

SUMMARY
β-Amino acids offer attractive opportunities to develop biologically active peptidomimetics, either employed alone or in conjunction with natural α-amino acids. Owing to their potential for unique conformational preferences that deviate considerably from α-peptide geometries, β-amino acids greatly expand the possible chemistries and physical properties available to polyamide foldamers. Complete in silico support for designing new molecules incorporating non-natural amino acids typically requires representing their side-chain conformations as sets of discrete rotamers for model refinement and sequence optimization. Such rotamer libraries are key components of several state-of-the-art design frameworks. Here we report the development, incorporation in to the Rosetta macromolecular modeling suite, and validation of rotamer libraries for β3-amino acids.
INTRODUCTION
The explicit treatment of the conformation of side-chain atoms distinguishes low- and high-resolution models of protein structure (Renfrew et al., 2014). A common low-resolution approach is to instead approximate the side chain with a single pseudoatom located at the center of mass (Levitt, 1976); such methods require less physically realistic energy functions (Kussell et al., 2002; Feig et al., 2000) but permit faster sampling over a smoother energy landscape (Simons et al., 1999; Bonneau et al., 2001). Explicit (all atom) side-chain conformations may instead be efficiently sampled using a rotameric approximation, where discrete sets of side-chain χ angles are chosen from well-validated conformations that are meant, ideally, to correspond to the conformational energy minima for the side chain in question (Dunbrack, 2002). These sets of side-chain conformations are called ‘‘rotamer libraries.’’ Sampling side-chain conformations from rotamer libraries is especially efficient when there is substantial interdependence between side-chain degrees of freedom, and when only a small fraction of the plausible local minima are near the global minimum. Rotamer sampling requires rotamer libraries from which to sample conformations and recent works have demonstrated that these rotamer libraries should be dependent (to further improve computational efficiency) on backbone conformation (Kothiwale et al., 2015).
Prior development of non-canonical rotamer libraries for use in Rosetta has employed side-chain coordinate scans and energy optimization, either through quantum mechanics software or a molecular mechanics force field, followed by k-means clustering to generate rotamer libraries for non-canonical amino acids (NCAAs) and peptoids (Renfrew et al., 2012). For canonical 03b1-branched amino acids, the Dunbrack and Richardson labs have curated the most widely used rotamer libraries. In both cases, these libraries are derived from a statistical treatment of high-quality experimental structures (Lovell et al., 2000; Hintze et al., 2016; Shapovalov and Dunbrack, 2011; Dunbrack and Karplus, 1993; Bower et al., 1997; Dunbrack, 2002). The Daggett lab has recently taken an alternate approach and employed molecular dynamics simulations (Scouras and Daggett, 2011; Towse et al., 2016). Work on these molecular dynamics-based rotamer libraries points to several limitations to statistics-derived rotamers, and also demonstrates that simulations can model side-chain configurational clusters and minima when statistical methods are not applicable, as in the case of NCAAs (Jacobson et al., 2002; Renfrew et al., 2008).
Both this work and prior work by Kuhlman and Renfrew suggest that there exists a valuable role for rotamer libraries derived from simulations as well as experimental structures. Furthermore, since structural data on non-natural residues is very limited, general rotamer libraries for these moieties must be simulation derived. Peptides entirely composed of or containing β-amino acids (Figure 1) possess useful pharmacological properties, particularly slow proteolytic degradation and a curbed immune response (Cheloha et al., 2015). Such molecules have been used to mimic apolipoprotein (Werder et al., 1999) and as antimicrobial compounds (Raguse et al., 2003); they may target mdm2 (Kritzer et al., 2004), the GLP-1 receptor (Denton et al., 2013), or HIV fusion (Bautista et al., 2009). Detailed efforts to target BCL-2 family proteins (Smith et al., 2013) and GLP-1 (Johnson et al., 2014) have been described.
Figure 1. Schemes of Chemical Species Referred to in the Text.

(A) The α- and β-amino acids are marked with two main-chain torsions that were considered equivalent in prior representations of β-amino acids in Rosetta. While this work only considers rotamer libraries for β3-amino acids, the expansions to the Rosetta code enables representation of any β-amino acid or, indeed, any polyamide, using the same framework.
(B) The 20 β3-amino acid side chains, 18 of which are described with rotamer libraries developed in this study. β3-Glycine and β3-alanine do not possess side-chain degrees of freedom.
(C) The g+, g−, and a conformations described here, for χ1: N-Cβ-Cγ-X dihedrals of 60°, −60°, and 180°.
These biological applications are enabled by the conformational preferences of β-amino acids. Their two backbone carbons provide four stereoisomers and six disubstituted isomers, leading to considerable conformational diversity. Helical structures achievable by β3-amino acids have been extensively studied. These include either the 314 or 14 helix, characterized by a helical axis opposite the α helix, and a 14-atom hydrogen bonding macrocycle, and the 2.512 or 12 helix, whose handedness is like the α helix (Seebach et al., 1996; Appella et al., 1996). Helical bundles similar to α-helical α-amino acid coiled coils are accessible to 314helices as well (Qiu et al., 2006), and one has been designed to have catalytic activity (Wang et al., 2014). β-Amino acids also form local structures that are ideal for mimicking parallel (Seebach et al., 1999, 2002; Langenhan et al., 2003) or antiparallel sheets (Seebach and Gardiner, 2008; Chung et al., 2000; Gademann and Seebach, 1999; Cheng et al., 2001). Such extended conformations may be adopted by cyclic β-peptide oligomers, but steric effects from multiple substitutions can provide a similar conformational bias. For example, similar extended conformations are favored by trans-2,3-disubstituted residues by encouraging an anti θ. In contrast, 2,3-disubstituted cyclic sidechains stabilize gauche θ conformers that favor turns and helices (Cheng et al., 2001), while achiral β2,2- or β3,3-disubstituted residues stabilize reverse turns (Seebach et al., 1998).
Although β-amino acids can form diverse conformations on their own, the accessible chemical space may be expanded further by using them in chimeric peptides, i.e., in combination with α-amino acids. Replacing an α-amino acid with a β-amino acid on both strands of a β hairpin extends the length of the hairpin by one methylene unit but maintains the hydrogen bond network and so only tertiary contacts must adjust to accommodate the perturbation (Lengyel et al., 2011; Haase et al., 2012).
