

Abstract
Background
MYC overexpression is associated with poor prognosis in breast tumors (BCa). The objective of this study was to determine the prevalence of MYC amplification and associated markers in BCa tumors from African American (AA) women and determine the associations between MYC amplification and clinico-pathological characteristics.
Methods
We analyzed 70 cases of well characterized archival breast ductal carcinoma specimens from AA women for MYC oncogene amplification. Utilizing immune histochemical analysis estrogen receptor (ER), progesterone receptor (PR), and (HER2/neu), were assessed. Cases were Luminal A (ER or PR+, Ki-67 < 14%), Luminal B (ER or PR+, Ki-67 = > 14% or ER or PR+ HER2+), HER2 (ER-, PR-, HER2+), and Triple Negative (ER-, PR-, HER2-) with basal-like phenotype. The relationship between MYC amplification and prognostic clinico-pathological characteristics was determined using chi square and logistic regression modeling.
Results
Sixty-five (97%) of the tumors showed MYC gene amplification (MYC: CEP8 > 1). Statistically significant associations were found between MYC amplification and HER2-amplified BCa, and Luminal B subtypes of BCa (p < 0.0001), stage (p < 0.001), metastasis (p < 0.001), and positive lymph node status (p = 0.039). MYC amplification was associated with HER2 status (p = 0.01) and tumor size (p = 0.01). High MYC amplification was seen in grade III carcinomas (MYC: CEP8 = 2.42), pre-menopausal women (MYC: CEP8 = 2.49), PR-negative status (MYC: CEP8 = 2.42), and ER-positive status (MYC: CEP8 = 2.4).
Conclusions
HER2 positive BCas in AA women are likely to exhibit MYC amplification. High amplification ratios suggest that MYC drives HER2 amplification, especially in HER2 positive, Luminal B, and subtypes of BCa.
Keywords: MYC, FISH, Gene amplification, Breast cancer subtypes
Background
A significant racial disparity exists in the presentation and outcome of breast cancer (BCa) between African American (AA) women and non-Hispanic white women in the United States. Despite the lower incidence of BCa among AAs (124.3 vs 128.1 per 100,000), the death rate is higher in non-Hispanic AA women (31.0 vs 21.9 per 100,000) [1]. Many Biological and non-biological factors are thought to contribute to these disparities, including access to health care, socioeconomic factors, cultural issues, hormones, reproductive influences, tumor characteristics, growth factors, cell cycle proteins [2], tumor suppressor genes, and chromosomal abnormalities [3, 4].
BCa is a heterogeneous disease consisting of different genetic, cellular, and molecular subtypes, distinct biological entities with unique clinical characteristics [5, 6]. On the basis of intrinsic gene-based signatures using transcriptional profiling, BCa has generally been subdivided into 4 major subtypes: luminal, HER2-enriched, basal-like, and normal breast tumors. Many BCa with basal-like phenotype tumors are ER negative, PR negative, and HER2 negative or triple negative (TNBC) [5.6]. TNBC account for 10 to 20% of all BCa cases and are more prevalent among AA, premenopausal women and women with the BRCA1 mutation. TNBCs tend to be aggressive tumors with poor prognosis in part because no effective targeted therapies have been identified for this BCa subtype [7].
MYC, a multifunctional oncogene located on human chromosome 8q24.21, has been shown to be amplified and overexpressed in many types of human cancers, including ovarian cancer, esophageal cancer, neuroblastoma, sarcoma, lung cancer, and BCas [8]. Depending upon the type of malignancy, the frequency of alterations in MYC varies between 1 and 94% at the cryptogenic level [9]. In a meta-analysis of 29 studies of breast ductal carcinomas, MYC amplification, defined as 2-fold increase in gene copy number, was found in approximately 16% of cases [10]. Amplification of MYC is associated with poor prognosis, high-grade BCas, and early relapse [11, 12].
