

Abstract
The switch from a motile, planktonic existence to an attached biofilm is a major bacterial lifestyle transition that is often mediated by complex regulatory pathways. In this report, we describe a CheY-like protein required for control of the motile-to-sessile switch in the plant pathogen Agrobacterium tumefaciens. This regulator, which we have designated ClaR, possesses two distinct CheY-like receiver (REC) domains and is involved in the negative regulation of biofilm formation, through production of the unipolar polysaccharide (UPP) adhesin and cellulose. The ClaR REC domains share predicted structural homology with characterized REC domains and contain the majority of active site residues known to be essential for protein phosphorylation. REC1 is missing the conserved aspartate (N72) residue and although present in REC 2 (D193), it is not required for ClaR-dependent regulation suggesting that phosphorylation, which modulates the activity of many CheY-like proteins, appears not to be essential for ClaR activity. We also show that ClaR-dependent negative regulation of attachment is diminished significantly in mutants for PruA and PruR, proteins known to be involved in a pterin-mediated attachment regulation pathway. In A. tumefaciens, pterins are required for control of the intracellular signal cyclic diguanylate monophosphate through the DcpA regulator, but our findings suggest that pterin-dependent ClaR control of attachment can function independently from DcpA, including dampening of c-di-GMP levels. This report of a novel CheY-type biofilm regulator in A. tumefaciens thus also adds significant details to the role of pterin-mediated signalling.
Keywords: CheY, Biofilm, Agrobacterium tumefaciens, unipolar polysaccharide, pterin
Introduction
Bacteria often reside in complex heterogeneous communities of surface-attached cells known as biofilms [1]. Biofilm growth confers several advantages, in part due to the production of an extracellular matrix that can be composed of polysaccharides, protein and nucleic acid [2]. The extracellular matrix provides protection from the harmful effects of desiccation [3], antibiotics and other toxic substances [4, 5]. In addition to their protective properties, biofilms can facilitate both nutrient [6] and genetic exchange [7, 8]. Pathogenic biofilm infections are often difficult to treat, largely due to substantial antibiotic tolerance [9], and novel therapeutics are needed. Therefore, gaining a better understanding of the signalling pathways, molecules and key proteins that regulate bacterial attachment and biofilm formation may provide insights into targeted control strategies.
Agrobacterium tumefaciens is a member of the Alphaproteobacteria and is a facultative plant pathogen that causes the neoplastic disease crown gall through inter-kingdom gene transfer to plants [10–12]. A. tumefaciens can attach to both biotic and abiotic surfaces [13, 14] and has been developed into an informative model of the motile-to-sessile transition in bacteria [15, 16]. Utilizing a surface contact-dependent [17] unipolar adhesin known as the unipolar polysaccharide (UPP), A. tumefaciens stably attaches to surfaces via a single pole [16, 18]. In addition to the UPP, cellulose can also play a role in attachment [19, 20].
A. tumefaciens attachment and biofilm formation are controlled by multiple environmental and regulatory factors [15] such as low pH [21, 22], metal and nutrient availability [18, 23] and redox conditions [13]. Motility and chemotaxis have been shown to play a role in attachment to surfaces [24], with aflagellate mutants, those with unpowered flagella and those with defects in chemotaxis exhibiting severe deficiencies in biofilm formation. However, not all non-motile A. tumefaciens mutants are decreased for biofilm formation. For example, mutants in the flagellar master regulators visN/visR are aflagellate, but exhibit dramatically increased levels of biofilm formation and UPP production, revealing the complex connections between the motile and sessile states [25].
One of the key regulators of attachment in many bacteria is the second messenger cyclic-diguanylate monophosphate (c-di-GMP) [26–28]. In A. tumefaciens, c-di-GMP was described in early reports as controlling cellulose biosynthesis [29], and has been subsequently shown also to be involved in regulation of UPP production [25], with enhanced levels of c-di-GMP both increasing UPP levels and uncoupling UPP formation from surface attachment [25]. We have recently shown that the diguanylate cyclase and phosphodiesterase activities of the DcpA regulator can drive both synthesis and degradation of c-di-GMP, respectively, and that the protein is an important regulator of UPP production and biofilm formation [30]. DcpA enzymatic activity is controlled by a signalling pathway that requires the function of small metabolites related to folate, known as pterins. A specific monapterin type(s) influences DcpA activity via the pteridine reductase, PruA and the predicted pterin-binding protein, PruR.