Chimeric α/β-peptides can adopt helical conformations (Hayen et al., 2004; Schmitt et al., 2004, 2006; Sadowsky et al., 2005; Price et al., 2007). Optimized compositions ultimately involved organizing all the β residues on a single helical face by employing β3-amino acids every third or fourth position (Horne et al., 2007). Due to the high therapeutic value of mimicking an α helix (Modell et al., 2016; Moon and Lim, 2015; Cromm et al., 2015) and the conceptual simplicity of homo-oligomers, peptides composed of or containing β-amino acids have been applied primarily as α helix mimetics (Johnson and Gellman, 2013). Consequently, β-amino acid rotamer libraries have been developed only for all-β conformations that form helical folds. There are high-resolution rotamer libraries for β3- and β2-amino acids available for individual backbone conformations: those of 314 and 312 helices (Shandler et al., 2010). Such rotamer libraries, while valuable, are not suited to a general peptidomimetic context in two ways. First, they do not cover backbone conformations that are entirely favorable for β-amino acids, but do not form homopolymer secondary structures, and so they are limited in application. Second, rotamer libraries fit to the rotamers assumed by an entire secondary structure element do not necessarily apply to other polymeric contexts. For example, the dihedrals assumed by a β-amino acid in a 314 helix are only 20° from those assumed by a β-amino acid in an α3β helix, but the α3β helix has an opposite dipole moment and each β-residue is flanked by two α-residues. The resulting bias on rotamer libraries can be substantial; long-range effects from adjacent helical turns are known to bias PDB-based rotamer libraries for at least valine, leucine, and isoleucine (Renfrew et al., 2008). As a concrete example, the (a, g+) rotamer common in α-helical α-leucine residues creates no clashes in the all-β 314 helix depicted in Figure 2, but is incompatible with a common conformation of the chimeric α3β helix. Thus, to apply to a general peptidomimetic context, rotamer libraries must be constructed independent of non-local influences. These prior rotamer libraries have not been available for use in macromolecular modeling programs such as Rosetta, which has long assumed that rotamer libraries were dependent only on two main-chain dihedrals (Watkins and Arora, 2016). Prior work that attempted to model β3-peptides in Rosetta instead employed a limiting approximation: that the first two main-chain torsions of a β3-amino acid could be treated analogously to the φ and ψ torsions of an α-amino acid (Figure 1) (Molski et al., 2013). Backbone dihedral angles typical for a 314 helix such as (−140°, 60°, −120°) are described by a high-energy of Ramachandran space such as (−140°, 60°) and as a result are drastically undersampled.
Figure 2. Rotamer Steric Compatibility Depends Not Just on the Local Backbone Dihedrals but on the Entire Polymer Context.

The (a, g+) rotamer imposed on every fourth residue (a leucine) in the 314 helix (A) and the α3β helix (B). Molprobity clash analysis reveals that the 314 helix is free of clashes (C), while the α3β helix (D) has a clash score of 114, far worse, in 12 residues, than low-quality electron microscopy structures of hundreds of residues. The distinct clash, shown in red dots, is with the i + 3 alanine methyl group.
Here, we have developed and incorporated into Rosetta backbone-dependent rotamer libraries for β3-amino acids (Figure 3). Furthermore, we generalized Rosetta’s treatment of rotamers and rotamer libraries such that it can create and use rotamer libraries with any number of backbone dihedral angles. We validated this approach both computationally and experimentally. Our results are in good agreement with quantum mechanical calculations on several side chains, which demonstrate that we recover the rotamer wells and their approximate populations. Our results also recover the rotamers found in high-quality crystal structures in the PDB. Furthermore, use of these rotamer libraries in conjunction with Rosetta recovers trends in experimental binding affinity in experiments with β-peptide helices. We anticipate that this rotamer library system will constitute an important step toward the direct, accurate rotamer-based design of structures containing arbitrary oligomers in Rosetta.
Figure 3. The Process by Which Rotamer Libraries Are Constructed.
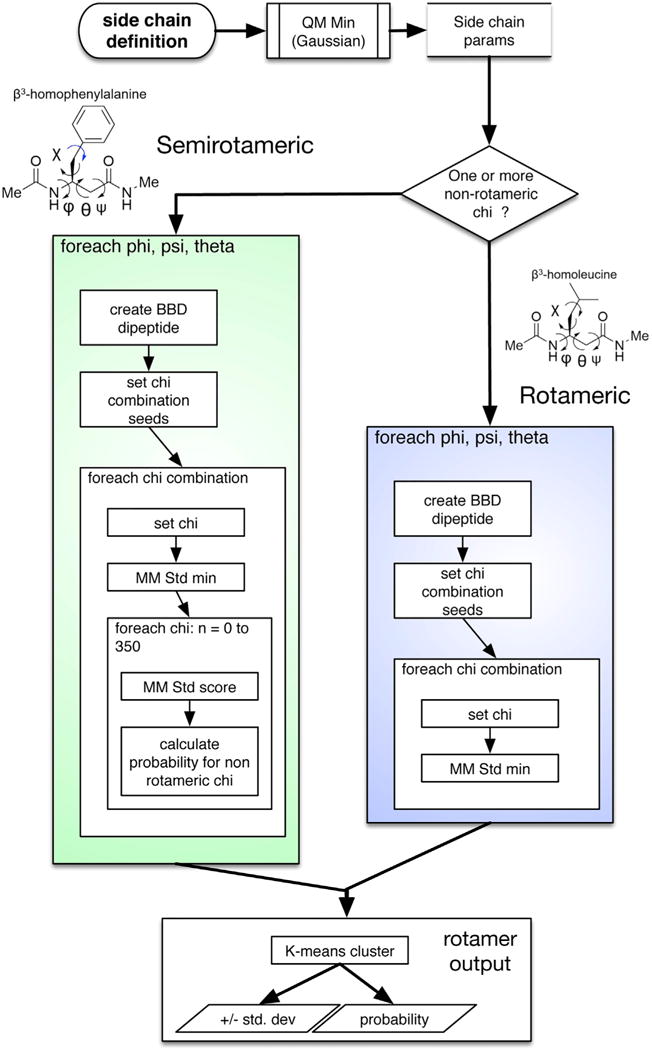
To construct rotamer libraries for β-amino acids, we create parameters for the residue by geometry optimization and charge fitting by quantum mechanics, as described previously. We seed backbone conformations and, for each one, we create hundreds or thousands of χ-combination seed conformations (omitting the final non-rotameric χ for semi rotameric amino acids). We minimize each χ combination (and, for semirotameric residues, obtain a probability distribution for the terminal χ). Finally, we cluster, obtain an SD in each dimension for each resulting rotamer well, and convert the minimized energies to probabilities via the Boltzmann distribution.
