

Abstract
We report secondary cutaneous infections in the mouse papillomavirus (MmuPV1)/mouse model. Our previous study demonstrated that cutaneous MmuPV1 infection could spread to mucosal sites. Recently, we observed that mucosal infections could also spread to various cutaneous sites including the back, tail, muzzle and mammary tissues. The secondary site lesions were positive for viral DNA, viral capsid protein and viral particles as determined by in situ hybridization, immunohistochemistry and transmission electron microscopy analyses, respectively. We also demonstrated differential viral production and tumour growth at different secondarily infected skin sites. For example, fewer viral particles were detected in the least susceptible back tissues when compared with those in the infected muzzle and tail, although similar amounts of viral DNA were detected. Follow-up studies demonstrated that significantly lower amounts of viral DNA were packaged in the back lesions. Lavages harvested from the oral cavity and lower genital tracts were equally infectious at both cutaneous and mucosal sites, supporting the broad tissue tropism of this papillomavirus. Importantly, two secondary skin lesions on the forearms of two mice displayed a malignant phenotype at about 9.5 months post-primary infection. Therefore, MmuPV1 induces not only dysplasia at mucosal sites such as the vagina, anus and oral cavity but also skin carcinoma at cutaneous sites. These findings demonstrate that MmuPV1 mucosal infection can be spread to cutaneous sites and suggest that the model could serve a useful role in the study of the viral life cycle and pathogenesis of papillomavirus.
Keywords: Mouse papillomavirus, mucosal infection, secondary infection, skin carcinoma, viral copy number, Q-PCR
Introduction
Human papillomaviruses (HPVs) are associated with both cutaneous and mucosal infections, as well as cancer development [1–3]. Because of the species-specificity of papillomaviruses, no laboratory animals support HPV infections [4]. Several animal models, including the cottontail rabbit papillomavirus (CRPV) model, have played a crucial role in elucidating viral pathogenesis and virus–host interactions in recent decades [4–9]. The CRPV/rabbit system mimics high-risk HPV infection [10, 11]. However, the domestic rabbit is not a natural host for CRPV infection, and minimal levels of infectious virions are present in the infected tissues [12–14]. A natural infection animal model that mimics high-risk HPV disease would be useful to examine the natural history of viral infection and cancer development [4].
Recently, a novel mouse papillomavirus (MmuPV1) was identified [15]. MmuPV1 induces lesions at both cutaneous and mucosal sites in immunocompromised mice and in some immunocompetent mice [16–23]. Interestingly, ultraviolet irradiation suppressed host immunity and accelerated MmuPV1- associated ear skin carcinoma development [18]. This finding suggests that MmuPV1 may be a highly oncogenic virus in immunocompromised hosts. Our previous studies demonstrated for the first time that infection initiated at cutaneous sites could spread to mucosal sites [24]. Later we confirmed that viral shedding in the oral cavity, as well as in the vaginal and anal tracts, could be tracked by Q-PCR. To follow disease progression in these mucosally infected mice, we have monitored our animals up to 10 months post-primary infection. We observed secondary cutaneous infections at multiple sites, appearing at about 8 weeks post-primary mucosal infections. In this study, we report that the mucosally infected mice displayed secondary cutaneous papillomas at various skin sites including the muzzle, tail, back, forearm, axilla (armpit) and mammary gland. The virus shed at mucosal sites (oral and vaginal) is infectious and able to induce tumour growth at multiple cutaneous and mucosal sites. Importantly, secondary forearm lesions of two different mice developed into micro-invasive squamous cell carcinomas at about 9.5 months following mucosal infections. These findings further confirm the malignant potential of MmuPV1 infections at cutaneous sites and therefore suggest that the mouse papillomavirus model can be used as a skin cancer model.
Results
Mucosal infection in the oral cavity, the anus and lower genital tract
Two anally infected, four orally infected and four vaginally infected nude mice housed in pairs in separate cages were tracked for viral infection as described in our previous study. Viral DNA was detected at infected mucosal sites by Q-PCR [22]. These mice were maintained for up to 10 months post-infection to follow disease progression and to monitor secondary infections. We observed that oral lavage via pipette was not optimal for DNA detection, as described in our previous study [22]. In the current experiments, we used a swab to collect the oral samples and this collection method has improved the consistency of viral DNA detection in the oral cavity. To repeat the oral infections, we infected four additional nude mice (14–6L and R and 15–6L and R) at the back of the tongue and monitored them for viral presence (Fig. 1). This new sampling method has shown increased sensitivity and, as a result, viral DNA could be detected in the oral cavity as early as week 11 post-infection; high titres were reached by week 18 post-infection (Fig. 1a). We also confirmed the presence of viral DNA at the base of the tongue and involving the circumvallate papilla (CVP) and adjacent ducts of von Ebner’s glands, as shown in a previous study [25]. This determination was made by in situ hybridization following the termination of the experiment at week 23 post-infection (Fig. 1b). Viral DNA copy numbers in the oral cavity were found to be comparable to those in the anal tract but lower than those in the vaginal tract in the infected animals at a given time post-infection (Fig. 1c).
Fig. 1.

Viral detection in orally infected mice. Viral DNA was detected by SYBR green Q-PCR from oral swabs collected at different time points post-infection (a) and by in situ hybridization (b) (post sacrifice), showing involvement of the circumvallate papilla (CVP) and adjacent ducts of von Ebner’s glands at the base of the tongue as reported previously [25]. An example of viral DNA copy numbers detected from the vaginal (V), anal (A) and oral (O) sites at week 10 post-primary infection (c). Much higher DNA copies were detected in the vaginal lavages when compared with those at the anal and oral sites.