A previous report from our laboratory [25] described a transposon screen that isolated several negative regulators of UPP production and biofilm formation due to differences in colony pigmentation in the presence of the dye Congo Red, known to bind certain polysaccharides and proteins with β-amyloid folds [31]. In mutants disabled for cellulose production (a polysaccharide that also binds Congo Red), the intensity of Congo Red colony pigmentation is remarkably proportional to UPP production. Standard solid bacteriological media (1.5 % agar) do not induce UPP production, and thus colonies pigment only lightly when Congo Red is supplemented in the medium. Defined growth medium supplemented with Congo Red was used to isolate mutants with elevated Congo Red pigmentation (the ECR phenotype), and these mutants uniformly had increased UPP production. This screen isolated four main mutant classes, including the visNR locus, which encodes the master regulators of motility in A. tumefaciens [25]. Transposon insertions were also discovered in the pruR-dcpA operon and separately in the distal pruA locus, which we now know comprise the pterin-mediated regulatory pathway described above [30]. The remaining class of ECR mutants consisted of transposon insertions in the Atu1631 gene [25]. UPP production was uncoupled from surface contact in Atu1631-disrupted strains, closely mimicking the phenotype associated with an elevated c-di-GMP state.
In this report, we find that Atu1631 negatively regulates attachment and biofilm formation in A. tumefaciens and, because of its similarity to the ubiquitous chemotaxis regulator CheY, we name it ClaR (CheY-like attachment regulator). In contrast to CheY, however, ClaR possesses two predicted CheY-like receiver (REC) domains. We show that despite the structural homology and conservation of several key active site residues, the negative regulatory activity of ClaR does not require phosphorylation of the conserved aspartate residues in either domain for full activity. ClaR does require, however, the presence of the PruA-PruR pterin-regulatory module for proper function. In addition to expanding pterin regulation in A. tumefaciens, this work illustrates a novel mechanism for regulation of attachment and biofilm formation in A. tumefaciens.
Methods
Reagents, strains and plasmids
All strains, plasmids and oligonucleotides used in this study are listed in the Supplementary Materials, Tables S1 and S2 (available in the online Supplementary Material), respectively. Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA). DNA sequencing was performed on an ABI 3730 Sequencer (Indiana Molecular Biology Institute, Bloomington, IN) or by ACGT Inc. (Wheeling, IL). Plasmids were introduced into E. coli via heat-shock transformation with chemically competent cell preparations and into A. tumefaciens via either conjugation or electroporation [32]. The E. coli strains used for conjugation of plasmids or plasmid DNA transformation were grown in LB broth (Difco Bacto tryptone at 10 g l−1, Difco yeast extract at 10 g l−1 and NaCl at 5 g l−1) with or without 1.5 % (w v−1) agar. A. tumefaciens strains were grown on either LB or AT minimal medium [33] supplemented with 0.5 % (w v−1) glucose and 15 mM ammonium sulfate (ATGN). To prevent accumulation of iron oxide precipitate, the FeSO4 prescribed in the referenced AT recipe was omitted, with no effect on growth. For biofilm cultures, 22 µM FeSO4·7H2O was added to ATGN medium directly before inoculation. For sacB counter-selection, 5 % (w v−1) sucrose (Suc) replaced glucose as the sole carbon source (ATSN). Antibiotics, chemicals and culture media were obtained from Sigma-Aldrich and Fisher Scientific. When necessary, antibiotics were added to the medium as follows: for E. coli, 100 µg ml−1 ampicillin, 50 µg ml−1 gentamicin (Gm) and 50 µg ml−1 kanamycin (Km); and for A. tumefaciens, 300 µg ml−1 Gm and 300 µg ml−1 Km. Isopropyl-β-d-thiogalactopyranoside (IPTG) was used at 400 µM when required. For Congo Red plates, the dye was dissolved in methanol at 20 mg ml−1 and passed through 0.2 µm syringe filters immediately before use to remove aggregates. Congo Red was added to ATGN medium for a final concentration of 100 µg ml−1 to generate ATGN-CR agar medium. For calcofluor plates, a 200 µg ml−1 stock of calcofluor white (fluorescent white) dye was added to LB plates to yield a final concentration of 20 µg ml−1. Plates were kept in the dark until use to avoid degradation of the dye.