RESULTS
β-Amino Acids Exhibit Diverse Backbone Conformations
To quantify the need for a general, fully backbone-dependent rotamer library for β3-amino acids, we explored the backbone conformations that they can adopt. Rather than setting an energetic threshold and exploring candidate energy functions, we simply sought out conformations that lacked intramolecular clashes. Table 1 compares the volume of backbone dihedral space that each β side chain may adopt without clashes versus its α-amino acid equivalent. The volume within 40° of the 314 helix or α3β helix dihedral angles is only 1.1% of the total backbone dihedral space of a β3-amino acid, illustrating that β3-amino acids may adopt far more than the two homopolymer conformations that have received most attention. The generous standard of clash-free conformations is useful to assess the intrinsic flexiblity of β-amino acids, in contrast to homopolymer-preferred conformations, which might require regular, repeating stabilizing interactions. These conformations are important to multiple β-amino acid use cases, such as the design of substituted macrocycles, where the cyclic constraint enforces the local conformation and there is no requirement for regularity. In addition to the well-studied helical conformations, in the process of this investigation we found a range of extended conformations available to β-amino acids, which have not been previously investigated in any systematic way. Such extended conformations possess φ between −130° and −180°, with θ and ψ positive and summing to 30° −50° less than |φ|.
Table 1.
Proportion of Backbone Dihedral Space (‘‘Ramachandran Space’’ for α-amino Acids) that Does Not Result in Any Clashes
| AA | α Torsion (%) |
β3 Torsion (%) |
AA | α Torsion (%) |
β3 Torsion (%) |
|---|---|---|---|---|---|
| ALA | 19.9 | 21.5 | MET | 18.5 | 22.7 |
| CYS | 19.1 | 28.9 | ASN | 18.4 | 23.1 |
| ASP | 18.5 | 25.3 | PROa | 8.7 | 17.7 |
| GLU | 18.7 | 22.4 | GLN | 18.3 | 22.5 |
| PHE | 18.0 | 21.4 | ARG | 18.0 | 19.1 |
| GLY | 46.6 | 47.2 | SER | 19.3 | 30.2 |
| HIS | 18.7 | 24.3 | THR | 17.1 | 24.0 |
| ILE | 12.3 | 13.7 | VAL | 11.8 | 18.9 |
| LYS | 14.0 | 19.4 | TRP | 18.2 | 20.3 |
| LEU | 16.2 | 19.6 | TYR | 18.2 | 21.4 |
A clash is defined by atoms closer than 60% of their summed van der Waals radii.
For proline, only dihedral space that kept the pyrrolidine ring closed was considered.
The Molecular Mechanics Energy Function Reproduces the Energy Wells of Quantum Mechanics Studies
The molecular mechanics scoring function employed in Rosetta, which we used to generate these rotamer libraries, reproduces the minima identified by quantum mechanical calculations. We evaluated four common β3-amino acid side chains, β3W, β3L, β3I, and β3S, restrained to two backbone conformations: a helical conformation reminiscent of both the 314 helix (−140°, 60°, −120°), and the α3β helix and an extended conformation that would form a pleated sheet (−140°, 75°, 65°). We conducted constrained geometry optimization followed by single-point energy calculation at the B3LYP level of theory on side-chain conformations sampled every 10°. The resulting energy surface as a function of χ1 and χ2 compares favorably to the molecular mechanics-derived energy surfaces for each residue type, showing that we capture the same rotamers as the considerably more computationally expensive method (Figure 4). To quantify the accuracy with which the two heatmaps agree, we computed the Bhattacharyya distance for each residue and backbone conformation (Table 2; STAR Methods). The results verify the significant similarity of the energy landscapes, improving confidence in our results.
Figure 4. Side-Chain Probability Distributions as Computed by Quantum Mechanics and Molecular Mechanics.
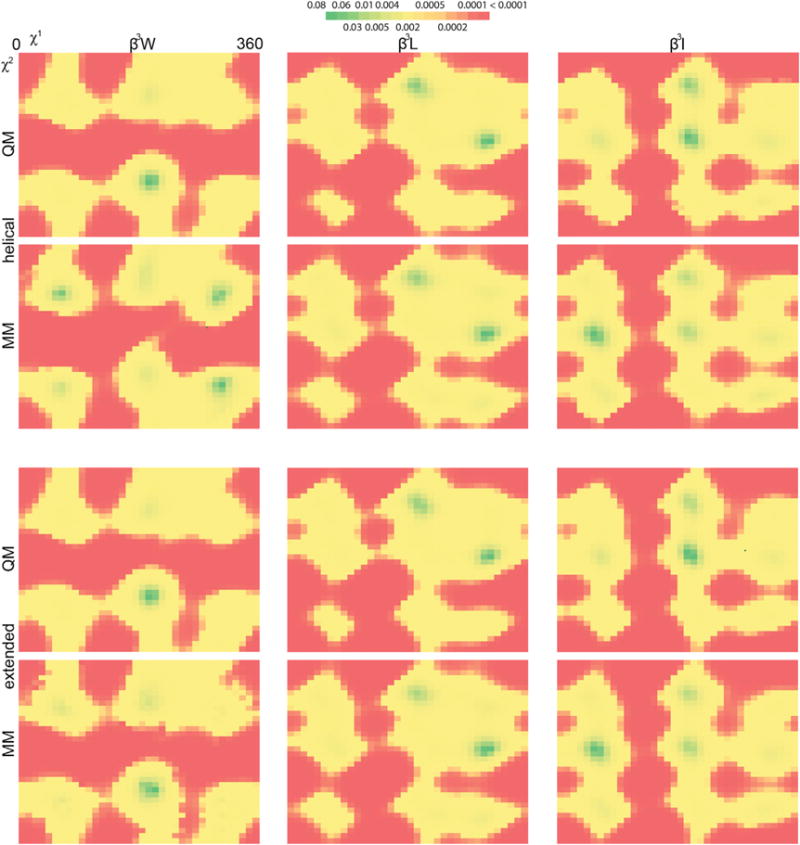
Two dimensional probability distributions for three residues (β3I, β3L, and β3W; columns) were determined by quantum mechanics (B3LYP optimization followed by Hartree-Fock energy; first and third row) and molecular mechanics (the energy function employed in the rotamer library protocol; second and fourth row). These distributions were generated for two distinct backbone conformations: one for a conformation common to the 314 and α3β helix (first and second rows) and one for an extended conformation (third and fourth rows). QM, quantum mechanical; MM, molecular mechanics.
Table 2.
The Bhattacharyya Distance for Each of Eight Pairs of Probability Distributions, Representing the Side-Chain Conformational Preferences of Four β3-Amino Acids in Two Conformations, Evaluated Either at the Quantum Mechanical or Molecular Mechanics Level of Theory
| AA | QM-MM Distance | |
|---|---|---|
| Helical | Extended | |
| β3I | 0.155 | 0.163 |
| β3L | 0.048 | 0.069 |
| β3S | 0.237 | 0.233 |
| β3W | 0.261 | 0.074 |
QM, quantum mechanical; MM, molecular mechanics.