Mice infected at mucosal sites developed lesions at numerous cutaneous sites
Progeny virus made at the productive stage of the viral life cycle arises within the terminally differentiating compartment of stratified squamous epithelia [26, 27]. Our previous study in nude mice demonstrated secondary infections of the mucosal sites following MmuPV1 infection at cutaneous sites [24]. We later demonstrated viral shedding at different mucosal sites including oral, anal and the lower genital tract [22]. The mucosally infected animals in the current study were kept for up to 10 months to assess disease progression and were monitored weekly. A photo log was maintained for each animal and regular lavages were carried out. In the course of our observations, we noted secondary cutaneous lesions at multiple sites including the back, antebrachium (forearm), hind leg, axilla (armpit) and mammary gland in addition to frequent infections of the muzzle and tail. These lesions began to appear in a subset of animals by week eight post-infection and were detected in all mucosally infected animals by week 18 (Fig. 2).
Fig. 2.
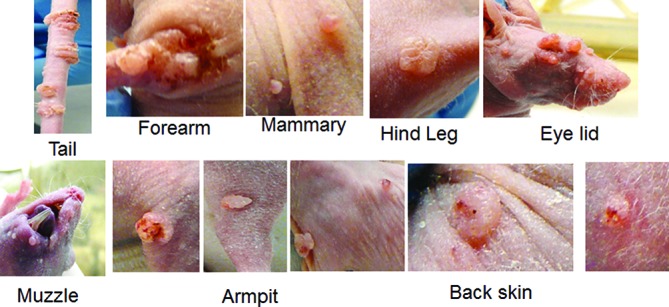
Secondary infection at various cutaneous sites of mucosally infected mice. Lesions were detected in the skin of the back, forearm, hind leg, armpit and tail as well as the mammary gland. Secondary muzzle lesions appeared at week nine post-infection in nude mice infected with MmuPV1 in the tongue and the anal tract, but not until week 18 post-infection in nude mice infected with MmuPV1 in the vaginal tract.
For experimental infection at cutaneous sites, we usually wound sites before viral infection [24]. It normally takes two to three weeks to visualize tumours at the infected tail and muzzle sites (our unpublished observations). For secondary cutaneous infections in these mucosally infected animals, one orally infected mouse showed a muzzle lesion at week nine post-infection, and the lesions increased in severity up until week 32 when the animal was sacrificed. (Fig. 3a, b). Similar results were observed in four other animals originally infected orally (Fig. 3c–e). Two anally infected mice also showed visible muzzle lesions at week nine post-infection (Fig. 3f). Interestingly, although no visible muzzle lesions were found in vaginally infected mice until week 18 post-infection, three out of four infected mice showed visible lesions on the tail by week 10 (Fig. 3g). Our more recent study again demonstrated secondary infections at both the muzzle and tail as early as week nine in mice originally infected in the anal canal and the vaginal tract (data not shown). In the limited number of animals tested in the current study we could not detect any definitive pattern of appearance of secondary infections at different cutaneous sites. Unlike experimental tail infections for which lesions usually appeared at the wounded sites within three weeks post-infection (Fig. 3h) and grew to become confluent, the secondary infections were always more scattered (Fig. 3g).
Fig. 3.
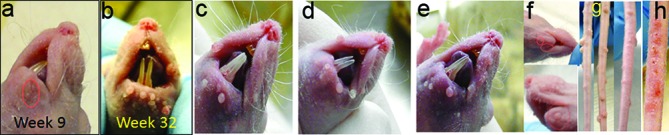
Secondary infections at muzzles and tails in mucosally infected mice. Secondary muzzle lesions were detected at week nine post-infection in a nude mouse infected with MmuPV1 in the tongue, and continued to grow up until sacrifice at week 32 post-infection (a, b). Muzzle lesions were found in three additional orally infected mice (c–e) and two anally infected mice (f). Scattered tail lesions were also found in three of four vaginally infected mice at week 18 post-infection (g). Localized lesions were found at the experimentally infected mouse tail at week three post-infection (h).
Differential virus production was observed in different cutaneous sites
Consistent with a previous study [28], we observed the back skin to be less susceptible to experimental infection and virus-induced tumour growth when compared to the tail and muzzle. The secondary back lesions were examined for viral presence by in situ hybridization and immunohistochemistry (Fig. 4). In contrast to a previous study in which L1 was reported to be present in the cytoplasm of back skin lesions [28], we detected L1 in the nucleus when using a commercially available anti-group-specific antibody (GSA) (Fig. 5). The same result was observed using an in-house specific anti-MmuPV1 L1 monoclonal antibody (MPV.B9) (Fig. 5a, b with insert). The vaginal samples clearly showed nuclear L1 expression, confirming the findings of our previous studies (Fig. 5c–d).
Fig. 4.
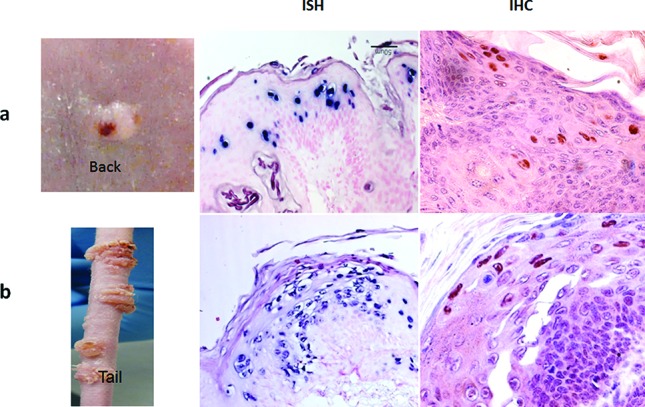
Viral detection at back sites following mucosal infection. Panel A (the back lesion) and panel B (the tail lesion) are shown here. These lesions were positive for viral DNA by in situ hybridization (ISH) and for viral capsid protein by immunohistochemistry (IHC). Virus can be identified predominantly in the nuclei by immunohistochemistry using a polyclonal antibody (anti-group-specific antigen, GSA).
Fig. 5.