Identified as a downstream target of HER2, MYC activates various kinase-signaling pathways that may be regulated by ER or PR [13–15]. Many studies have shown that MYC-amplified BCas also harbor HER2 amplification, suggesting a co-amplification [16]. These studies demonstrate that co-amplification of MYC and HER2 augments the development of aggressive tumors with enhanced self-renewal and tumor propagating characteristics. Nevertheless, the underlying mechanism of the interaction between HER2 and MYC that contributes to mammary oncogenesis remains poorly understood [17].
In light of the above, we analyzed a series of well characterized archival invasive ductal carcinoma specimens in order to better understand which BCa subtypes are more likely to be driven by MYC with potential co-amplification of HER2. The objective of the study was to determine the prevalence of MYC amplification in BCa tumors from African American (AA) women and determine the association between MYC amplification and BCa clinic-pathological characteristics.
Methods
Specimen acquisition
This retrospective study was based on anonymized, formalin-fixed, paraffin-embedded FFPE archival BCa tissues (e.g. triple negative BCa, Luminal A, Luminal B, and HER2 positive) were obtained from Howard University Hospital Department of Pathology in Washington, DC. They were collected over a ten year period of time ranging from 2000 to 2010. IRB approval for the conduct of this study was received from Howard University’s Institutional Review Board and designated as exempt 15CMED-53. A total of 70 samples were analyzed. Samples were selected based on the availability and adequacy of FFPE tissue blocks for further study. Five μm thick sections were cut and areas of well-preserved carcinoma in a Hematoxylin and Eosin (H&E) orienting slide from each block were marked for study through hybridization and microscopy. Demographic and clinic-pathological data were obtained through the Howard University Cancer Center Tumor Registry.
Fluorescence in situ hybridization (FISH)
Fluorescence in situ hybridization (FISH) techniques were used to assess MYC amplification in tumor specimens as previously described in our previous study (18). Briefly, the orange fluorophore directly-labeled MYC probe (CymoGen DX, NY USA) and a green labeled centromere 8 probe (Empire Genomics, NY, USA) were used to make a FISH probe mix. The slides were then baked overnight at 60 °C to adhere the tissues to the slides. Slides were subsequently deparaffinized by consecutive 10-min xylene washes before being rehydrated through 100% ethanol incubation for 10 min at room temperature. The tissue on the slides were then permeabilized and digested using the Abbott (Naperville, IL) tissue digestion kit containing pepsin, according to manufacturer’s instruction. The sections were washed with wash buffer after consecutive pretreatment and protease digestion. The probe was denatured at 74 °C for 10 min and immediately transferred to an ice bucket. The denatured probe was then added to the marked area on the slides and was sealed with rubber cement. The slides and the probe were co-denatured for 8 min in HYBrite heat plate (Vysis, Naperville, Illinois) at 85 °C for 8 min and then incubated for 16 to 24 h at 37 °C. Coverslips were removed and the slides were Post Washed in 2X SSC hybridization buffer for 2 min at 73 °C and transferred to 2X SSC at room temperature for 5 min. The slides were air dried in the dark for 1 h in an upright position. Lastly, the slides were counterstained with DAPI (4′, 6-diamidino-2-phenylindole) from Sigma Aldrich and were viewed with a Zeiss Axioscope fluorescence microscope and imaged with Applied Imaging (Pittsburgh, PA) Cytovision software.
Signal enumeration
FISH images were acquired using a Zeiss Axioscope fluorescence microscope equipped with a camera and multiple fluorescence filter sets with three color filter set DAPI, FITC and TRITC. The slides were screened with 100× oil objective lens through the DAPI filter to determine the cell area by observing the area of interest marked by the Pathologist. The centromeres were counted as green signals whereas the MYC signals were viewed as orange signals with the TRITC filter (Fig. 1). At least 50 cells were enumerated from a single slide when possible. The signal ratio of the centromere to MYC in normal cells was calculated. The normal control was 2:2; an increase in the ratio was considered abnormal or amplified. The results were expressed as number of MYC signals/number of chromosome 8 signals (MYC: CEP8) [18].
Fig. 1.