For assay plates containing either Congo Red or calcofluor, A. tumefaciens cultures were grown to mid-exponential phase, normalized to an OD600 of 0.5 and spotted (5 µl) onto ATGN-CR or LB calcofluor (with IPTG as required). After 48 h incubation at 28 °C, photographs were taken under white light for Congo Red, and under UV light exposure for calcofluor.
Controlled expression plasmids
For plasmid-borne expression, predicted coding sequences were inserted into the LacIQ encoding, IPTG-inducible expression vector pSRKGm [34], fusing the coding sequence to the lacZ start codon and Shine-Dalgarno sequence to create a Plac fusion. Sequences were PCR amplified from A. tumefaciens C58 genomic DNA using the corresponding primers for each gene (Table S2) and Phusion DNA polymerase (New England Biolabs, Beverly, MA). PCR products were ligated into pGEM-T Easy (Promega), their sequence confirmed, excised by restriction enzyme cleavage and ligated with appropriately cleaved pSRKGm. The presence of the correct, in-frame coding sequence was verified by restriction digestion prior to electroporation into competent A. tumefaciens cells.
Site-specific mutagenesis
An altered version of the QuikChange mutagenesis protocol (Agilent Technologies, Santa Clara, CA) was used to engineer site-specific mutations in claR. Briefly, two complementary primers were designed with the required base pair change flanked by ~15 bp of original wild-type sequence on each side. The oligos were utilized in a thermal cycler reaction (16 cycles) of the entire plasmid using the high-fidelity Phusion DNA polymerase to generate nicked plasmid derivatives with altered sequences. Vector amplification was confirmed via gel electrophoresis, 1 µl of the methylation-dependent restriction endonuclease Dpn I was added to digest unmodified/methylated wild type plasmid, and the digested mixture was transformed into competent E. coli. Plasmid clones were isolated and sequenced to certify introduction of the desired mutation.
Construction of in-frame markerless deletions
In-frame deletions of A. tumefacines genes were created using a previously described method [32]. Phusion DNA polymerase was used to amplify 500–750 bp of sequence upstream (P1 and P2) and downstream (P3 and P4) of the gene to be deleted. Extreme 5′ and 3′ ends of genes were left intact. Primers P2 and P3 were designed with 5′ sequences (lowercase in Table S2, 15–20 bp) with reverse complementarity to each other’s 3′ proximal sequence. These complementary sequences on the two primers enabled splicing by overlapping extension (SOE) of the two PCR products, as previously described [32]. Both flanking sequences were amplified and gel purified. A second PCR reaction, in which the two purified products were used as both templates and primers, generated the final spliced product, which was ligated into pGEM-T easy (Promega, Madison, WI). Subsequent DNA sequencing ensured proper splicing. The deletion construct was then removed using restriction enzymes and ligated into the suicide vector pNPTS138 cleaved with compatible restriction enzymes. The pNPTS138 plasmid confers Km resistance (KmR) and sucrose sensitivity (SucS). Derivatives of pNPTS138 were introduced into A. tumefaciens C58 by conjugation with pNPTS138 containing S17-1/λpir E. coli. Single-crossover integration into the chromosome is required to obtain KmR transformants, as the ColE1 origin of pNPTS138 does not replicate in A. tumefaciens. Plasmid integration was confirmed by patching onto ATGN-Km and ATSN-Km to identify SucS derivatives. Excision of the integrated plasmid was then facilitated by growing overnight cultures of SucS KanR derivatives and plating dilutions onto ATSN. Plasmid excision was confirmed by patching SucR clones onto ATSN and ATGN-Km to identify KanS derivatives. Diagnostic PCR, using primers flanking the deletion site, was used to confirm deletion of the target gene.