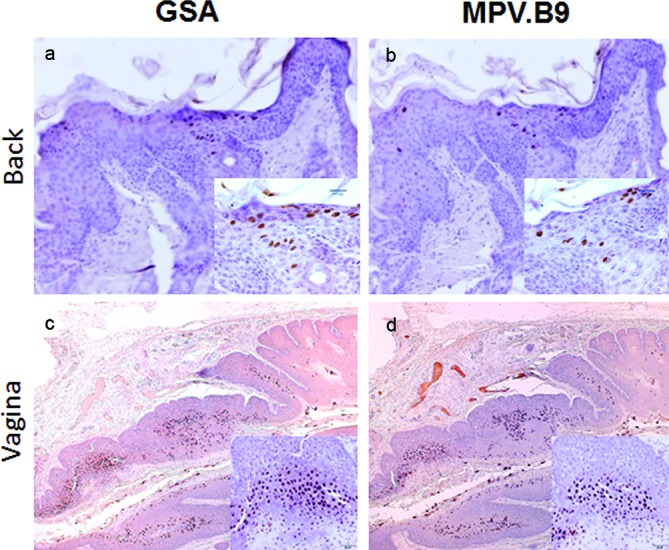
L1 protein was detected in the nuclei of cells of the back lesions. L1 capsid was stained in the back lesion with a polyclonal antibody (anti-group-specific antigen, GSA) and an in-house monoclonal antibody against MmuPV1 L1 (MPV.B9). Infected cells can be identified by the presence of L1 in the nuclei of the back lesion (a and b, 20×, and insert, 40×) and vaginally infected tissues (b and d, 4×, and insert, 20×) by immunohistochemistry.
We further examined viral particles in back, tail and muzzle lesions from secondary infections by transmission electronic microscopy (TEM) (Fig. 6a–c). We scanned many fields of view and identified only a few areas containing viral particles in the infected cells in back lesions when compared to those in muzzle and tail lesions (Fig. 6a). In muzzle and tail lesions, abundant viral particles were readily observed in infected cells (Fig. 6b–c). The viral DNA copy number was quantitated by SYBR Green Q-PCR according to the method described previously[22]. Interestingly, no significant difference was found in the viral DNA copy numbers between the tail and back lesions by Q-PCR analysis (Fig. 6d, P>0.05, one-way ANOVA).
Fig. 6.

Viral particle detection by transmission electron microscopy (TEM). From many fields of view, only a few infected cells, each containing relatively few viral particles, were identified in the back (a), while the muzzle (b) and the tail (c) lesions showed abundant infected cells filled with viral particles. No significant difference was found in the viral DNA copy numbers between the tail, the muzzle and the back lesions by Q-PCR analysis (d, P>0.05, one way ANOVA analysis). However, viral DNA packaging rates were significantly lower in the back lesions when compared to those in the muzzle and tail lesions (e, P<0.01, one way ANOVA analysis).
To investigate whether viral DNA packaging was impeded at the back sites, we homogenized selected lesions from back (N=2), tail (N=3) and muzzle (N=3) and divided each homogenate into two equal portions. One portion was treated with DNase I as described previously[29]. The other portion, without DNase I treatment, was used as the control. DNA was subsequently extracted from these samples and viral DNA was quantitated by Q-PCR [22]. The percentage of packaged DNA was calculated as the ratio between the viral DNA in DNase I treated and non-treated samples×100 %. Significantly less packaged viral DNA was detected in the back lesions when compared to the tail and muzzle lesions (Fig. 6e, P<0.01, one-way ANOVA). Therefore, viral DNA packaging was hindered in the back lesions.
Virus harvested from vaginal and oral lavage was infectious at cutaneous sites
To determine whether virus arising from mucosal sites was infectious at cutaneous sites, we harvested virus from oral and vaginal lavages of infected animals and tested them by inoculating two mice at both cutaneous and mucosal sites. Virus-induced tumours appeared at approximately week three post-infection at the muzzle and tail sites, an equivalent time observed for cutaneous infections using viral suspensions harvested from cutaneous (tail) lesions (Fig. 7a–d). Viral DNA copy numbers from mucosal (vaginal, anal and oral) sites infected with the viral suspension from either vaginal or oral lavages were also similar to those infected with tail lesion suspensions (Fig. 7e, f, P>0.05, one-way ANOVA analysis). Viral DNA was also detected at the vaginal, anal and oral tissues by in situ hybridization (Fig. 7g–i). Taken together, these findings demonstrate that the virus released from the mucosal sites is infectious in both mucosal and cutaneous sites.
Fig. 7.

Viral infection with vaginal or oral lavage samples. Representative muzzle (a, b) and tail (c, d) lesions induced by either vaginal lavage extracts (a, c) or oral lavage extract (b, d) in the outbred nude mouse at 8.5 months post-infection. The lesions became evident around week three post-infection and continued to grow over the period of observation. The lesions were comparable to those induced by the virus harvested from the tail lesions used in our previous studies. (e) Detection of viral DNA by SYBR Green Q-PCR analysis from mucosal [vaginal (V), anal (A) and oral (O)] sites infected with viral suspension from tail lesion (control), oral or vaginal lavages. Comparable viral DNA copy numbers were found in all three mucosal sites infected from the three sources of viruses (e and f, P>0.05, one-way ANOVA analysis). Lavage samples from naïve mice without MmuPV1 infection were used as the negative control. Viral DNA was detected at the vaginal (g), anal (h) and oral (i) infected sites by in situ hybridization.
Skin carcinoma developed at a secondarily infected skin site
A secondary cutaneous papilloma grew near the antebrachium (forearm) of one infected animal. When this mouse was sacrificed at 9.5 months post-primary infection (Fig. 2, forearm), histological analysis revealed that the lesion had progressed to micro-invasive squamous cell carcinoma (Fig. 8). The invasive cells were negative (Fig. 8f–h) for virus by both IHC and ISH, but some adjacent benign cells were positive (Fig. 8b–d). The malignant portion of the lesion displayed multiple foci of micro-invasion by chains of cells or single cells (Fig. 8f and supplementary Fig. 1). Abundant expression of viral transcripts was detected by RNA ISH using an RNA scope probe from the second exon of the transcript (Figs 8e and S2a, available in the online Supplementary Material). The cytoplasmic location of these transcripts can be appreciated with a higher magnification (Fig. S2a, 100×, the insert). We have also identified a region where mRNA signal was absent in normal tissue on one side while very strong in carcinomatous tissue on the other side (Fig. S2b). Increased P16INKa production has been used in the clinical diagnosis of HPV-associated cervical and oral cancers [30]. We examined P16INKa expression in the malignant lesion and in infected but untransformed tissue using immunohistochemistry. P16INKa expression was strong but no significant differences were noted (data not shown). When we used another biomarker, p16ARC, also known as the Actin-related protein 2/3 complex subunit 5 (ARPC5), a gene that contributes to cell migration and invasion [31, 32], we found significantly more p16ARC-positive cells at the tumour site (Fig. 8j) when compared to the adjacent normal skin site (Fig. 8k). However, we could not distinguish between malignant and infected but untransformed tissues.