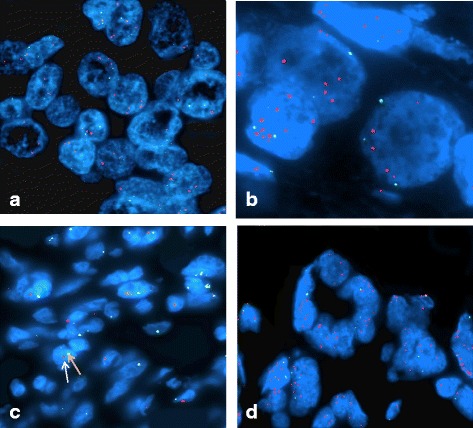
FISH hybridization of MYC in breast tumor tissues. The FISH probe for MYC is labeled with a red fluorochrome, and the normal control signal for the chromosome 8 centromere is labeled in green. The nuclei of the cells are visualized via DAPI counterstaining. a 1:3 ratio of MYC to centromere signals indicating low amplification is shown. b 1:5 ration of MYC to centromere signals indicating moderate amplification is shown. c 1:1 copy ratio of MYC to centromere signals indicating no amplification of the MYC gene is shown. d 1: 8 copy ratio of MYC to centromere signals indicating high amplification is shown in the panel
Statistical methods
The statistical analysis software, IBM SPSS Statistics for Windows(Version 22. Armonk, NY: IBM Corp) was used to perform descriptive analysis of clinic-pathological variables and determine the association between MYC amplification (dependent variable) and prognostic clinic-pathological characteristics such as ER, PR, and HER2 positivity, molecular BCa subtype, stage, grade, and tumor size (independent variables). The Chi-square (χ2) test or T test, as appropriate, was used to examine bivariate association between MYC-amplification, CK 5/6 expression and clinic-pathologic variables. Age was categorized into two groups with a cutoff of 50 years. The logistic regression analysis or ANOVA, as appropriate, was used to examine independent association between MYC amplification and clinic-pathological variables. Results were reported as odds ratios (OR) along with calculated 95% confidence intervals (CI). Pearson’s correlation was used to determine the correlation between amplification ratio, tumor size, and age of diagnosis.. Kaplan-Meier (K-M) estimate of overall survival were plotted and log-rank test was performed to compare estimates among groups.
Results
Clinicopathologic features
Clinicopathological characteristics of the study population are summarized in Table 1. Seventy paraffin-embedded archival tissues of BCa were studied. The average age at diagnosis was 56 years with a minimum and maximum age of 29 and 85 years, respectively. Approximately 37% of the cases were TNBCs; HER2-amplified tumors comprised 11.4% of the tumors in this study; and Luminal A and Luminal B tumors comprised 30% and 21% of the tumors, respectively. ER, PR, and HER2 were positive in 50%, 45.7%, and 18.6% of tumors, respectively. Most of the tumors were poorly differentiated (74%). The frequency of stage I, II, II, and IV tumors were 23.4%, 41.4%, 22.9%, and 5.7%, respectively.
Table 1.
Demographic and clinico-pathological characteristics of cases
| Characteristics | Mean | Range |
|---|---|---|
| Age of Diagnosis (Years) | ||
| 56.36 years | 29–85 years | |
| Tumor Size | 58.0 mm | 2.0–120.0 mm |
| Frequency | Percentage | |
| Breast Cancer Subtypes | ||
| HER2 | 8 | 11.4 |
| Luminal A | 21 | 30 |
| Luminal B | 15 | 21.4 |
| Luminal B (Ki-67 ≥ 14%) | 9 | 12.9 |
| Luminal B HER2+ | 6 | 8.6 |
| Triple Negative | 26 | 37.1 |
| ER Status | ||
| Positive | 35 | 50 |
| Negative | 35 | 50 |
| PR Status | ||
| Positive | 32 | 45.7 |
| Negative | 38 | 54.3 |
| HER2 Status | ||
| Amplified | 13 | 18.6 |
| Not Amplified | 57 | 81.4 |
| Stage | ||
| I | 15 | 23.4 |
| II | 29 | 41.4 |
| III | 16 | 22.9 |
| IV | 4 | 5.7 |
| Grade | ||
| Grade I: Well differentiated | 1 | 1.4 |
| Grade II: Moderately differentiated | 15 | 21.4 |
| Grade III: Poorly differentiated | 52 | 74.3 |
FISH analysis of gene amplifications and BCa subtypes
Figure 1 demonstrates representative FISH images with a red labeled MYC probe and a green labeled chromosome 8 centromere probe. Normal, intermediate, and high MYC amplification tissues for these studies are presented. The normal MYC: CEP8 ratio is 1. The average MYC amplification ratio was 2.25. Thirty-six percent of the cases had high amplification ratio (MYC: CEP 8 ratio > 2). High MYC amplification scores were observed in HER2 (3.97 ± 1.99) and Luminal B (3.34 ± 2.11) subtypes, whereas low amplification was seen in TNBC (1.65 ± 0.94) and Luminal A (1.65 ± 0.93) subtypes as shown in Table 2. The mean MYC amplification ratio of HER2, Luminal A, and TNBC differ significantly; however, HER2 and Luminal B do not significantly differ from each other (p < 0.001).