The New β3-Amino Acid Rotamer Libraries Reproduce Observed PDB Rotamers
A considerable number of structures containing β3-amino acids already exist in the PDB, although all but one is in a mixed α/βor 314 helix. To gauge the performance of these rotamer libraries against experimental data, we built the same rotamer set as employed in Rosetta’s packing algorithm, by performing tricubic interpolation between adjacent backbone dihedral bins (i.e., the eight vertices of the cube of backbone torsion space grid points for which explicit rotamers samples appear in the library). The resulting rotamer sets possess a rotamer within 15.5° of the experimental rotamer or better, with many closer than 10° (Figure 5).
Figure 5. Rotamer Libraries Accurately Capture Experimentally Determined Rotamers.

root-mean-square error in degrees for the nearest rotamer across all resolved β3-amino acid samples in the PDB. The number of examples of that β-amino acid are above each bar. No data exist on β3-homocysteine or β3-homohistidine, and rotamer libraries do not apply to β3-homoalanine or β3-homoglycine. Errors are comparable with those found in prior work on fitting peptoid and non-canonical amino acid rotamer libraries (Renfrew et al., 2012, 2014) and to the effective uncertainty of Dunbrack rotamer libraries (see the STAR Methods).
This performance in native rotamer recovery is comparable with the performance of the Dunbrack libraries on the equivalent α-amino acid side chain (see the STAR Methods; especially considering the narrow scope of the test data only one region of backbone torsion space) against the general model employed here, we find this result very encouraging. In the process of assessing the errors in PDB rotamer recovery, we found that several of the native rotamers we had been treating as ‘‘gold standards’’ inhabited structurally improbable conformations. Since they were not forming tightly packed interactions in a hydrophobic core, nor with a binding partner, nor with a crystal contact, we suspected that they might constitute structural errors. We examined several representative structures that possessed high-energy native rotamers and built maps from the publicly available structure factors; in doing so, we found that many β-amino acid residues with very little electron density supporting any particular side-chain conformation; for example, we highlight four examples from PDB: 3G7A and 4BPI in Figure 6, but others are found in PDB: 4KGR, 3O42, and more. We strongly suspect that many of the deviations observed here are due to rotamers that were built poorly in the absence of electron density constraints; these experimental samples are thus a poor reflection of physical ground truth. Since the total number of β-amino acid rotamers in the PDB is so small, we cannot perform this analysis on only those rotamers with sufficiently high-quality structural data supporting their conformations. Rather, we propose that, with the recent development of new structural refinement tools within Rosetta (Park et al., 2015; Chou et al., 2013), these and other β-amino acid containing structures may be further refined, particularly now that there exist general, backbone-dependent rotamer libraries for the task.
Figure 6. Many Crystal Structures Possess Poorly Resolved Density for β-Amino Acid Residues.

Many native side chains ‘‘missed’’ by these rotamer libraries are in very unlikely conformations in the crystal structure. Due to the challenges of fitting atomic coordinates to electron density, and due to the additional challenge of phasing diffraction data for novel heteropolymers, structural data might be particularly uninformative for these molecules. Indeed, maps frequently provided minimal density near β-amino acid residues, as in the four examples above (PDB: 3G7A and 4BPI).
The New Rotamer Libraries Improve Modeling of 314 Helix Ligands
Encouraged by the agreement with higher levels of theory and the recovery of native rotamers, we undertook a retroactive binding affinity prediction. We rewrote a substantial part of Rosetta’s core libraries so that rotamer libraries might possess any number of dihedral angles, and so that these rotamer libraries may be employed in the packer, Rosetta’s algorithm for concurrent side-chain optimization. To test the ability of this new code and these new libraries to deliver scientifically useful results, we revisited an effort to design improved 314 helix p53 mimetics from the Schepartz lab. Their dataset included computational predictions via Monte Carlo free-energy perturbation (Zwanzig, 1954) (MC/ FEP) in the OPLS-AA force field (Jorgensen and Tirado-Rives, 2005), as well as experimentally measured binding energies (Michel et al., 2009). We sought to reproduce the trend in point mutation energies (ΔΔG) versus the base sequence. We compared the original MC/FEP result (R2 = 0.79) to a simple, standard “relaxation” simulation carried out in Rosetta, using either the new or old rotamer libraries. Our rotamer libraries deliver comparable results (R2 = 0.80), without requiring any fit parameters. In contrast, with the prior rotamer libraries, which possessed the erroneous α-peptide-like dependence on only φ and θ, performed quite poorly in comparison (R2 = 0.40) (Figure 7). This improvement suggests that a dependence on all three backbone angles is likely necessary for useful binding simulations.
Figure 7. The Fully Backbone-Dependent Rotamer Libraries Perform Considerably Better than the Old Model in Real Applications.

We compared experimental ΔΔ G values (kcal/mol) for mutations in a series of 314 helical p53 mimetics, which were assayed against the proteins mdm2 and mdm4, to computational predictions. The original predictions made by Schepartz (A) provided a standard of comparison; our Rosetta protocol performed poorly with the old (B) rotamer libraries but quite well using the rotamer libraries developed in this work (C).
Relative Rotamer Prevalence across β3-Leucine Backbone Cross-Sections
To visualize three-dimensional rotamer libraries in a useful way, we suggest plotting rotamer probability against two of the dependent main-chain dihedral angles, holding the third fixed. Three of these plots can be analyzed in combination to understand the neighborhood surrounding a backbone configuration of interest (Figure S1). To make comparable plots, one may employ the following Mathematica code:
anti_gaucheplus = Import[‘‘B3L_a_gp.txt’’, ‘‘Table’’];
gaucheminus_anti = Import[‘‘B3L_gm_a.txt’’, ‘‘Table’’];
interpolated_a_gp = Interpolation[anti_gaucheplus];
interpolated_gm_a = Interpolation[gaucheminus_anti];
Plot3D[{interpolated_a_gp[phi, psi], interpolated_gm_a[phi, psi]}, {phi, −180, 180}, {psi, −180, 180}, AxesLabel –> {‘‘phi’’, ‘‘psi’’}, AspectRatio –> 1, Ticks –> {{−180, −120, −60, 0, 60, 120, 180}, {−180, −120, −60, 0, 60, 120, 180}, {0.2, 0.4, 0.6, 0.8, 1.0}}]
The above code imports text files containing the probability of the (a, g+) and (g−, a) rotamers, respectively, as a function of φ and ψ, holding θ fixed at −60°. Those three-column text files are imported as tables (lines 1 and 2), then interpolated as functions of two variables (lines 3 and 4), then finally plotted over the full range of φ and ψ.
Detailed Rotamer Recovery for a Structure of Interest
To provide some context about the methods employed, we provide a worked example of rotamer recovery for two particular PDB structures: PDB: 4BPI, a complex between a Puma BH3 α/β-peptide mimetic with Mcl-1, and PDB: 3G7A, an HIV gp41 bundle with an α/β-peptide mimic of the CHR domain. Neither was an example of particularly good performance; rather, both illustrate the challenges of working with relatively sparse native structural data.