Fig. 8.
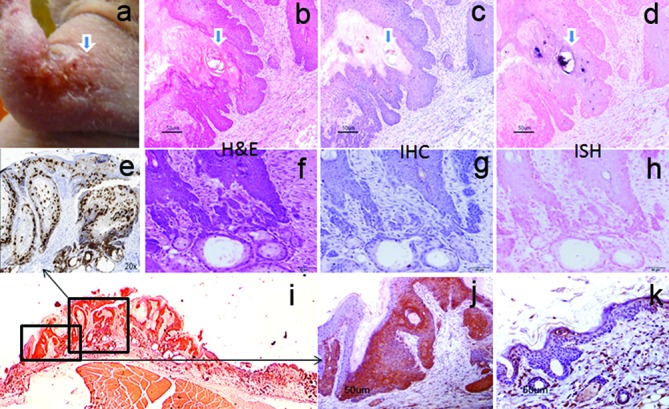
A micro-invasive squamous skin lesion in the athymic nude mouse skin at 9.5 months post-primary viral infection. Viral DNA and viral protein were detected at benign sites adjacent (c, d) to the malignant site where no viral DNA or viral protein was detected (g, h). However, viral transcript can be detected by RNA-ISH in all the sites (e, 20×). There are multiple foci of micro-invasion by chains of cells or single cells (squamous cell carcinoma, f, H and E, 20×). Significantly higher numbers of p16-ARC (ARPC5 (i, 1×), a gene that contributes to cell migration and invasion)-positive cells were identified in the tumour tissue (j, 20×) when compared to those at the adjacent normal skin (k, 20×).
Discussion
HPV infection at multiple anatomic sites in the same individual has been reported in human populations [33]. Most studies showed this co-infection to be in tissues of the same tropism, such as oral and anogenital sites [34]. However, some case studies have reported infections present in tissues of different tropisms [35–37]. For example, gamma HPV types were detected in both cutaneous and mucosal tissues [36, 38, 39]. However, due to the complicated nature of human studies, there is no well-documented evidence to determine whether these viruses spread from one site to another. Our MmuPV1 infection model, which displays a broad tissue tropism, is an ideal model for such studies. In an earlier study, we demonstrated that secondary mucosal infection resulted from the spread of the virus from primary cutaneous sites [24] despite the virus initially being reported to be cutaneotropic [15]. Later studies by another group confirmed our findings with respect to the ability of MmuPV1 to infect mucosal tissues [17, 40]. We have previously demonstrated that the vagina, anus and tongue are susceptible to MmuPV1 infection, and that infectious virus can be tracked longitudinally[21–23, 25]. For cutaneous infections, several laboratories have tested different sites including the back, tail, ear and muzzle [16, 17, 20, 28]. However, secondary cutaneous infections arising from primary mucosal or cutaneous infection have not been reported. We have followed our infected animals for more than 10 months post-infection to monitor potential malignant progression. This long-term follow-up strategy has allowed us to observe secondary infections, many of which did not appear until nine weeks post-infection or later.
Although simultaneous papillomavirus infection in multiple tissues is uncommon in the healthy human population, such infections can be found in immunocompromised patients indicating that host immunity plays a role in the control of viral disease [41, 42]. We speculate that the deficient adaptive T cell-mediated immunity present in the nude mice used in our study has allowed for relaxed tropism of viral infection and for enhanced tumour development [16, 17, 20]. However, the infectious life cycle is not equivalent at all anatomical sites in the same animal. For example, the back skin is least permissive among the cutaneous sites including muzzle and tail. In a different immune-compromised mouse strain (NOD/SCID), we detected no visible disease at cutaneous sites despite active infection at mucosal sites (unpublished observations). These observations suggest that, in addition to systemic host defence mechanisms, local host defence factors may play a role in the disease outcome at different sites and in different strains of mice.
We detected active viral shedding in the anogenital and vaginal tracts and the oral cavity in our previous studies[22]. In the current study, we noted that shedding from mucosal sites led to secondary cutaneous infections. These lesions appeared at different times in different locations, and we hypothesize that proximity to the primary infection site, grooming habits and viral titres may all have played a role. It should be noted that our Q-PCR analyses on vaginal lavage samples always yielded higher titres than did analyses on anal and oral lavages, indicating that more virus is shed at the lower genital tract when compared to the anal and oral sites (Fig. 1c).
In agreement with a previous study [28], we observed in earlier studies that the back skin is not the favoured site for primary MmuPV1 infection. However, in this study, we detected many secondary back lesions. This may be due to the possibility that our scarification protocol was not optimal for back skin infection. When we adapted our established pre-scarification method from the rabbit model to the mouse model, we observed that wound healing was much more rapid in mouse skin [43]. We therefore revised our mouse protocol to pre-wound 24 h before viral infection. This revised protocol has worked well for tail and muzzle infections but is less efficient for back skin infections. We postulate that simple scratching of back skin might be a more effective method to initiate back skin infections, since this is presumably how secondary infections are initiated in these mice.