Table 2.
C-MYC Amplification in various breast cancer subtypes
| Breast Cancer Subtype | Frequency | Mean | Standard Deviation | Min | Max | Tukey Grouping |
|---|---|---|---|---|---|---|
| HER2 | 8 | 3.63 | 2.27 | 1.16 | 7.08 | A |
| Luminal A | 21 | 1.65 | 2.12 | 0.94 | 4.7 | B |
| Luminal B | 15 | 3.19 | 0.67 | 1.00 | 9.28 | A |
| Triple negative | 26 | 1.52 | 2.27 | 0.76 | 3.56 | B |
ANOVA F-test =8.52; p < 0.0001 A = 0.625 B = 1.00
MYC amplification and HER2/PR/ER status
In all, 20% of the samples were HER2 positive (13% HER2 subtype, 7% Luminal B). The average MYC amplification ratio for HER2 positive BCas was 4.36 (±2.24) and 1.77 (±0.97) for HER2 negative/not amplified BCa cases as presented in Table 3. The mean difference between Her2 + and Her2- tumors was statistically highly significant with p = < 0.001 as detailed in Table 3. Average MYC amplification scores did not differ significantly between ER+ and ER-, as well as PR+ and PR- tumors. Regression analysis also revealed an association between the HER2+ subtype (OR = 29.75 95% CI: 2.80–315.56; p < 0.0001) and Luminal B HER2 positive subtype (OR = 10.63 95% CI 2.16–52.15; p < 0.0001), as well as with HER2 immuno-histochemical expression (OR = 40.62 95% CI 4.82–342.39; p < 0.00001). However, no statistically significant association was found between MYC amplification and cancer grade, origin of tumor, tumor size, or age of diagnosis.
Table 3.
MYC amplification with ER, PR and HER2
| Characteristic | N | Mean | Std Dev | Minimum | Maximum | p-value |
|---|---|---|---|---|---|---|
| ER | ||||||
| Negative | 35 | 2.18 | 1.57 | 0.76 | 7.08 | |
| Positive | 35 | 2.33 | 1.71 | 0.94 | 9.28 | 0.71 |
| PR | ||||||
| Negative | 38 | 2.47 | 1.94 | 0.76 | 9.28 | |
| Positive | 32 | 1.99 | 1.14 | 0.94 | 5.50 | 0.23 |
| HER2 | ||||||
| Negative | 57 | 1.77 | 0.97 | 0.76 | 5.00 | |
| Positive | 13 | 4.36 | 2.24 | 1.28 | 9.28 | < 0.001 |
BCa survival analysis and BCa subtypes
In addition, Table 4 shows the survival analysis data in relation to MYC amplification and clinic-pathological variables. Statistically significant differences in overall survival were found when stratifying by stage (p < 0.001), having metastatic disease (p < 0.001), and lymph node positivity (p = 0.039). However, no statistically significant difference in overall survival was observed among BCa subtypes in this study (Log rank test p = 0.201). The mean overall survival was 71.00 months for HER2 subtype (95% CI = 44.09–97.91), 94 months (95% CI = 84.56–103.44) for Luminal B, 67.71 months (95% CI = 52.05–83.36) for Luminal A, and 100.07 months (95% CI = 76.57–123.57) for TNBC subtype.
