subunits and differing interactions with A Kinase Anchoring Proteins (AKAPs) allows it to play essential regulatory roles in cells including cell division, metabolism and apoptosis.3-5
The PKA R-subunit serves as the prime effector of cAMP and is expressed as four different isoforms (RIα, RIβ, RIIα, RIIβ) and directly interacts with the C-subunit of PKA.6 R-subunit expression varies by cell- and tissue-type7 and while the isoforms share many overlapping roles in cells, each can also perform unique, non-redundant functions.8 The R-subunits form homodimers via the dimerization/docking (D/D) domain,9, 10 but the structure of the tetrameric holoenzyme complex varies considerably in an R-subunit isoform-specific manner.8 A hydrophobic groove is formed in the D/D domain of the R-subunit, and this groove serves as a docking site for AKAPs where an amphipathic α-helix derived from an AKAP binds to R-subunits in an isoform-specific fashion.9-11
AKAPs bind a homodimer of R-subunits of PKA (R) to localize PKA and form small signaling complexes in cells (Figure 1a).12 Currently, over 40 AKAPs have been identified and all cell types express approximately 10-15 different AKAPs with varying expression levels.13, 14 The relationship between expression and interaction of these proteins provides context-specific signaling effects. This family of scaffold proteins functions to enhance the precision and directionality of signaling events by assembling complexes of signaling effectors to precisely control where and when PKA is activated. Remarkably, almost all AKAPs demonstrate specificity towards the PKA-RII isoform,14, 15 although there are several dual-specific AKAPs that bind both the RI and RII isoforms.16, 17 More recently, some AKAPs were identified that were highly specific for only one of the four PKA isoforms such as neurochondrin which is highly selective for RIIα.18
Figure 1.

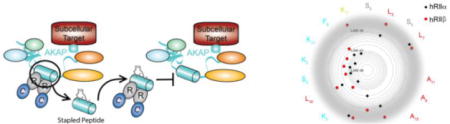
Targeted disruption of AKAP signaling complexes with constrained peptides. a) AKAP signaling complexes can scaffold a variety of different proteins in addition to PKA including adenylyl cyclases (ACs), phosphodiesterases (PDEs), substrates and protein phosphatases (PPases). The tetrameric holoenzyme complex interacts with an AKAP-derived helix through its regulatory domains. b) A hydrocarbon stapled peptide, STAD-2, was previously shown to selectively inhibit RII:AKAP complexes and was used as a template for library design. c) The constrained peptide is designed to inhibit the binding groove formed in the docking/dimerization (D/D) domain of the R-subunit of PKA (gray) that serves as a docking site for AKAPs (teal). In the STAD-2 sequence above, amino acids facing the D/D domain are depicted in red and solvent exposed amino acids are shown in teal.
In order to investigate isoform specificity in PKA:AKAP interactions, many peptides have been developed that were designed from the AKAP-derived α-helix that interacts with the D/D domain of the R-subunits (Figure 1b).19 While numerous peptides were identified or designed to have selectivity for RI over RII or vice versa (reviewed in19), it has proven a challenge to identify sequences that are specific for the R-subunit subtypes (α versus β). In order to determine the contribution of specific residues to RII subunit specificity, a library of constrained protein-protein interaction disruptors was developed based on the RII-selective disruptor of AKAP binding termed STAD-2 which was derived from the AKAP220 docking sequence (Figure 1c, Supplementary Figure S6).20 To study the contribution of each peptide residue on R-subunit binding, STAD-2 was used as a template where alanine substitutions were introduced at each position while leaving the hydrocarbon staple placement unaffected. Each analog was then assessed for its ability to bind each of the R-subunit isoforms of PKA. Based upon these findings, several positions were identified that were critical for both RIIα and RIIβ while other positions caused a significant loss of affinity for RIIβ while having little to no effect on RIIα interactions. Interestingly, non-native lysine residues that were added to both the N- and C-terminals of the sequence to facilitate water solubility appear to contribute to isoform specificity for RIIβ over RIIα. To our knowledge, this is the first identification of constrained peptide sequences that can selectively distinguish between the α and β subtype isoforms for RII. This study contributes to our understanding of AKAP engagement to PKA-R and may facilitate the design of future isoform specific disruptors.