The former structure contains four β3 residues: β3D, β3R, β3Q, and β3E; while the latter contains two β3-homoglutamate residues. First, we had to revise atom and residue names in Rosetta to match Rosetta’s nomenclature, to make sure that no atoms were wrongly detected as missing, which would have resulted in coordinates being built from ideal. In fact, we discard the β3E residue from PDB: 4BPI on these grounds, as it is involved in binding a cadmium atom in the native structure, and one of the β3E residues from PDB: 3G7A, as its Cγ and Oδ atoms are missing. Second, we built rotamer sets for each residue. The closest interpolated rotamers to each experimental rotamer are given in Table S1.
Native conformations so far from the theoretical sp3-sp3 optimum, especially in mostly surface-exposed positions, seem surprising. Reproducing them perfectly would be an inappropriate burden for a rotamer library; this library’s attempts to do so largely hinge on very low-probability rotamers, which would likely be rebuilt in most simulations. Indeed, their distance from optima suggests that they could reflect structural errors that remain un-corrected simply because of the limited knowledge surrounding the preferred side-chain conformations of β-amino acids. The B factors for each β3 residue in these structures are quite high, from 45 to 70. Examining maps derived from the deposited structure factors, experimental evidence for these particular conformations seems either weak or absent entirely (Figure 6). Ironically, more side-chain electron density is present for the lower-resolution structure (PDB: 3G7A, at 2.8 Å, at least able to secure the relative position of the α and γ carbon atoms) than for the higher-resolution structure (PDB: 4BPI, at 1.98 Å).
Distinguishing Features of β-Amino Acid Rotamer Libraries versus Corresponding α-Amino Acid Rotamer Libraries
More than any other feature, rotamers for α- and β-amino acids are distinguished by steric clashes. As described throughout, many common rotamers compatible with an α-amino acid backbone are sterically incompatible with any β-amino acid backbone that shares φ and θ. A more minor influence is the absence of particular attractive interactions. For example, α-serine is well-known to form a hydrogen bond between its γ-H and its carbonyl oxygen in some backbone conformations, while that feature is largely absent in β3-serine: the six-membered hydrogen bonding ‘‘macrocycle’’ is replaced with a much less-favored seven-membered ‘‘ring.’’ In principle, β2-serine rotamer libraries would retain this backbone-dependent bias.
DISCUSSION
We developed and tested a model of backbone-dependent libraries of β-amino acid side-chain conformations. This model showed excellent agreement to simulations conducted at a higher level of theory as well as accurate recovery of experimental data, especially compared with previous models that borrowed α-amino acid conformations, without fitting additional scoring function parameters. We incorporated new code and new residue parameters into the Rosetta software suite. Integration with Rosetta enables our contributions to open up new possibilities in design, modeling, and structure determination protocols. Furthermore, users of the software may parameterize new β-amino acid side chains of interest and obtain their rotamer libraries. We anticipate an eventual need for higher-resolution rotamer libraries (with more backbone bins, especially in highly populated regions). Doing so demands particular attention for β-amino acids because their backbone-dependent libraries are dramatically larger, in both memory and on disk. To this end, superior β-amino acid rotamer libraries will require more sophisticated interpolation methods. Further efforts to create a framework suited for general heteropolymer modeling and design will include scoring function optimization to include alternative solvation and electrostatics models (Haberthur and Caflisch, 2008; Bazzoli et al., 2015), reference energies for design, and better methods for modeling hydrogen and halogen bonds. Re-refinement of structures containing β-amino acids will provide an experimental basis against which that scoring function might be optimized. Taken together, these efforts will create the groundwork for designing arbitrary polymers of α- and β-amino acids.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and Algorithms | ||
| Gaussian 09 Revision D.01 | Frisch et al., 2009 | https://gaussian.com |
| Open Babel | O’Boyle et al., 2011 | https://openbabel.org |
| PyMOL Molecular Graphics System, Version 1.8 | Schrödinger, LLC | https://pymol.org |
| Rosetta | Leaver-Fay et al., 2011 | https://rosettacommons.org |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rich Bonneau (rb133@nyu.edu).
METHOD DETAILS
β-peptide Rotamer Library Creation
β-peptide rotamer libraries were created using the MakeRotLib protocol in Rosetta (Figure 3). For each backbone bin (from −150° to 180°, i.e. 123 = 1728 combinations), acetylated and methylamidated amino acid structures were initialized with varied side chain conformations. Residues with one χ(β3Ser, β3Thr, β3Val) were sampled every degree; residues with two χ were sampled every 10° (1296 samples); residues with three (β3Met; β3Glu, β3Gln) every 15° (13824 samples) and four (β3Arg, β3Lys) χ angles every 30° (20736 samples).
The resulting conformations were minimized, holding all backbone bond geometry fixed but allowing all sidechain geometry, including bond lengths and angles, to vary, using a linear, monotone mini-mizer. The force field employed was the pre-existing mm_std energy function (Renfrew et al., 2012) augmented with previously developed terms to score bond lengths and angles. All bonded andnonbonded parameters employed in the mm_std energy function are derived from CHARMM; like in the standard Rosetta energy functions employed for canonical amino acids, mm_std uses the Lazaridis-Karplus solvation model (Lazaridis and Karplus, 1999). We clustered the resulting conformations using k-means clustering, with initial cluster centroids chosen to correspond to physical intuition and to the choices made by the 2010 Dunbrack rotamer libraries where possible (Shapovalov and Dunbrack, 2011) Specifically, every rotameric χ was given a centroid initialized to g-, a, or g+, while nonrotameric χ angles had centroids every 30°. (Symmetric nonrotameric χ angles such as aspartate’s second χ had six wells, 0° through 150°, while asymmetric nonrotameric χ angles such as asparagine’s second χ had twelve.) For each cluster, the energies for the rotamer sample closest to the cluster center are converted to Boltzmann factors and thereby to probabilities.
To provide complete support for scoring and sampling, each rotamer well is approximated as a Gaussian, as in the Dunbrack rotamer libraries. The dimensions of the cluster that results from optimizing these conformations are quite small and not particularly physically meaningful. Rather, we manually search around each rotamer center to find the angle deviation necessary to increase the conformational energy by 0.5 Rosetta energy units (Renfrew et al., 2012), which produces physically realistic rotamer wells with standard deviations on the same scale as the Dunbrack libraries. We also implemented functionality to provide a continuous treatment of terminal nonrotameric χ angles, as is possible in the 2010 Dunbrack libraries, thus permitting a smoother surface for energy minimization. In such rotamer libraries, created for β3D, β3E, β3F, β3H, β3N, β3Q, β3W, and β3Y, only the rotameric χ angles are clustered, while the final χ is sampled at 36 evenly spaced positions (5° increments for symmetric χ; 10° increments otherwise) and the energies in question are converted to probabilities similarly. These rotamer libraries may be visualized to evaluate how well the major rotamers preferred at familiar backbone conformations correspond to physical intuition (Supplemental Information, Figure S1).