Table 4.
Regression analysis of MYC expression and clinico-pathological variables
| C-MYC Amplification | Odds | 95% CI | ||||||
|---|---|---|---|---|---|---|---|---|
| < 2 | > 2 | Ratio | Lower | Upper | p-value | |||
| Breast Cancer Molecular Subtype | ||||||||
| Luminal A | 17 | 81.0% | 4 | 19.0% | ref. | |||
| Luminal B | 4 | 28.6% | 10 | 71.4% | 10.63 | 2.16 | 52.15 | < 0.0001 |
| HER 2+ | 1 | 12.5% | 7 | 87.5% | 29.75 | 2.80 | 315.56 | < 0.0001 |
| Triple Negative Breast Cancer | 23 | 85.2% | 4 | 14.8% | 0.74 | 0.16 | 3.38 | 0.72 |
| Estrogen Receptor Status | ||||||||
| Positive | 21 | 60.0% | 14 | 40.0% | ref. | |||
| Negative | 24 | 68.6% | 11 | 31.4% | 0.69 | 0.26 | 1.84 | 0.31 |
| Progesterone Receptor Status | ||||||||
| Positive | 21 | 65.6% | 11 | 34.4% | ref. | |||
| Negative | 24 | 63.2% | 14 | 36.8% | 1.11 | 0.42 | 2.98 | 0.52 |
| HER2 Status | ||||||||
| Negative | 44 | 77.2% | 13 | 22.8% | ref. | |||
| Positive | 1 | 7.7% | 12 | 92.3% | 40.62 | 4.82 | 342.39 | < 0.0001 |
Table 5 demonstrates that differences in mean survival were not found when stratifying by MYC amplification (p = 0.265). .
Table 5.
Survival analysis of clinico-pathological variables and MYC amplification
| 95% CI | |||||
|---|---|---|---|---|---|
| Mean Months | Std. Error | Lower Bound | Upper Bound | p-value | |
| Stage | |||||
| 1 | 119.59 | 9.62 | 100.73 | 138.45 | |
| 2 | 92.03 | 7.75 | 76.85 | 107.21 | |
| 3 | 98.25 | 15.12 | 68.61 | 127.89 | |
| 4 | 18.25 | 6.02 | 6.45 | 30.05 | p < 0.001 |
| Size (T) | |||||
| T1 | 89.35 | 4.53 | 80.47 | 98.23 | |
| T2 | 100.60 | 9.98 | 81.04 | 120.17 | |
| T3 | 82.50 | 16.74 | 49.69 | 115.32 | p = 0.06 |
| Distant Metastases | |||||
| No Metastatic Disease (0) | 114.88 | 7.06 | 101.05 | 128.71 | |
| Metastatic Disease (1) | 38.78 | 13.97 | 11.40 | 66.16 | p < 0.001 |
| Lymph Nodes | |||||
| Lymph Node Negative (0) | 124.65 | 8.29 | 108.40 | 140.90 | |
| Lymph Node Positive (1) | 86.88 | 10.33 | 66.64 | 107.11 | p = 0.039 |
| Breast Cancer Molecular Subtype | |||||
| Luminal A | 67.71 | 7.99 | 52.05 | 83.36 | |
| Luminal B | 94.00 | 4.82 | 84.56 | 103.44 | |
| HER 2+ | 71.00 | 13.73 | 44.09 | 97.91 | |
| Triple Negative Breast Cancer | 100.07 | 11.99 | 76.57 | 123.57 | p = 0.201 |
| C-MYC Amplification | |||||
| < 2 | 93.96 | 8.96 | 76.41 | 111.52 | |
| > 2 | 117.03 | 9.98 | 97.47 | 136.59 | p = 0.265 |
Discussion
An association between MYC amplification and HER2 overexpressing tumors in AAs was found in this pilot study of AA patients. Importantly, cases of Luminal B with positive HER2 expression exhibited the highest MYC amplification ratios. Specifically, HER2/neu receptor positive cases had 4.5 more copies of MYC than HER2/neu negative cases, a difference that suggests a significant association between MYC amplification and hormone receptor positivity in HER2 positive Luminal B BCas (p = 0.01). Luminal A HER2 positive BCas did not demonstrate high MYC amplification. We did not observe any association between MYC amplification and tumor grade, tumor size, or age at diagnosis similar to other studies [18–20].