2. Results and Discussion
While AKAP disruptors are extremely useful tools to study PKA signaling in cells, it would be highly desirable to identify disruptors with greater specificity for each of the individual R-subunit isoforms. Toward this goal, we used an AKAP disruptor, STAD-2,20 as a template to explore whether analogs could be designed to achieve isoform-specific disruption of AKAP complexes. STAD-2 is a cell-permeable, constrained peptide scaffold that exhibits high-affinity towards both RII isoforms (RIIα and RIIβ).20 Previously, we showed that STAD-2 binds RII and selectively disrupts the interaction between RII and AKAPs while having weak affinity interactions with RI. In this study, we developed a library of STAD-2 analogs where an alanine residue was introduced in each position of the amphipathic helix to interrogate the effects on binding affinity and isoform specificity (Figure 2). The hydrocarbon peptide staple, originally optimized for this scaffold,20 was kept in the same position and remained unmodified for each analog. Ring-closing metathesis was performed on-resin prior to N-terminal addition of a PEG3 moiety and labeling.
Figure 2.
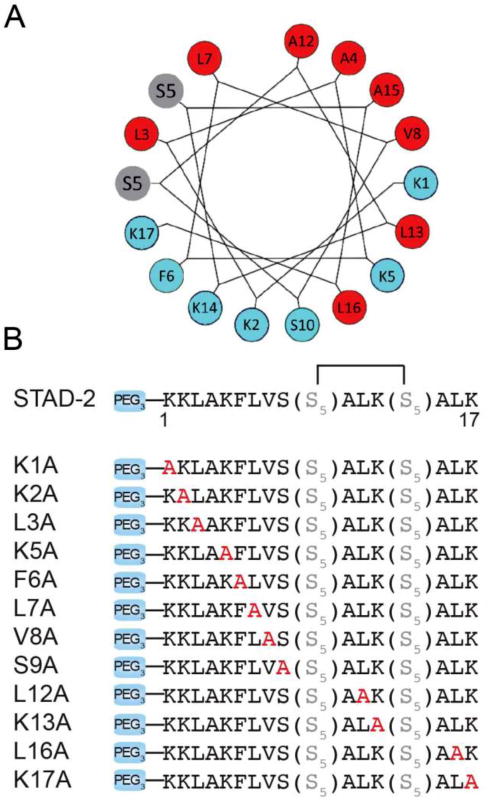
Design of STAD-2 analogs. a) A helical wheel representation of STAD-2 demonstrates an amphipathic helix. b) In order to determine which residues may be critical for distinguishing between RIIα and RIIβ, STAD-2 was used as a template and alanine substitutions were introduced at each position (shown in red). The staple position was unmodified for all analogs.
The affinities of each analog toward all R-subunit isoforms was subsequently investigated by to determine the significance of each position on PKA-R:AKAP interactions using STAD-2 as a control. Binding affinities were measured for STAD-2 and for each analog peptide employing direct fluorescence polarization (FP) assays20 as a fast and reliable screening method. Recombinant human R-subunits were purified to homogeneity as described in the Methods section (Supplementary Figure S4). At least two independent protein preparations were used for each assay and each assay was performed in duplicate. KD values were determined for each peptide using full-length constructs of human RIIα and RIIβ subunits (Figure 3, Table 1 and Supplementary Figures S1 and S2). The R-subunits were assayed over a concentration range of 5 μM to 200 pM. When using protein concentrations above 5 μM, considerable viscosity effects occurred and thereby hindered accurate KD value measurements for weak binding peptides (Supplementary Figure S5). Fluorescently labeled (5(6)-FAM) STAD-2 or each analog were plated at a final concentration of 0.5 nM. Although STAD-2 demonstrated a weak micromolar range affinity for the RI isoforms, we wanted to ensure that all variants also remained RII-selective. Therefore, STAD-2 and each analog were screened for binding by FP using a single high concentration (5 μM) of human full-length RIα and RIβ subunits (Supplementary Figure 3) and 20 nM of peptide. None of the analogs demonstrated an increase in affinity for the RI isoforms as compared to STAD-2 and thus remained RII-selective compounds.