The authors of a previous study reported that L1 expression was predominantly cytoplasmic in back lesions [28]. When we investigated L1 localization in back lesions from the secondary infections, we found that L1 expression was nuclear rather than cytoplasmic (Figs 4 and 5). This nuclear localization was demonstrated with a polyclonal antibody against group-specific antigen (GSA) and confirmed with an in-house monoclonal antibody against MmuPV1 L1. We did note, however, by transmission electron microscopy, that there were fewer viral particles in the back lesions when compared to those in muzzle and tail lesions, even though viral DNA replication at the back sites was comparable to that of the other skin sites. This led us to measure encapsidated and unencapsidated DNA in our samples and we found that significantly less viral DNA was packaged in back lesions when compared to that in the muzzle and tail lesions (Fig. 6e). These findings suggest that viral assembly might be compromised in the back skin. Interestingly, our first viral stock was generated from lesions generated by naked MmuPV1 DNA scratched into the back. We were able to harvest infectious virus from these small lesions and successfully use it to infect other sites, indicating that back sites are capable of producing infectious virions. Further investigation is planned to pinpoint factors that are involved in the observed site-specific differences.
Our previous studies demonstrated epithelial dysplasia in both the lower genital tract and oral cavity of MmuPV1-infected animals [23, 25]. In this study, we analysed two sections of a forearm lesion and found multiple foci of micro-invasion by squamous cells. Viral DNA and viral capsid protein were undetectable in the carcinoma cells, although DNA and capsid proteins were present in the adjacent non-invasive tumour cells. A similar observation was made by Uberoi et al. in a study in which MmuPV1-induced ear skin carcinomas developed following UVB irradiation and concomitant immunosuppression [18]. These lesions contained very little viral DNA. The authors commented that the demonstration of viral transcripts would be useful to confirm the viral origin of the tumour. However, that analysis was deferred for the future. In our study, we did demonstrate the presence of abundant transcripts thus confirming the correlation between viral infection and cancer development in our lesion. Investigation of E6/E7 transcripts may be undertaken at a later date. However, as virus integration may trigger or block host promoter activity at the integration site, the chimeric host/viral transcripts could be more or less depending on how and where the integration takes place. Thus, assuming integration has occurred, it is not definitive that the carcinoma would have a higher viral oncogene mRNA level vis-à-vis benign regions. We should note that while we did not check integration status in this specimen, RNA sequencing results on other samples from related mouse research have shown some virus–host fusions (data not shown).
The cancer discussed here, as well as a second nearly identical lesion from a different animal (data not shown), were evaluated by a board-certified veterinary pathologist and confirmed by others in his group. We wished to use an immunohistochemical marker to further distinguish the lesion and initially studied P16INKa, whose increased expression is commonly used to confirm oral and cervical cancers (31, 47). However, we could not detect increased expression in our tissues (data not shown). We then turned to p16ARC (ARPC5), a gene associated with cell migration and cell invasion. We found that it was significantly upregulated in the lesion and absent in the normal skin tissues (Fig. 8). However, we could not distinguish between the malignant and benign tissue on the basis of p16ARC expression. We note that Uberoi et al. used cytokeratin 14 to track cell invasion in their ear skin carcinomas [18]. We will consider investigating this marker in future studies. Taken together, our findings suggest that the MmuPV1/mouse model is a useful model not only to study benign disease but also to explore the mechanisms leading to both cutaneous and, potentially, mucosal PV-associated cancers.
Secondary infections have been reported in HPV patients [44, 45]. It is still unclear how long it takes for a subclinical infection to manifest as visible disease and what factors trigger the process. A major question remaining to be investigated relates to how the secondary infection occurs. While we have been inclined to favour such factors as grooming, another possibility is a systemic spread within the infected organism. We normally house the animals in pairs in our experiments; therefore cross-infections between the two cage mates are possible. However, we have also found secondary infections in mice housed singly, supporting both the hypothesis that self-grooming may contribute to secondary infections and that systemic spread may be the source of secondary infections.
Methods
Animals and viral infection
All mouse studies were approved by the Institutional Animal Care and Use Committee of Pennsylvania State University’s College of Medicine. Female outbred athymic nude mice (4–5 weeks old) were obtained from ENVIGO (Hsd: NU-Foxn1nu/nu, code 069). Mice were housed in pairs in sterile cages within sterile filter hoods and were fed sterilized food and water in the BL2 animal core facility. For mucosal infection, mice were synchronized at the dioestrus stage with Depo-medroxy progesterone acetate (Depo-Provera) 3–5 days before viral infection. Anal, vaginal and oral sites were wounded briefly as described in a previous publication [25]. Animals were monitored weekly and photos were recorded. Infection in mice hdas been shown to be most efficient one day post-scarification, and all infections followed this protocol [23]. Mice were sedated i.p. with 0.1 ml/10 g body weight with ketamine/xylazine mixture (100 mg/10 mg in 10 ml ddH2O), and 1×108 viral DNA genome equivalents were used for each infection[22].
Collection of lavages from mucosal sites for infection
Vaginal and anal lavages were collected using 25 µl of sterile 0.9 % NaCl introduced into the vaginal vault and the anal canal with a disposable filter tip. The rinse was gently pipetted in and out of the sites and stored at −20 °C. For the oral cavity, swabs wet with 25 µl of sterile 0.9 % NaCl were used to harvest infected cells (Puritan purflock Ultra, Puritan Diagnostics LLC). This method, rather than oral lavage, has been shown to generate higher DNA yields and better consistency in viral tits in our experiments.
Detection of viral DNA by SYBR Green Q-PCR
Biopsies were harvested from the cutaneous sites where the tumours were visible. DNA was extracted from these biopsies as well as from mucosal lavages using the DNeasy kit (QIAGEN). Viral copy number was quantitated by a SYBR green Q-PCR assay [22]. In brief, the linearized MmuPV1 genome DNA was used to generate the standard curve (FastStart Universal SYBR Green Master (Rox), Roche). The primer pairs (5′GCCCGA AGACA ACACCG CCACG3′ and 5′CCTCCGCCTC GTCCCCA AATGG 3′) that amplify E2 were used. Viral copy numbers in 1 µl of 50 µl DNA extract from a lavage sample were converted into equivalent DNA load using the formula 1 ng viral DNA=1.2×108 copy number (http://cels.uri.edu/gsc/cndna.html). The Q- PCR reactions were run in an Mx Pro-Mx3000P (Stratagene). Each reaction consisted of 7.5 µl of ultra-pure water, 7.5 µl of SYBR Green-PCR Master Mix (Roche) including 5 pmol of each primer and 2 µl of DNA template. PCR conditions were: initial denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s, 55 °C for 1 min and 72 °C for 1 min. All samples were tested in at least duplicates. Viral titres were calculated according to the standard curve [22]. We also calculated the difference in the cycle (Ct) between the 18S RNA gene and viral DNA (ΔCt). Fold change (2ΔCt) demonstrates the relative viral DNA load in some samples as described previously [22].