In BCas, amplification of MYC is associated with poor prognosis, high-grade, and early relapse [11–13]. The distribution of MYC amplification varies by BCa subtypes and MYC amplification has been shown to be associated with BCa molecular subtypes and hormonal receptor status. High MYC amplification (MYC: CEP8 ratio > 2) has been reported in HER2 and Luminal B subtypes of BCa whereas low MYC amplification scores were observed in Luminal A and TNBC subtypes [19, 20]. Another study showed high MYC amplification in TNBCs, particularly incases demonstrating a basal-like phenotype [21–23].
The majority of analyses have shown that MYC-amplified BCas also harbor HER2 amplification, suggesting a co-amplification mechanism (16). Consistent with these studies, we also demonstrated that HER2 positive BCas in this population of AA women were more likely to exhibit MYC amplification. High amplification ratios may suggest that increases in MYC activity drives HER2 amplification, especially in HER2 positive and Luminal B HER2-amplified subtypes of BCa. Therefore, MYC may play a significant role in steroid receptor dependent as well as independent tumorigenesis.
This study shows low MYC amplification scores in Luminal A and TNBCs. Another recent study measured MYC gene copy number in 94 breast tumors of all subtypes with semi-quantitative multiplex PCR. They did not find any association of amplification with age, tumor size, ER and PR status, local metastases, or histologic grading, but noted that MYC amplification was present in early tumors but not adjacent normal tissues [11]. Laboratory test systems, FISH probe choices, and microscopy factors affect study sensitivity, particularly with low levels of amplification which may not be resolved adequately.
To further explore the relationship between MYC amplification and steroid receptor status, we evaluated the association between MYC: CEP8 ratio and the presence/absence of PR and ER. Our study yielded mixed results consistent with those obtained in previous studies [15, 24]. The mean MYC amplification ratio in PR negative tumors was 0.5 times higher than in tumor tissues that were PR positive; ER positive cases had slightly higher MYC amplification ratios than ER negative cases. However, there was no significant relationship between the PR/ ER receptor status and MYC amplification (p = 0.50, 0.57, respectively). Notably, the MYC amplification score was high in tumors that metastasized to brain or bone. Although the precise nature of the ER and PR signaling pathway and its relationship with MYC amplification remain unclear, PR was reported to influence proliferation and cell differentiation, and to exhibit a biphasic effect on cell growth and MYC expression [25]. A more recent study conducted by Wang et al. indicated that MYC expression could be induced by estrogen, involving the integration of MYC between ER and AP-1 at the distal enhancer element suggesting that AP-1 may play a role in induction of the MYC oncogene [26].
Future studies to investigate the role of MYC as a downstream mediator of HER2 may assist in understanding the role MYC gene on growth regulatory genes in different BCa subtypes. Also, studies evaluating differences in MYC amplification between primary breast tumors and metastatic sites in bone, lung, and brain would be useful to determine whether lethality of metastatic disease is related to MYC amplification, especially in minority populations.
Conclusion
We demonstrate that HER2+ breast cancer tissues from AA women that demonstrate also have a high likelihood to show MYC amplification. High amplification ratios suggest that MYC drives HER2 amplification, especially in HER2 positive BCas including both the HER2 subtype and HER2 + Luminal B subtype. This provides support the concept that MYC plays a part on steroid dependent and independent tumor development.
Acknowledgements
We thank Will Mumford for his assistance with manuscript preparation.
Funding
The MYC probe used in this study was generously supplied by CymoGenDx, New York, USA. Partial funding for AG was provided by a Susan Komen Foundation grant Graduate Training in Breast Cancer Disparities at Lombardi Cancer Center (GTDR15330383).
Availability of data and materials
The datasets and selected material analyzed in the current study are available from the corresponding author upon reasonable request.