Figure 3.
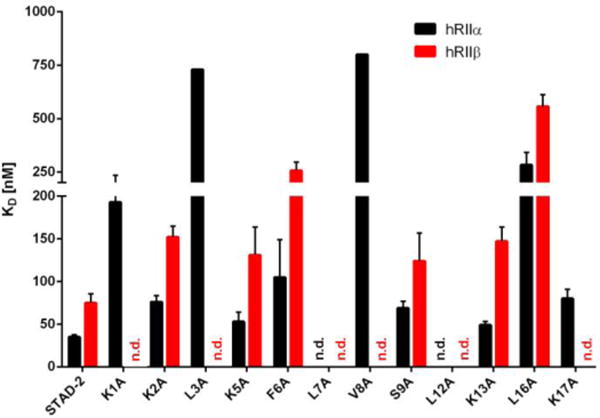
Bar diagram representing KD values ± SD from fluorescence polarization binding assays (hRIIα is shown in black and hRIIβ is shown in red). Substitutions at position K1 and K17 are tolerated by hRIIα but not by the hRIIβ isoform. (n.d.: no KD value determined; see Table 1 legend.)
Table 1.
Summary of KD values measured in fluorescence polarization assays using full-length human RIIα and RIIβ. KD values were determined from two independent protein preparations in duplicate. The KD values are presented as a mean of each preparation ± SD. While substitutions at the core hydrophobic residues (L3, L7, V8, L12) caused a loss off affinity for both RII isoforms, substitutions at positions K1 and K17 also showed a significant loss in affinity for RIIβ while having less effects on affinity for RIIα. (n.d.: no KD value determined due to viscosity effects at high protein concentrations.)
| KD ± SD [nM] | ||
|---|---|---|
| hRIIα | hRIIβ | |
| STAD-2 | 35 ± 3 | 75 ± 11 |
|
| ||
| K1A | 193 ± 41 | n.d. |
|
| ||
| K2A | 76 ± 8 | 152 ± 13 |
|
| ||
| L3A | > 730 | n.d. |
|
| ||
| K5A | 53 ± 11 | 131 ± 33 |
|
| ||
| F6A | 105 ± 44 | 257 ± 40 |
|
| ||
| L7A | n.d. | n.d. |
|
| ||
| V8A | > 800 | n.d. |
|
| ||
| S9A | 69 ± 8 | 124 ± 33 |
|
| ||
| L12A | n.d. | n.d. |
|
| ||
| K13A | 49 ± 4 | 147 ± 17 |
|
| ||
| L16A | 283 ± 59 | 557 ± 55 |
|
| ||
| K17A | 80 ± 11 | n.d. |
Table 1 summarizes the KD values of STAD-2 and STAD-2 analogs using human RIIα and RIIβ. As expected, peptide analogs bearing alanine substitutions in the hydrophobic register that is anticipated to make key contacts at the hydrophobic protein-protein interface between the AKAP sequence and the D/D domain of RII (L3A, L7A, V8A, L12A) caused a significant decrease in the binding affinity towards both RII isoforms. In some cases such as L3A and V8A, alanine substitutions caused a loss of binding affinity for RIIβ beyond 1 μM while still weakly binding RIIα in the high nM range (>700 nM). However, in other cases such as L7A and L12A, the loss of affinity was comparable for both RII isoforms where binding affinity was greater than 1 μM. This result underlines the importance of the hydrophobic nature of the AKAP:R-subunit interaction interface where critical contact points are necessary for AKAP engagement. Another analog was also identified that caused intermediate effects on binding: L16A. In this case, L16A was found to bind both RII isoforms with a considerably weaker affinity as compared to STAD-2 (283 nM versus 35 nM for RIIα and 557 nM versus 75 nM and RIIβ). This hydrophobic position at the C-terminal end of the amphipathic helix is situated more on the far side of the D/D domain and appears to serve as an additional interaction point for binding. This finding correlates with recent results that suggest an extended conserved hydrophobic core may be critical for the AKAP:RII interaction interface.21 On the other hand, the hydrophilic face of the amphipathic helix which is expected to be solvent exposed, including positions K2, K5, F6, S9 and K13 results in minimal perturbation of KD values for both RII isoforms, thereby highlighting that these positions are not crucial for AKAP binding on the RII surface as expected.