Evaluation of Clash-Free Conformations
We sampled each amino acid at 5° intervals for every backbone dihedral angle. We minimized the sidechain degrees of freedom to ensure that the sidechain was not making any unavoidable clashes (For a fair comparison to residues with one or more χ angles, we provided alanine and β3A with a ‘fake’ χ to model the rotation of their methyl sidechains.) Then, we evaluated the distance between every pair of atoms and determined whether they were making a severe clash by comparing to 0.6 times the sum of the two atoms’ van der Waals radii. For proline, we also disqualified conformations whose five-membered rings had been broken. Thus, the value for α-amino acids represents a percentage, area-over-area, while for β-amino acids, the corresponding value is a percentage, volume-over-volume.
Structure Optimization via Gaussian and Comparison to the Molecular Mechanics Scoring Function
Using Gaussian09 revision D, we initialized acetylated and N-methylamidated structures of β3Trp, β3Leu, β3Ile, and β3Ser to two distinct and structurally valuable backbone conformations: (−140°, 60°, −120°), intended to be near stable conformations for the 314 helix and the chimeric α3β-helix, and (65°, 75°, −140°), an extended state that can form pleated sheets like anα-amino acid forms β-sheets. We initialized a 10° grid of side chain conformations (i.e. 1296 points per two-χ residue; 36 points for β3Ser) and carried out energy optimization at the HF/6-311+G(2d,p) level of theory, followed by single point energy calculation at B3LYP/6-311+G(2d,p). Subsequently, we used OpenBabel to generate PDB format files from the resulting Gaussian output and scored each structure using the molecular mechanics scoring function within Rosetta. To analyze the similarity of the probability distributions, we evaluated the Bhattacharyya distance (see STAR Methods). Some deviations are, in fact, desirable and expected, as gas-phase quantum mechanics ought to deviate from molecular mechanics models that include solvation terms. The worst fit is for the helical conformation of β3Trp, where the MM scoring function appears to favor a g- χ1 dihedral. This result may partially result from Rosetta’s smaller van der Waals radii for hydrogen, as the g- rotamer may feature some minor clashes. The next worst fit is for β3Ser, whose affinity for the g- rotamer in both backbone conformations relies in part upon a seven-membered intramolecular hydrogen bond that mm_std does not score and that the QM method may over-value due to the vacuum state. Importantly, even in the cases where there is disagreement, the best QM rotamers are always represented in the MM rotamer set and therefore will be sampled in simulations.
Comparison to PDB Rotamers
For each crystal structure rotamer, we apply Rosetta’s tricubic interpolation to the nearest back-bone bins to obtain an interpolated set of rotamers for the exact backbone dihedrals from the crystal structure. We measure the minimum RMS distance in degrees from a rotamer built for that position to the rotamer in the crystal structure and average that value for each residue type. Figures from PDB structures are ray-traced in PyMOL. Electron density is generated from structure factors using phenix.maps or downloaded from the Uppsala Electron Density Server and visualized in PyMOL using the isomesh command at two standard deviations within 1.6Å of atoms.
Rosetta Simulations to Predict Binding Affinity
We conducted simulations of the 314 helices studied experimentally beginning with the same crystal structures of mdm2 and mdm4 as employed in the original computational study (PDB codes 2GV2 and 2Z5T, respectively). We generated point mutant structures of each 314 helix ligand by mutating a position then repacking and minimizing an 8Å shell around the mutated residue. We conducted a simple relaxation simulation, generating 1000 structures, to diversify the ensemble of β-peptide/protein complex structures, followed by interface scoring using the dGcross score term computed by the InterfaceAnalyzerMover. Separate sets of simulations were conducted with the old rotamer libraries (the canonicalα-amino acid rotamers applied to the first two mainchain torsions) and with those developed in this work.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparing MM- and QM-derived Probability Distributions by Evaluating the Bhattacharyya Distance
The Bhattacharyya distance
where p and q are two probability distributions and X is their domain (Bhattacharyya, 1943) is a generalization of the Mahalanobis distance that does not assume equal standard deviations; its range is [0, ∞), where zero reflects identical distributions and where entirely distinct distributions (two different delta functions, for example) have unbounded distance. Values from 0.04 to 0.27, as observed for these rotamer libraries, represent extremely similar distributions.
Comparing the Accuracy of These Rotamer Libraries to Dunbrack Rotamer Libraries for the α-amino Acids
It is challenging to directly compare two rotamer library models of different methodology and generality. This challenge, however, does not preclude the assessment of the fits of Figure 5. The Dunbrack libraries model rotamer wells as Gaussians with some standard deviation. We can employ these standard deviations to provide a natural scale for our observed average RMS errors. Let us assume that a Dunbrack rotamer is exactly centered on the ‘‘true’’ rotamer centroid for each χ angle. A typical standard deviation for such a Gaussian rotamer well is 8°. The expected RMS deviation from that rotamer well for one to four χ angles is 6.4°, 10°, 12.8°, and 15.1°. The standard deviation for this RMS deviation statistic is around 5° in each case. Thus, the performance we observe reflects the same underlying variability of protein and peptide structures that the Dunbrack libraries do.
Please describe here all of the statistical analysis and software used. We ask authors to indicate in this section where all of the statistical details of experiments can be found (e.g., in the figure legends, figures, Results, etc.), including the statistical tests used, exact value of n, what n represents (e.g., number of animals, number of cells, etc.), definition of center, and dispersion and precision measures (e.g., mean, median, SD, SEM, confidence intervals). Also please summarize in this section how significance was defined, the statistical methods used to determine strategies for randomization and/or stratification, sample size estimation, and inclusion and exclusion of any data or subjects, as well as any methods used to determine whether the data met assumptions of the statistical approach.
DATA AND SOFTWARE AVAILABILITY
Applications to create amino acid rotamer parameter files and rotamer libraries, as well as the resulting rotamer libraries, are distributed as part of the Rosetta software suite, which is free for academic use (https://rosettacommons.org).
The rotamer libraries generated are available as supplemental data (Data S1).
Supplementary Material
Highlights.
Constructed rotamer libraries for β-amino acids
Validated against quantum mechanical calculations
Good reproduction of experimental side-chain conformations
Compatible with the Rosetta software suite
Acknowledgments
A.M.W. is supported by an NYU Dean’s Dissertation Fellowship. P.S.A. thanks the NIH (R01GM073943) for financial support of this work. R.B. and P.D.R are supported by the Simons Foundation, Center for Computational Biology.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes one figure, one table, and one data file and can be found with this article online at https://doi.org/10.1016/j.str.2017.09.005.