Author’s contributions
Study idea, design, and workflow supervision: JKB, AEK, RD and TN. Laboratory analysis: AG, MA, LRS, YK, RD and JKB. Data analysis and interpretation: KM, AEK, LSR, and GA. Technical assistance in manuscript preparation: TN, LRS, and JKB. All authors read and approved the final manuscript.
Abbreviations
- AA
African American
- ANOVA
Analysis of Variants
- BCa
Breast cancer
- CEP
Centromere Enumeration Probe
- CI
Confidence Interval
- DAPI
4′,6-diamidino-2-phenylindole
- ER
Estrogen Receptor
- FFPE
Formalin fixed paraffin embedded
- FISH
Fluorescence in situ Hybridization
- FITC
Fluorescein isothiocyanate
- H & E
Hematoxylin and Eosin
- HER2/neu
Receptor tyrosine-protein kinase erbB-2
- IRB
Institutional Review Board
- MYC
c-Myc gene
- OR
Odds Ratio
- PR
Progesterone Receptor
- TNBC
Triple Negative Breast Cancer
- TRITC
Tetramethylrhodamine
Ethics approval and consent to participate
This study was based on existing de-identified data and pathologic specimens. It was not deemed to be human subjects’ research, but determined to be of exempt status. Approval for this research protocol was given by the Howard University School of Medicine IRB, designated as reference number 15CMED-53.
Consent for publication
Subject consent was not required for this study. It is bound by the ruling of the US Health and Human Services Secretary’s Advisory Committee on Human Research Protections (SACHRP) under the Common Rule for exempt research and the Helsinki guidelines for establishment of IRB committees. IRB approval #15C MED 53 was obtained from the Howard University IRB for the study of de-identified materials.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tammey J. Naab, Email: tnaab@huhosp.org
Anita Gautam, Email: Anita.Gautam@umassmed.edu.
Luisel Ricks-Santi, Email: luisel.rickssanti@hamptonu.edu.
Ashwini K. Esnakula, Email: Esnakula@ufl.edu
Yasmine M. Kanaan, Email: ymkanaan@howard.edu
Robert L. DeWitty, Email: rdewitty@howard.edu
Girmay Asgedom, Email: Girmay.Asgedom@howard.edu.
Khepher H. Makambi, Email: khm33@georgetown.edu
Massih Abawi, Email: mabawi@Genedx.com.
Jan K. Blancato, Email: blancatj@georgetown.edu
References
- 1.Copeland G LA, Firth R, et al. Cancer in North America: 2008–2012. North American Association of Central Cancer Registries, Inc, Springfield, IL. June 2015.;Volume One: Combined Cancer Incidence for the United States, Canada and North America.
- 2.Lund MJ, Trivers KF, Porter PL, et al. Race and triple negative threats to breast cancer survival: a population-based study in Atlanta, GA. Breast Cancer Res Treat. 2009;113(2):357–370. doi: 10.1007/s10549-008-9926-3. [DOI] [PubMed] [Google Scholar]
- 3.Loo LW, Wang Y, Flynn EM, et al. Genome-wide copy number alterations in subtypes of invasive breast cancers in young white and African American women. Breast Cancer Res Treat. 2011;127(1):297–308. doi: 10.1007/s10549-010-1297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danforth DN., Jr Disparities in breast cancer outcomes between Caucasian and African American women: a model for describing the relationship of biological and nonbiological factors. Breast Cancer Res. 2013;15(3):208. doi: 10.1186/bcr3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100(14):8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2011;16(Suppl 1):61–70. doi: 10.1634/theoncologist.2011-S1-61. [DOI] [PubMed] [Google Scholar]
- 8.Dang CVMYC. On the path to cancer. Cell. 2012;149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao DJ, Dickson RB. C-Myc in breast cancer. Endocr Relat Cancer. 2000;7(3):143–164. doi: 10.1677/erc.0.0070143. [DOI] [PubMed] [Google Scholar]
- 10.Deming SL, Nass SJ, Dickson RB, et al. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83(12):1688–1695. doi: 10.1054/bjoc.2000.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chrzan P, Skokowski J, Karmolinski A, et al. Amplification of c-myc gene and overexpression of c-Myc protein in breast cancer and adjacent non-neoplastic tissue. Clin Biochem. 2001;34(7):557–562. doi: 10.1016/S0009-9120(01)00260-0. [DOI] [PubMed] [Google Scholar]