Changing the two N-terminal basic amino acids of the STAD-2 peptide sequence caused a profound loss of affinity for RIIβ (Figure 4, positions K1 and K17). While the alanine substitutions at these positions were tolerated by RIIα, binding affinities to RIIβ strongly affected, however, due to experimental limitations an accurate KD value could not be obtained. Based upon these findings, it appears that these basic, hydrophilic side chains may serve as critical contact points for binding both RII isoforms. Further, these positions may contribute to isoform specificity for RIIα versus RIIβ where they may make critical contact interactions that are specific for RIIβ. In this line the K1A and K17A analogs may be a starting point for developing RII subtype-specific AKAP disruptor peptides.
Figure 4.
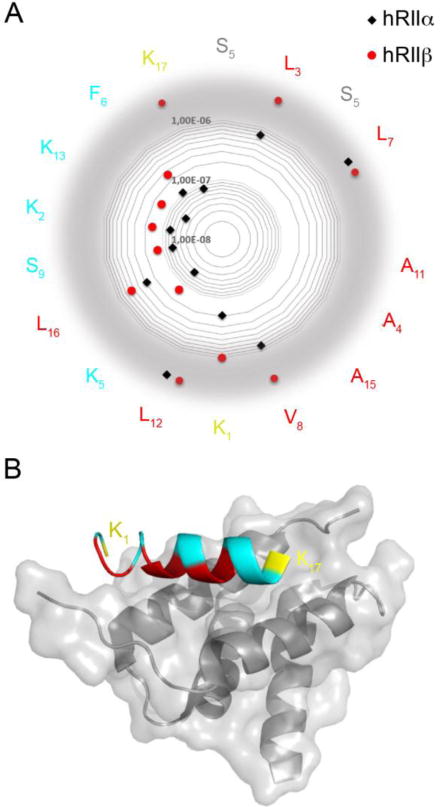
a) Representation of KD values on a radar plot of the STAD-2 peptide sequence. KD values for hRIIα are shown in black and hRIIβ are shown in red. The amino acids facing the D/D domain are depicted in red, the solvent exposed in teal. The hydrocarbon staple is illustrated in grey and the flanking lysines are yellow. While substitutions at many positions were largely inconsequential and indicate that they are not major contributors to the AKAP:PKA binding interface (K2, F6, S9, K13), substitutions at position K1 and K17 were better tolerated by hRIIα than by hRIIβ, indicating that these terminal residues may perform key contacts to confer isoform selectivity between RIIα and RIIβ. Values out of the scale are displayed on a gray border. b) Structural representation highlighting the terminal positions 1 and 17 (yellow) of an AKAP peptide bound to the D/D domain of RIIα. The critical hydrophobic residues that are important for binding to both RIIα and RIIβ are shown in red. The structure was rendered using PDB 2hwn.
3. Conclusions
In a systematic attempt, isoform-selectivity of all human PKA R-subunits were tested with a previously developed disruptor for AKAPs, STAD 2, employing an alanine scan. As expected, alanine substitutions in the hydrophobic register, providing key contacts at the hydrophobic interface between the AKAP and the D/D domain, showed the strongest decrease in binding affinity towards both RII isoforms. The type I isoforms were not targeted with any of the variants tested. Interestingly, Lys residues positioned at the N- and C-terminal ends of the conserved AKAP interaction surface caused a considerable loss in affinity for RIIβ. This finding may lead to the development of constrained peptide sequences that can distinguish between the RIIα and RIIβ isoforms.