AUTHOR CONTRIBUTIONS
Conceptualization, A.M.W. and R.B.; Methodology, A.M.W., P.D.R., and R.B.; Investigation, A.M.W.; Writing – Original Draft, A.M.W.; Writing – Review & Editing, A.M.W., P.D.R., R.B., and P.S.A.; Funding Acquisition, P.S.A. and R.B.; Resources, P.S.A. and R.B.; Supervision, P.S.A. and R.B.
References
- Appella DH, Christianson LA, Karle IL, Powell DR, Gellman SH. β-Peptide foldamers: robust helix formation in a new family of β-amino acid oligomers. J Am Chem Soc. 1996;118:13071–13072. [Google Scholar]
- Bautista AD, Stephens OM, Wang L, Domaoal RA, Anderson KS, Schepartz A. Identification of a β3-peptide HIV fusion inhibitor with improved potency in live cells. Bioorg Med Chem Lett. 2009;19:3736–3738. doi: 10.1016/j.bmcl.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoli A, Kelow SP, Karanicolas J. Enhancements to the Rosetta energy function enable improved identification of small molecules that inhibit protein-protein interactions. PLoS One. 2015;10:1–18. doi: 10.1371/journal.pone.0140359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A. On a measure of divergence between two statistical populations defined by their probability distributions. Bulletin of the Calcutta Mathematical Society. 1943;35:99–109. [Google Scholar]
- Bonneau R, Tsai J, Ruczinski I, Chivian D, Rohl C, Strauss CE, Baker D. Rosetta in CASP4: progress in ab initio protein structure prediction. Proteins. 2001;(Suppl 5):119–126. doi: 10.1002/prot.1170. [DOI] [PubMed] [Google Scholar]
- Bower MJ, Cohen FE, Dunbrack RL., Jr Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool. J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- Cheloha RW, Sullivan JA, Wang T, Sand JM, Sidney J, Sette A, Cook ME, Suresh M, Gellman SH. Consequences of periodic α-to-β3 residue replacement for immunological recognition of peptide epitopes. ACS Chem Biol. 2015;10:844–854. doi: 10.1021/cb500888q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng RP, Gellman SH, DeGrado WF. β-Peptides: from structure to function. Chem Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods. 2013;10:74–76. doi: 10.1038/nmeth.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YJ, Huck BR, Christianson LA, Stanger HE, Krauthäuser S, Powell DR, Gellman SH. Stereochemical control of hairpin formation in β-peptides containing dinipecotic acid reverse turn segments. J Am Chem Soc. 2000;122:3995–4004. [Google Scholar]
- Cromm PM, Spiegel J, Grossmann TN. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem Biol. 2015;10:1362–1375. doi: 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- Denton EV, Craig CJ, Pongratz RL, Appelbaum JS, Doerner AE, Narayanan A, Shulman GI, Cline GW, Schepartz A. A β-peptide agonist of the GLP-1 receptor, a class B GPCR. Org Lett. 2013;15:5318–5321. doi: 10.1021/ol402568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL, Jr, Karplus M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- Feig M, Rotkiewicz P, Kolinski A, Skolnick J, Brooks CL., 3rd Accurate reconstruction of all-atom protein representations from side-chain-based low-resolution models. Proteins. 2000;41:86–97. doi: 10.1002/1097-0134(20001001)41:1<86::aid-prot110>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, et al. Gaussian 09 Revision D.01. Gaussian Inc; 2009. [Google Scholar]
- Gademann K, Seebach D. Preparation and NMR structure of the cyclo-β-tripeptide [β3-HGlu]3 in aqueous solution: a new class of enterobactintype C3-symmetrical ligands?, Preliminary Communication. Helv Chim Acta. 1999;82:957–962. [Google Scholar]
- Haase HS, Peterson-Kaufman KJ, Lan Levengood SK, Checco JW, Murphy WL, Gellman SH. Extending foldamer design beyond α-helix mimicry: α/β-peptide inhibitors of vascular endothelial growth factor signaling. J Am Chem Soc. 2012;134:7652–7655. doi: 10.1021/ja302469a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberthur U, Caflisch A. FACTS: fast analytical continuum treatment of solvation. J Comput Chem. 2008;29:701–715. doi: 10.1002/jcc.20832. [DOI] [PubMed] [Google Scholar]
- Hayen A, Schmitt MA, Ngassa FN, Thomasson KA, Gellman SH. Two helical conformations from a single foldamer backbone: ‘‘split personality’’ in short α/β-peptides. Angew Chem Int Ed. 2004;43:505–510. doi: 10.1002/anie.200352125. [DOI] [PubMed] [Google Scholar]
- Hintze BJ, Lewis SM, Richardson JS, Richardson DC. MolProbity’s ultimate rotamer-library distributions for model validation. Proteins. 2016;84:1177–1189. doi: 10.1002/prot.25039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne WS, Price JL, Keck JL, Gellman SH. Helix bundle quaternary structure from α/β-peptide foldamers. J Am Chem Soc. 2007;129:4178–4180. doi: 10.1021/ja070396f. [DOI] [PubMed] [Google Scholar]
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Johnson LM, Gellman SH. α-Helix mimicry with α/β-peptides. Methods Enzymol. 2013;523:407–429. doi: 10.1016/B978-0-12-394292-0.00019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LM, Barrick S, Hager MV, McFedries A, Homan EA, Rabaglia ME, Keller MP, Attie AD, Saghatelian A, Bisello A, et al. A potent α/β-peptide analogue of GLP-1 with prolonged action in vivo. J Am Chem Soc. 2014;136:12848–12851. doi: 10.1021/ja507168t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Tirado-Rives J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc Natl Acad Sci USA. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothiwale S, Mendenhall JL, Meiler J. BCL::Conf: small molecule conformational sampling using a knowledge based rotamer library. J Cheminform. 2015;7:47. doi: 10.1186/s13321-015-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. Helical β-peptide inhibitors of the p53-hDM2 interaction. J Am Chem Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- Kussell E, Shimada J, Shakhnovich EI. A structure-based method for derivation of all-atom potentials for protein folding. Proc Natl Acad Sci USA. 2002;99:5343–5348. doi: 10.1073/pnas.072665799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenhan JM, Guzei IA, Gellman SH. Parallel sheet secondary structure in β-peptides. Angew Chem Int Ed. 2003;42:2402–2405. doi: 10.1002/anie.200350932. [DOI] [PubMed] [Google Scholar]
- Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel GA, Frank RC, Horne WS. Hairpin folding behavior of mixed α/β-peptides in aqueous solution. J Am Chem Soc. 2011;133:4246–4249. doi: 10.1021/ja2002346. [DOI] [PubMed] [Google Scholar]