Detection of virus by in situ hybridization, immunohistochemistry and transmission electron microscopy
Biopsies were fixed in 10 % neutral buffered formalin and embedded in paraffin. Adjacent sequential sections were cut for haematoxylin and eosin (H and E) staining, in situ hybridization and immunohistochemistry [46]. A sub-genomic fragment of MmuPV1 (3913 bp EcoRV/BamHI fragment) was used as an in situ hybridization probe for the detection of MmuPV1 DNA in tissues. The probe was biotinylated using the random priming method and diluted in a hybridization cocktail described in previous work [25]. Access to target DNA was obtained with incubation with 0.2 mg ml−1 pepsin in 0.1 N HCl at 37 °C for 8 min. After thorough washing, the biotinylated probe was applied and heated to 95 °C for 5 min to achieve dissociation of target and probe DNA. Re-annealing was allowed to occur for 2 h at 37 °C. Target-bound biotin was detected using a streptavidin AP conjugate followed by colorimetric development in BCIP/NBT. For immunohistochemical (IHC) detection of L1 capsid, an in-house mouse monoclonal antibody against MmuPV1 L1 (MPV.B9) and a goat polyclonal anti-group-specific antibody (GSA) to a conserved region of L1 (ViroStat #5001) were used. Detection was achieved using enzyme-conjugated anti-mouse secondary or the ImmPRESS anti-goat IgG polymer system (Vector #MP-7405), respectively [23].
For transmission electron microscopy (TEM), the tissue was immersion-fixed for 24 h in Karnovsky's fixative (5 % glutaraldehyde/4 % paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.3). Following fixation the tissue was washed in 0.1 M sodium cacodylate buffer, post-fixed in buffered 1% osmium tetroxide/1.5 % potassium ferrocyanide, washed again with buffer, dehydrated in a graded series of ethanol, transferred to propylene oxide and embedded in Spurr low-viscosity resin. Sections (70–90 nm) were cut with a diamond knife, mounted on copper grids and stained with 2 % aqueous uranyl acetate and lead citrate. The sections were examined under a JEOL JEM 1400 electron microscope. The pictures were recorded [22].
Lesion homogenization, DNase treatment and viral DNA isolation
Two lesions from the back and three each from the muzzle and tail were retrieved from the liquid nitrogen tank where they had been stored for future projects. Each approximately 50 mg piece was cut into small pieces and homogenized in 400 µl PBS in 2 ml Eppendorf tubes with conical bottoms using a OMNI International homogenizer for hard tissue. Homogenization was carried out at 30 000 r.p.m. with ice bath cooling for 40 s. Fifty microlitres of each homogenate was transferred to 500 µl tubes and 5 µl of Turbo DNA-free 10× buffer (Ambion) was added to one of each of the pairs of tubes. The other tube was sealed and retained as control for the sample. To the tubes containing the buffer was added 2 µl TURBO DNase. The tubes were incubated at 37 °C in a water bath for 1 h to allow for digestion of any free DNA. DNase inactivation reagent (0.2 vols) from the kit were added and tubes were vortexed and left at room temperature on the bench for 10 min with occasional mixing. DNA was extracted from treated and untreated lesions using the DNeasy kit (QIAGEN) as per the protocol for DNA extraction from tissue. DNAs were quantitated and subjected to Q-PCR as per ‘Detection of viral DNA by SYBR Green Q-PCR’ above. Percentage of encapsidated DNA in each sample was determined by measuring the ratio of viral DNA in the DNase-treated sample vs. total viral DNA in the untreated sample [22].
RNA in situ hybridization (RNA ISH)
Viral RNA expression in formalin-fixed, paraffin-embedded (FFPE) tissues was detected by RNAscope technology (Advanced Cell Diagnostics, Inc.) using custom probes mapping within the second exon of the ORF (nt 3139–3419) following the manufacturer’s protocol. After hybridization the bound probes were detected by colorimetric staining using RNAscope 2.5 HD Assay – BROWN. Before mounting, the slides were counterstained with 50 % haematoxylin Gill’s No. 1 solution (Sigma-Aldrich) and 0.02 % ammonium hydroxide solution (Sigma-Aldrich).
Statistical analysis
The data were statistically analysed with one-way ANOVA analysis using Sigmaplot 12 software. Differences were considered to be significant at P<0.05.
Funding information
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, under Award Number R21AI121822 (Christensen and Hu) and the Jake Gittlen Memorial Golf Tournament.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
Footnotes
Abbreviations: ARPC5, Actin-related 2/3 complex subunit 5; BSL2, biosafety level 2; CRPV, cottontail rabbit papillomavirus; CVP, circumvallate papilla; FFPE, formalin fixed parafin embedded tissues; GSA, group-specific antigen; H HPV, human papillomavirus; IHC, immunohistochemistry; ISH, in situ hybridization; MmuPV1, mouse papillomavirus 1; Q-PCR, quantitative polymerase chain reaction; RNA-ISH, RNA in situ hybridization; TEM, transmission electron microscope.
Two supplementary figures are available with the online Supplementary Material.