Interestingly, although STAD-2 was designed based on the AKAP docking sequence derived from AKAP220 (Supplementary Figure S6),20 the lysine residues at positions 1 and 2 of STAD-2 are not part of the native sequence AKAP220 sequence which bears and S and G at the corresponding positions. While the lysine residues were originally introduced into STAD-2 to improve both cell permeability and solubility of the inhibitor peptide, it appears that at least for position 1, lysine is a determinant for RII isoform specificity. Several AKAPs contain charged polar residues in the N-terminus such as AKAP18 which contains a glutamate at the first two N-terminal positions that correlate with STAD-2.21 The crystal structure of AKAP18 and RIIα reveal that one of these glutamate residues (Glu 74) is engaged in salt bridges with Arg 22 of the D/D domain for RIIα. Indeed, it will be interesting to test future derivatives of STAD-2 such as those lacking amino acids in positions 1 and 17 or longer derivatives that contain additional or differential N- and C-terminal residues such as glutamate to determine whether isoform specificity can be further refined.
4. Methods
4.1 General Information
All standard N-α-Fmoc amino acids and Rink Amide MHBA resin were purchased from Novabiochem. Unless otherwise noted all other reagents and solvents were purchased from Fisher Scientific. HPLC-grade methanol, acetonitrile and trifluoroacetic acid were used for solutions involving peptide purification or analysis.
4.2 Peptide Synthesis
Peptide synthesis was performed on Rink Amide MHBA resin using 9-fluorenylmethoxycarbonyl (Fmoc) solid phase synthesis. Each deprotection step was performed using 25% (v/v) piperidine in 1-methyl-2-pyrrolidinone (NMP) for 25 minutes with agitation by house air. Each standard N-α-Fmoc amino acids was coupled using 10 equivalents (0.25 M final concentration in NMP) followed by the addition of 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU, 0.23 M final concentration in NMP) and 8% (v/v) N,N-diisopropyl ethylamine (DIEA) for 45 minutes with agitation. Each (S)-N-Fmoc-2-(4′-pentenyl)alanine (S5; Okeanos Tech (Beijing, China)) or 11-amino-3,6,9-trioxaundecanoic acid (PEG3, ChemPep) was coupled using 4 equivalents and half the volume of used with standard residue couplings of HCTU and DIEA for 1 hour 30 minutes, with the addition of another half volume of DIEA after the first 45 minutes, with agitation. Ring closing metathesis was performed prior to addition of the N-terminal PEG3 linker using 0.4 equivalents bis(tricyclohexylphosphine) benzylidene ruthenium(IV) dichloride (Grubbs Catalyst, 1st generation; Sigma). The reaction was performed two times for one hour each at room temperature with agitation. Addition of a N-terminal fluorescein label was performed using 2 equiv 5(6)-carboxy-fluorescein (FITC; Acros) in dimethylformamide (DMF) with the addition of 0.046M HCTU and 2% (v/v) DIEA overnight at room temperature with agitation. Completed peptides were cleaved from resin using trifluoroacetic acid/water/triisopropylsilane (95:2.5:2.5; Sigma) for 4 hours at room temperature, mixed by inversion. Cleaved peptides were then precipitated in 4°C methyl-tert-butyl ether and following centrifugation the supernatant was discarded and the precipitate was lyophilized. Peptides were purified via reverse phase High Performance Liquid Chromatography (HPLC) and verified using electro spray injection mass spectrometry (ESI-MS; results summarized in Supplementary Table 1). Peptides were quantified via the N-terminal FITC label by measuring absorbance at 495 nm using a Synergy2 microplate reader (Bio-Tek).