Growth and analysis of static biofilms
A. tumefaciens biofilms were grown and analysed basically as described previously [13]. In summary, trimmed PVC coverslips were placed upright in 12-well polystyrene culture plates (Corning Inc.) and UV sterilized. Mid-to-late exponential phase cultures were then subcultured into ATGN (with added IPTG, 400 µM) to an OD600 of 0.05 and incubated statically at room temperature for approximately 48 h. For crystal violet (CV) quantification, coverslips were rinsed with ddH20, stained with 0.1 % (w v−1) CV and rinsed once more with ddH20. CV-stained biomass was then quantified by immersing coverslips in 1 ml of 33 % acetic acid to solubilize CV. Data collection was performed by reading absorbance of soluble CV at 600 nm (A600) on a Biotek Synergy HT microplate reader. Values were normalized for planktonic growth by dividing the CV A600 by the OD600 of the remaining culture in the biofilm well.
Soft agar motility assay
A. tumefaciens swimming motility was assayed as previously described [35]. Briefly, A. tumefaciens cultures were grown to mid- to late exponential phase and spotted (5 µl) into the centre of ATGN plates with 0.3 % (w v−1) Bacto agar. Plates were dried on a bench for approximately 1 h and then placed in a container at room temperature alongside a beaker of saturated potassium sulfate. Plates were incubated for 5 days, at which time pictures were taken.
LC-MS/MS analysis of c-di-GMP levels
A. tumefaciens derivatives were grown in ATGN medium (plus 400 µM IPTG if needed) at 28 °C to stationary phase (>OD600 : 1.0). Culture densities were normalized so that all samples had the same cell density once resuspended in extraction buffer. Culture aliquots were spun for 5 min at 10 000 g at 4 °C and the pellet was immediately resuspended in 250 µl extraction buffer (methanol/acetonitrile/dH2O 40 : 40 : 20+0.1 N formic acid cooled at −20 °C) by vigorous pipetting and vortexing. After incubation at −20 °C for 30 min, the extractions were transferred to new microfuge tubes on ice. Cell debris was removed by centrifugation at 10 000 g for 3 min., after which 200 µl of supernatant was transferred into new tubes on ice. Samples were neutralized within 1 h of preparation by adding 4 µl of 15 % NH4HCO3 per 100 µl of sample, aimed to set a pH of 7~7.5.
Before analysis, the sample was vacuum centrifuged to remove the extraction buffer and resuspended in an equal volume of water. Ten microlitres of each sample was then analysed using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) on a Quattro Premier XE mass spectrometer (Waters Corporation) coupled with an Acquity Ultra Performance LC system (Waters Corporation). Detection and quantification of c-di-GMP was performed as previously demonstrated [36]. To calculate the c-di-GMP concentration, chemically synthesized c-di-GMP (Axxora) was dissolved in water at concentrations of 250, 125, 62.5, 31.2, 15.6, 7.8, 3.9 and 1.9 nM and analyzed using LC-MS/MS, in the process generating a standard curve.
Statistical analysis
Statistical significance between data sets was determined by performing paired, two-tailed Student’s t-tests using Microsoft Excel software. A statistically significant difference was represented by P-values ≤0.05.
Results
Transposon mutants in a CheY homologue result in UPP overproduction
Five independent A. tumefaciens transposon mutants with the ECR phenotype had insertions that mapped to Atu1631, four within the predicted Atu1631 coding sequence (Fig. 1a) and a fifth insertion located upstream in the presumptive promoter region. All of these transposon mutants have previously been shown to exhibit elevated UPP labelling, implicating Atu1631 as a UPP-negative regulator [25]. The annotated Atu1631 coding sequence is 747 base pairs (249 aa), and is located on the A. tumefaciens circular chromosome 243 bp downstream of Atu1632 (Fig. 1a), a gene predicted to encode a dimethylglycine dehydrogenase. Downstream and convergently transcribed with Atu1631 is Atu1630, with a gene product homologous to a phospholipase D family protein.
Fig. 1.

A biofilm regulator with two CheY-like receiver domains. (a) Diagram of claR genetic locus (Atu1631). Atu1632 is a predicted dimethylglycine dehydrogenase and Atu1630 is a predicted phospholipase D family protein. Gene sizes are to scale. ECR screen transposon insertions are notated by lines capped with reverse arrowheads (b) Gene topology of ClaR. Domains predicted using Pfam database (http://pfam.xfam.org/). All domains are to scale. (c) Alignment of ClaR REC domains to known REC domains. Red boxes notate essential active site residues known to be required for phosphorylation. Aligned REC domain amino acid residues are as follows: ClaR REC1 : 21–121; ClaR REC2 : 144–252; A. tumefaciens CheY1 : 1–121; A. tumefaciens CheY2 : 1–129; E. coli CheY: 1–129; E. coli OmpR: 7–117; E. coli NtrC: 6–115. Alignment performed with ClustalX software (www.clustal.org) using multiple alignment mode.