References
- 1.Stier EA, Sebring MC, Mendez AE, Ba FS, Trimble DD, et al. Prevalence of anal human papillomavirus infection and anal HPV-related disorders in women: a systematic review. Am J Obstet Gynecol. 2015;213:278–309. doi: 10.1016/j.ajog.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang JW, Hung CF, Huh WK, Trimble CL, Roden RB. Immunoprevention of human papillomavirus-associated malignancies. Cancer Prev Res. 2015;8:95–104. doi: 10.1158/1940-6207.CAPR-14-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moerman-Herzog A, Nakagawa M. Early defensive mechanisms against human papillomavirus Infection. Clin Vaccine Immunol. 2015;22:850–857. doi: 10.1128/CVI.00223-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doorbar J. Model systems of human papillomavirus-associated disease. J Pathol. 2016;238:166–179. doi: 10.1002/path.4656. [DOI] [PubMed] [Google Scholar]
- 5.Christensen ND, Cladel NM, Hu J, Balogh KK. Formulation of cidofovir improves the anti-papillomaviral activity of topical treatments in the CRPV/rabbit model. Antiviral Res. 2014;108:148–155. doi: 10.1016/j.antiviral.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fausch SC, da Silva DM, Eiben GL, Le Poole IC, Kast WM. HPV protein/peptide vaccines: from animal models to clinical trials. Front Biosci. 2003;8:s81–s91. doi: 10.2741/1009. [DOI] [PubMed] [Google Scholar]
- 7.Breitburd F, Kirnbauer R, Hubbert NL, Nonnenmacher B, Trin-Dinh-Desmarquet C, et al. Immunization with viruslike particles from cottontail rabbit papillomavirus (CRPV) can protect against experimental CRPV infection. J Virol. 1995;69:3959–3963. doi: 10.1128/jvi.69.6.3959-3963.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng X, Knouse JA, Hernon KM. Rabbit models for studying human infectious diseases. Comp Med. 2015;65:499–507. [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, Peng X, Schell TD, Budgeon LR, Cladel NM, et al. An HLA-A2.1-transgenic rabbit model to study immunity to papillomavirus infection. J Immunol. 2006;177:8037–8045. doi: 10.4049/jimmunol.177.11.8037. [DOI] [PubMed] [Google Scholar]
- 10.Brandsma JL. The cottontail rabbit papillomavirus model of high-risk HPV-induced disease. Methods Mol Med. 2005;119:217–235. doi: 10.1385/1-59259-982-6:217. [DOI] [PubMed] [Google Scholar]
- 11.Breitburd F, Salmon J, Orth G. The rabbit viral skin papillomas and carcinomas: a model for the immunogenetics of HPV-associated carcinogenesis. Clin Dermatol. 1997;15:237–247. doi: 10.1016/S0738-081X(97)00009-6. [DOI] [PubMed] [Google Scholar]
- 12.Watts SL, Ostrow RS, Phelps WC, Prince JT, Faras AJ. Free cottontail rabbit papillomavirus DNA persists in warts and carcinomas of infected rabbits and in cells in culture transformed with virus or viral DNA. Virology. 1983;125:127–138. doi: 10.1016/0042-6822(83)90069-7. [DOI] [PubMed] [Google Scholar]
- 13.Hu J, Cladel NM, Balogh K, Budgeon L, Christensen ND. Impact of genetic changes to the CRPV genome and their application to the study of pathogenesis in vivo. Virology. 2007;358:384–390. doi: 10.1016/j.virol.2006.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Budgeon LR, Cladel NM, Culp TD, Balogh KK, et al. Detection of L1, infectious virions and anti-L1 antibody in domestic rabbits infected with cottontail rabbit papillomavirus. J Gen Virol. 2007;88:3286–3293. doi: 10.1099/vir.0.82879-0. [DOI] [PubMed] [Google Scholar]
- 15.Ingle A, Ghim S, Joh J, Chepkoech I, Bennett Jenson A, et al. Novel laboratory mouse papillomavirus (MusPV) infection. Vet Pathol. 2011;48:500–505. doi: 10.1177/0300985810377186. [DOI] [PubMed] [Google Scholar]
- 16.Handisurya A, Day PM, Thompson CD, Bonelli M, Lowy DR, et al. Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1. PLoS Pathog. 2014;10:e1004314. doi: 10.1371/journal.ppat.1004314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundberg JP, Stearns TM, Joh J, Proctor M, Ingle A, et al. Immune status, strain background, and anatomic site of inoculation affect mouse papillomavirus (MmuPV1) induction of exophytic papillomas or endophytic trichoblastomas. PLoS One. 2014;9:e113582. doi: 10.1371/journal.pone.0113582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uberoi A, Yoshida S, Frazer IH, Pitot HC, Lambert PF. Role of ultraviolet radiation in papillomavirus-induced disease. PLoS Pathog. 2016;12:e1005664. doi: 10.1371/journal.ppat.1005664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang RT, Wang JW, Peng S, Huang TC, Wang C, et al. Spontaneous and vaccine-induced clearance of mus musculus papillomavirus 1 infection. J Virol. 2017;91:e00699-17. doi: 10.1128/JVI.00699-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang JW, Jiang R, Peng S, Chang YN, Hung CF, et al. Immunologic control of mus musculus papillomavirus type 1. PLoS Pathog. 2015;11:e1005243. doi: 10.1371/journal.ppat.1005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cladel NM, Budgeon LR, Balogh KK, Cooper TK, Hu J, et al. Mouse papillomavirus MmuPV1 infects oral mucosa and preferentially targets the base of the tongue. Virology. 2016;488:73–80. doi: 10.1016/j.virol.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu J, Budgeon LR, Cladel NM, Balogh K, Myers R, et al. Tracking vaginal, anal and oral infection in a mouse papillomavirus infection model. J Gen Virol. 2015;96:3554–3565. doi: 10.1099/jgv.0.000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cladel NM, Budgeon LR, Balogh KK, Cooper TK, Hu J, et al. A novel pre-clinical murine model to study the life cycle and progression of cervical and anal papillomavirus infections. PLoS One. 2015;10:e0120128. doi: 10.1371/journal.pone.0120128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cladel NM, Budgeon LR, Cooper TK, Balogh KK, Hu J, et al. Secondary infections, expanded tissue tropism, and evidence for malignant potential in immunocompromised mice infected with Mus musculus papillomavirus 1 DNA and virus. J Virol. 2013;87:9391–9395. doi: 10.1128/JVI.00777-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cladel NM, Budgeon LR, Balogh KK, Cooper TK, Hu J, et al. Mouse papillomavirus MmuPV1 infects oral mucosa and preferentially targets the base of the tongue. Virology. 2016;488:73–80. doi: 10.1016/j.virol.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow LT, Broker TR, Steinberg BM. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS. 2010;118:422–449. doi: 10.1111/j.1600-0463.2010.02625.x. [DOI] [PubMed] [Google Scholar]