4.3 Expression and Purification of the Recombinant PKA-R Subunits
Plasmids for expression of human PKA regulatory subunits RIα and RIIα were kind gifts from Prof. Kjetil Tasken (U. Oslo). Plasmids for RIβ and RIIβ were previously published.22, 23 Recombinant human PKA regulatory subunits hRIα, hRIβ, hRIIα and hRIIβ were expressed in E. coli BL21DE3 RIL. The hRIα, hRIβ and hRIIβ isoforms were purified by an ammonium sulfate precipitation24 and subsequent anion exchange chromatography.25 This purification method offers a high protein yield in combination with high purity (>95%). The hRIIα isoform yielded the best results using affinity chromatography with Sp-8-AEA-cAMPS agarose as described previously.26 Cell lysis was performed as previously described in26 using lysis buffer containing 20 mM MOPS, 150 mM NaCl and 5 mM β-mercaptoethanol at pH 7 for hRIα and hRIβ. The hRIIα isoform was lysed in 25 mM MES, 100 mM NaCl, 5 mM EDTA, 5 mM EGTA and 5 mM β-mercaptoethanol at pH 6.5. For the RIIβ isoform, the lysis buffer contained 20 mM MES, 100 mM NaCl, 2 mM EDTA, 2 mM EGTA and 2 mM β-mercaptoethanol at pH 6.5. After centrifugation of the cell debris, a saturated ammonium sulfate solution was slowly added to the supernatant until a concentration of 40 % (hRIα) or 50 % (hRIβ, hRIIβ) was reached. After 1 h, the precipitated protein was recovered by centrifugation at 10,000 × g for 10 min, re-dissolved in lysis buffer, and dialyzed against running buffer (25 mM HEPES, pH 8 and 25mM NaCl for hRIβ or 50 mM NaCl for hRIα and hRIIβ) prior to anion exchange chromatography (ResourceQ, GE Healthcare). The purified R-subunits were eluted using a linear gradient of 0-20 % (hRIα and hRIβ), or 0-30 % (hRIIβ) running buffer that additionally contained 1 M NaCl. SDS-PAGE was used to monitor protein expression and purity (Supplementary Figure S5).
4.4 Fluorescence Polarization (FP)
Binding affinity of STAD-2 and analog peptides were measured against full-length hRIIα and hRIIβ using FP in a direct assay format.20 Increasing concentrations (200 pM to 5 μM final concentrations) of both PKA R-subunit isoforms were mixed with 0.5 nM of fluorescently labeled STAD-2 or STAD-2 analogs in buffer containing 20 mM MOPS pH 7, 150 mM NaCl and 0.005% (v/v) CHAPS.
Due to the low affinity of the full-length hRIα and hRIβ to STAD-220 and the STAD-2 peptides, single concentration FP screenings were performed. For this, 5 μM of the respective R-isoforms were mixed with 20 nM fluorescently labeled peptide. All data were obtained in duplicates using a CLARIOstar (BMG LABTECH) plate reader at room temperature and a data acquisition of 0.1 s at Ex 482 nm/Em 520 nm in a 384 well microtiter plate (BRANDplate, BRAND GMBH CO+KG). Equilibrium dissociation constants (KD) for hRIIα and hRIIβ were calculated with a nonlinear regression dose-response curve using GraphPad Prism 6. At least two independent protein preparations were measured two times and the KD-values are presented ± SD.
As a control for viscosity effects, the FP signal of 2 nM STAD-2 peptides was measured in the presence of increasing concentrations of bovine serum albumin (BSA, 1.4 μM – 25 μM).
Supplementary Material
Acknowledgments
We would like to thank M.J. Knape for helpful discussions and M. Hansch for technical support. This research was generously funded by the National Institutes of Health (CA154600 and CA188439 to EJK). This work was supported by the Deutsche Forschungsgemeinschaft (He1818/10) and the funding line Future (PhosMOrg) to FWH. EM was supported by Kassel Graduate School “Clocks”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
Supplementary figures and methods can be found in the online version of this article.