- 12.Blancato J, Singh B, Liu A, et al. Correlation of amplification and overexpression of the c-myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br J Cancer. 2004;90(8):1612–1619. doi: 10.1038/sj.bjc.6601703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouchalova K, Cizkova M, Cwiertka K, et al. Triple negative breast cancer--current status and prospective targeted treatment based on HER1 (EGFR), TOP2A and C-MYC gene assessment. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2009;153(1):13–17. doi: 10.5507/bp.2009.002. [DOI] [PubMed] [Google Scholar]
- 14.Hynes NE, Myc LHA. Mammary cancer: Myc is a downstream effector of the ErbB2 receptor tyrosine kinase. J Mammary Gland Biol Neoplasia. 2001;6(1):141–150. doi: 10.1023/A:1009528918064. [DOI] [PubMed] [Google Scholar]
- 15.Todorovic-Rakovic N, Neskovic-Konstantinovic Z, Nikolic-Vukosavljevic D. C-myc as a predictive marker for chemotherapy in metastatic breast cancer. Clin Exp Med. 2012;12(4):217–223. doi: 10.1007/s10238-011-0169-y. [DOI] [PubMed] [Google Scholar]
- 16.Park K, Kwak K, Kim J, et al. C-myc amplification is associated with HER2 amplification and closely linked with cell proliferation in tissue microarray of nonselected breast cancers. Hum Pathol. 2005;36(6):634–639. doi: 10.1016/j.humpath.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Nair R, Roden DL, Teo WS, et al. C-Myc and Her2 cooperate to drive a stem-like phenotype with poor prognosis in breast cancer. Oncogene. 2014;33(30):3992–4002. doi: 10.1038/onc.2013.368. [DOI] [PubMed] [Google Scholar]
- 18.Blancato JK, Williams MS, Dickson RB. Fluorescence in situ hybridization assessment of c-myc gene amplification in breast tumor tissues. Methods Mol Med. 2006;120:297–307. doi: 10.1385/1-59259-969-9:297. [DOI] [PubMed] [Google Scholar]
- 19.Li C, Bai J, Hao X, et al. Multi-gene fluorescence in situ hybridization to detect cell cycle gene copy number aberrations in young breast cancer patients. Cell Cycle. 2014;13(8):1299–1305. doi: 10.4161/cc.28201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasojima H, Shimomura A, Naoi Y, et al. Association between c-myc amplification and pathological complete response to neoadjuvant chemotherapy in breast cancer. Eur J Cancer. 2011;47(12):1779–1788. doi: 10.1016/j.ejca.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 21.Horiuchi D, Kusdra L, Huskey NE, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209(4):679–696. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singhi AD, Cimino-Mathews A, Jenkins RB, et al. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol. 2012;25(3):378–387. doi: 10.1038/modpathol.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alles MC, Gardiner-Garden M, Nott DJ, et al. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the "basal" breast cancer subgroup. PLoS One. 2009;4(3):e4710. doi: 10.1371/journal.pone.0004710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez EA, Jenkins RB, Dueck AC, et al. C-MYC alterations and association with patient outcome in early-stage HER2-positive breast cancer from the north central cancer treatment group N9831 adjuvant trastuzumab trial. J Clin Oncol. 2011;29(6):651–659. doi: 10.1200/JCO.2010.30.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong MS, Murphy LC. Differential regulation of c-myc by progestins and antiestrogens in T-47D human breast cancer cells. J Steroid Biochem Mol Biol. 1991;39(1):39–44. doi: 10.1016/0960-0760(91)90010-3. [DOI] [PubMed] [Google Scholar]
- 26.Wang C, Mayer JA, Mazumdar A, et al. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol Endocrinol. 2011;25(9):1527–1538. doi: 10.1210/me.2011-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and selected material analyzed in the current study are available from the corresponding author upon reasonable request.