- 27.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci. 2006;110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 28.Handisurya A, Day PM, Thompson CD, Buck CB, Pang YY, et al. Characterization of Mus musculus papillomavirus 1 infection in situ reveals an unusual pattern of late gene expression and capsid protein localization. J Virol. 2013;87:13214–13225. doi: 10.1128/JVI.02162-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmgren SC, Patterson NA, Ozbun MA, Lambert PF. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J Virol. 2005;79:3938–3948. doi: 10.1128/JVI.79.7.3938-3948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dediol E, Sabol I, Virag M, Grce M, Muller D, et al. HPV prevalence and p16INKa overexpression in non-smoking non-drinking oral cavity cancer patients. Oral Dis. 2016;22:517–522. doi: 10.1111/odi.12476. [DOI] [PubMed] [Google Scholar]
- 31.Kinoshita T, Nohata N, Watanabe-Takano H, Yoshino H, Hidaka H, et al. Actin-related protein 2/3 complex subunit 5 (ARPC5) contributes to cell migration and invasion and is directly regulated by tumor-suppressive microRNA-133a in head and neck squamous cell carcinoma. Int J Oncol. 2012;40:1770–1778. doi: 10.3892/ijo.2012.1390. [DOI] [PubMed] [Google Scholar]
- 32.Liu C, Liu R, Zhang D, Deng Q, Liu B, et al. MicroRNA-141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro-metastasis genes. Nat Commun. 2017;8:14270. doi: 10.1038/ncomms14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gimenes F, Medina FS, Abreu AL, Irie MM, Esquiçati IB, et al. Sensitive simultaneous detection of seven sexually transmitted agents in semen by multiplex-PCR and of HPV by single PCR. PLoS One. 2014;9:e98862. doi: 10.1371/journal.pone.0098862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Hang D, Deng Q, Liu M, Xi L, et al. Concurrence of oral and genital human papillomavirus infection in healthy men: a population-based cross-sectional study in rural China. Sci Rep. 2015;5:15637. doi: 10.1038/srep15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi HJ, Lee JH. Multiple human papilloma virus 16 infection presenting as various skin lesions. J Craniofac Surg. 2016;27:e379. doi: 10.1097/SCS.0000000000002642. [DOI] [PubMed] [Google Scholar]
- 36.Torres M, Gheit T, Mckay-Chopin S, Rodríguez C, Romero JD, et al. Prevalence of beta and gamma human papillomaviruses in the anal canal of men who have sex with men is influenced by HIV status. J Clin Virol. 2015;67:47–51. doi: 10.1016/j.jcv.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Paolini F, Cota C, Amantea A, Curzio G, Venuti A. Mucosal alpha-papillomavirus (HPV89) in a rare skin lesion. Virol J. 2015;12:105. doi: 10.1186/s12985-015-0336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolatti EM, Chouhy D, Casal PE, Pérez GR, Stella EJ, et al. Characterization of novel human papillomavirus types 157, 158 and 205 from healthy skin and recombination analysis in genus γ-Papillomavirus. Infect Genet Evol. 2016;42:20–29. doi: 10.1016/j.meegid.2016.04.018. [DOI] [PubMed] [Google Scholar]
- 39.Oštrbenk A, Kocjan BJ, Hošnjak L, Li J, Deng Q, et al. Identification of a novel human papillomavirus, type HPV199, Isolated from a nasopharynx and anal canal, and complete genomic characterization of papillomavirus species gamma-12. PLoS One. 2015;10:e0138628. doi: 10.1371/journal.pone.0138628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joh J, Ghim SJ, Chilton PM, Sundberg JP, Park J, et al. MmuPV1 infection and tumor development of T cell-deficient mice is prevented by passively transferred hyperimmune sera from normal congenic mice immunized with MmuPV1 virus-like particles (VLPs) Exp Mol Pathol. 2016;100:212–219. doi: 10.1016/j.yexmp.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Hošnjak L, Kocjan BJ, Pirš B, Seme K, Poljak M. Characterization of two novel gammapapillomaviruses, HPV179 and HPV184, isolated from common warts of a renal-transplant recipient. PLoS One. 2015;10:e0119154. doi: 10.1371/journal.pone.0119154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bottalico D, Chen Z, Dunne A, Ostoloza J, Mckinney S, et al. The oral cavity contains abundant known and novel human papillomaviruses from the betapapillomavirus and gammapapillomavirus genera. J Infect Dis. 2011;204:787–792. doi: 10.1093/infdis/jir383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cladel NM, Hu J, Balogh K, Mejia A, Christensen ND. Wounding prior to challenge substantially improves infectivity of cottontail rabbit papillomavirus and allows for standardization of infection. J Virol Methods. 2008;148:34–39. doi: 10.1016/j.jviromet.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koskimaa HM, Waterboer T, Pawlita M, Grénman S, Syrjänen K, et al. Human papillomavirus genotypes present in the oral mucosa of newborns and their concordance with maternal cervical human papillomavirus genotypes. J Pediatr. 2012;160:837–843. doi: 10.1016/j.jpeds.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 45.Uken RB, Brummer O, von Schubert-Bayer C, Brodegger T, Teudt IU. Oral HPV prevalence in women positive for cervical HPV infection and their sexual partners: a German screening study. Eur Arch Otorhinolaryngol. 2016;273:1933–1942. doi: 10.1007/s00405-016-3953-1. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Cladel NM, Budgeon L, Balogh KK, Christensen ND. Papillomavirus DNA complementation in vivo. Virus Res. 2009;144:117–122. doi: 10.1016/j.virusres.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.