References
- 1.Cohen P. Eur J Biochem. 2001;268:5001. doi: 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- 2.Taylor SS, Buechler JA, Yonemoto W. Annu Rev Biochem. 1990;59:971. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 3.Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. Biochim Biophys Acta. 2004;1697:259. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 4.Turnham RE, Scott JD. Gene. 2016;577:101. doi: 10.1016/j.gene.2015.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch EJ, Jones BW, Scott JD. Mol Interv. 2010;10:86. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott JD. Pharmacol Ther. 1991;50:123. doi: 10.1016/0163-7258(91)90075-w. [DOI] [PubMed] [Google Scholar]
- 7.Skalhegg BS, Tasken K. Front Biosci. 2000;5:D678. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- 8.Taylor SS, Ilouz R, Zhang P, Kornev AP. Nat Rev Mol Cell Biol. 2012;13:646. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure. 2010;18:155. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. Mol Cell. 2006;24:397. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newlon MG, Roy M, Hausken ZE, Scott JD, Jennings PA. J Biol Chem. 1997;272:23637. doi: 10.1074/jbc.272.38.23637. [DOI] [PubMed] [Google Scholar]
- 12.Langeberg LK, Scott JD. Nat Rev Mol Cell Biol. 2015;16:232. doi: 10.1038/nrm3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lester LB, Coghlan VM, Nauert B, Scott JD. J Biol Chem. 1996;271:9460. doi: 10.1074/jbc.271.16.9460. [DOI] [PubMed] [Google Scholar]
- 14.Esseltine JL, Scott JD. Trends Pharmacol Sci. 2013;34:648. doi: 10.1016/j.tips.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E. Int Rev Cell Mol Biol. 2010;283:235. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- 16.Huang LJ, Wang L, Ma Y, Durick K, Perkins G, Deerinck TJ, Ellisman MH, Taylor SS. J Cell Biol. 1999;145:951. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K. J Mol Biol. 2000;298:329. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- 18.Hermann JS, Skroblin P, Bertinetti D, Hanold LE, von der Heide EK, Wagener EM, Zenn HM, Klussmann E, Kennedy EJ, Herberg FW. Biochim Biophys Acta. 2015;1854:1667. doi: 10.1016/j.bbapap.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy EJ, Scott JD. Methods Mol Biol. 2015;1294:137. doi: 10.1007/978-1-4939-2537-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GC, Schendowich LP, Sukhu A, Spelts RC, Zhang J, Herberg FW, Kennedy EJ. ACS Chem Biol. 2014;9:635. doi: 10.1021/cb400900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gotz F, Roske Y, Schulz MS, Autenrieth K, Bertinetti D, Faelber K, Zuhlke K, Kreuchwig A, Kennedy EJ, Krause G, Daumke O, Herberg FW, Heinemann U, Klussmann E. Biochem J. 2016;473:1881. doi: 10.1042/BCJ20160242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solberg R, Tasken K, Keiserud A, Jahnsen T. Biochem Biophys Res Commun. 1991;176:166. doi: 10.1016/0006-291x(91)90904-l. [DOI] [PubMed] [Google Scholar]
- 23.Solberg R, Sandberg M, Natarajan V, Torjesen PA, Hansson V, Jahnsen T, Tasken K. Endocrinology. 1997;138:169. doi: 10.1210/endo.138.1.4864. [DOI] [PubMed] [Google Scholar]
- 24.Saraswat LD, Filutowicz M, Taylor SS. J Biol Chem. 1986;261:11091. [PubMed] [Google Scholar]
- 25.Buechler YJ, Taylor SS. J Biol Chem. 1991;266:3491. [PubMed] [Google Scholar]
- 26.Bertinetti D, Schweinsberg S, Hanke SE, Schwede F, Bertinetti O, Drewianka S, Genieser HG, Herberg FW. BMC Chem Biol. 2009;9:3. doi: 10.1186/1472-6769-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.