- Levitt M. A simplified representation of protein conformations for rapid simulation of protein folding. J Mol Biol. 1976;104:59–107. doi: 10.1016/0022-2836(76)90004-8. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- Michel J, Harker EA, Tirado-Rives J, Jorgensen WL, Schepartz A. In silico improvement of β3-peptide inhibitors of p53·hDM2 and p53·hDMX. J Am Chem Soc. 2009;131:6356–6357. doi: 10.1021/ja901478e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell A, Blosser S, Arora P. Systematic targeting of protein-protein interactions. Trends Pharmacol Sci. 2016;37:702–713. doi: 10.1016/j.tips.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molski MA, Goodman JL, Chou FC, Baker D, Das R, Schepartz A. Remodeling a β-peptide bundle. Chem Sci. 2013;4:319–324. [Google Scholar]
- Moon H, Lim H. Synthesis and screening of small-molecule α-helix mimetic libraries targeting protein-protein interactions. Curr Opin Chem Biol. 2015;24:38–47. doi: 10.1016/j.cbpa.2014.10.023. [DOI] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminformatics. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, DiMaio F, Baker D. CASP11 refinement experiments with ROSETTA. Proteins. 2015;84:314–322. doi: 10.1002/prot.24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Horne WS, Gellman SH. Discrete heterogeneous quaternary structure formed by α/β-peptide foldamers and α-peptides. J Am Chem Soc. 2007;129:6376–6377. doi: 10.1021/ja071203r. [DOI] [PubMed] [Google Scholar]
- Qiu JX, Petersson EJ, Matthews EE, Schepartz A. Toward β-amino acid proteins: a cooperatively folded β-peptide quaternary structure. J Am Chem Soc. 2006;128:11338–11339. doi: 10.1021/ja063164+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raguse TL, Porter EA, Weisblum B, Gellman SH. Environment-independent 14-helix formation in short β-peptides: striking a balance between shape control and functional diversity. J Am Chem Soc. 2003;125:5592–5593. doi: 10.1021/ja0341485. [DOI] [PubMed] [Google Scholar]
- Renfrew PD, Butterfoss GL, Kuhlman B. Using quantum mechanics to improve estimates of amino acid side chain rotamer energies. Proteins. 2008;71:1637–1646. doi: 10.1002/prot.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrew PD, Choi EJ, Bonneau R, Kuhlman B. Incorporation of noncanonical amino acids into rosetta and use in computational protein-peptide interface design. PLoS One. 2012;7:e32637. doi: 10.1371/journal.pone.0032637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrew PD, Craven TW, Butterfoss GL, Kirshenbaum K, Bonneau R. A rotamer library to enable modeling and design of peptoid foldamers. J Am Chem Soc. 2014;136:8772–8782. doi: 10.1021/ja503776z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky JD, Schmitt MA, Lee HS, Umezama N, Wang S, Tomita Y, Gellman SH. Chimeric (α/β+α)-peptide ligands for the BH3-recognition cleft of Bcl-XL: critical role of the molecular scaffold in protein surface recognition. J Am Chem Soc. 2005;127:11966–11968. doi: 10.1021/ja053678t. [DOI] [PubMed] [Google Scholar]
- Schmitt MA, Weisblum B, Gellman SH. Unexpected relationships between structure and function in α, β-peptides: antimicrobial foldamers with heterogeneous backbones. J Am Chem Soc. 2004;126:6848–6849. doi: 10.1021/ja048546z. [DOI] [PubMed] [Google Scholar]
- Schmitt MA, Choi SH, Guzei IA, Gellman SH. New helical foldamers: heterogeneous backbones with 1:2 and 2:1 α:β-amino acid residue patterns. J Am Chem Soc. 2006;128:4538–4539. doi: 10.1021/ja060281w. [DOI] [PubMed] [Google Scholar]
- Scouras AD, Daggett V. The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci. 2011;20:341–352. doi: 10.1002/pro.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach D, Gardiner J. β-peptidic peptidomimetics. Acc Chem Res. 2008;41:1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- Seebach D, Ciceri PE, Overhand M, Jaun B, Rigo D, Oberer L, Hommel U, Amstutz R, Widmer H. Probing the helical secondary structure of short-chain β-peptides. Helv Chim Acta. 1996;79:2043–2066. [Google Scholar]
- Seebach D, Abele S, Sifferlen T, Hänggi M, Gruner S, Seiler P. Preparation and structure of β-peptides consisting of geminally disubstituted β2,2- and β3,3-amino acids: a turn motif for β-peptides. Helv Chim Acta. 1998;81:2218–2243. [Google Scholar]
- Seebach D, Abele S, Gademann K, Jaun B. Pleated sheets and turns of β-peptides with proteinogenic side chains. Angew Chem Int Ed. 1999;38:1595–1597. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1595::AID-ANIE1595>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Seebach D, Sifferlen T, Bierbaum DJ, Rueping M, Jaun B, Schweizer B, Schaefer J, Mehta AK, O’Connor RD, Meier BH, et al. Isotopically labelled and unlabelled β-peptides with geminal dimethyl substitution in 2-position of each residue: synthesis and NMR investigation in solution and in the solid state. Helv Chim Acta. 2002;85:2877–2917. [Google Scholar]
- Shandler SJ, Shapovalov MV, Dunbrack RL, Jr, DeGrado WF. Development of a rotamer library for use in β-peptide foldamer computational design. J Am Chem Soc. 2010;132:7312–7320. doi: 10.1021/ja906700x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalov MV, Dunbrack RL., Jr A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19:844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999;37:171–176. doi: 10.1002/(sici)1097-0134(1999)37:3+<171::aid-prot21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- Smith BJ, Lee EF, Checco JW, Evangelista M, Gellman SH, Fairlie WD. Structure-guided rational design of α/β-peptide foldamers with high affinity for BCL-2 family prosurvival proteins. ChemBioChem. 2013;14:1564–1572. doi: 10.1002/cbic.201300351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towse CL, Rysavy SJ, Vulovic IM, Daggett V. New dynamic rotamer libraries: data-driven analysis of side-chain conformational propensities. Structure. 2016;24:187–199. doi: 10.1016/j.str.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PS, Nguyen JB, Schepartz A. Design and high-resolution structure of a β3-peptide bundle catalyst. J Am Chem Soc. 2014;136:6810–6813. doi: 10.1021/ja5013849. [DOI] [PubMed] [Google Scholar]
- Watkins A, Arora P. Side-chain conformational preferences govern protein-protein interactions. J Am Chem Soc. 2016;138:10386–10389. doi: 10.1021/jacs.6b04892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werder M, Hauser H, Abele S, Seebach D. β-Peptides as inhibitors of small-intestinal cholesterol and fat absorption. Helv Chim Acta. 1999;82:1774–1783. [Google Scholar]
- Zwanzig RW. High-temperature equation of state by a perturbation method. J Chem Phys. 1954;22:1420–1426. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.