The annotated Atu1631 protein contains tandem predicted CheY-type receiver or REC domains, common in two-component response regulators (Fig. 1b). REC1 (Pfam PF00072, E-value 4.6×10−6) is defined by residues 21–121, while the C-terminal REC2 domain (Pfam PF00072, E-value 2.6×10−22) is defined by residues 144–252. No other annotated domains are predicted on Atu1631. Therefore, Atu1631 was re-designated ClaR (CheY-like attachment regulator). There is no sensor kinase homologue within close proximity (<10 kb) of the claR gene.
REC domains typically contain phospho-accepting aspartate residues that are phosphorylated by cognate sensor kinases as part of two-component systems [37], one of the most prominent bacterial signalling mechanisms. The well-studied, canonical CheY proteins of E. coli and Salmonella typhimurium are composed of a single REC domain, and these proteins are central to the modified two-component system that controls bacterial chemotaxis [38]. After accepting a phosphate group from the histidine kinase CheA [39], CheY interacts with the flagellar switch protein FliM [40] to modulate the speed and/or direction of flagellar rotation. The crystal structures of S. typhimurium and E. coli CheY were two of the first available crystal structures of bacterial response regulators [41, 42]. These studies, along with others, have uncovered specific amino acid residues in addition to general secondary and tertiary structures of CheY, defining their roles in the phosphorylation signalling cascade [43].
The two REC domains of ClaR were aligned to several characterized CheY proteins in addition to the REC domains from the well-studied E. coli NtrC and OmpR multi-domain response regulators (Fig. 1c). All REC domains contain the conserved phosphor-accepting aspartate (D57 in E. coli CheY) [41, 44]. In contrast to REC2 (D193), REC1 of ClaR does not contain this aspartate residue, and instead has an asparagine residue substituted at this position (N72). Both the REC1 and REC2 domains contain the conserved D/E-D/E motif (D12D13 in E. coli) that has been shown to be important for coordinating the Mg+2 ion necessary for phosphorylation [44, 45]. The REC1 and REC2 domains of ClaR also both possess a conserved threonine/serine residue (Fig. 1c, T87 in E. coli, T98 ClaR REC1 and S223 ClaR REC2), hypothesized to play a role in the coupling of the phosphorylation event to the conversion of CheY into an activated, signalling-proficient form [46]. Lastly, both REC1 and REC2 contain a positively charged residue (K109 in E. coli, K120 ClaR REC1 and R245 ClaR REC2) that is thought to stabilize the phosphorylated active site by forming a salt bridge with Asp12 in CheY [47]. Therefore, both the REC domains of ClaR contain some (REC1) or all (REC2) key residues necessary for receiver domain function.
ClaR negatively regulates biofilm formation in A. tumefaciens
A markerless, in-frame deletion mutant of claR was constructed and tested for the ability to form biofilms on PVC coverslips using a 12-well plate format [18, 25]. The ΔclaR mutant exhibits a more than two-fold increase in biofilm formation compared to the wild-type strain (Fig. 2a, P-value of <0.05 by paired t-test vs WT). This elevation is consistent with the enhanced levels of biofilm formation previously seen in the pruA and dcpA genetic backgrounds (Fig. 2a, [25]). As expected, UPP production is absolutely required for biofilm formation in the claR mutant (Fig. S1). Disabling the ability to synthesize cellulose (Δcel) diminishes biofilm formation substantially, suggesting that both cellulose and the UPP contribute to the increased attachment of the claR mutant, although prior work clearly shows that cellulose is not involved in wild-type A. tumefaciens biofilm formation under laboratory conditions [18]. Unlike the pruA mutant, deletion of claR does not affect motility or production of the acidic polysaccharide succinoglycan (Fig. S2a, b). Unlike the pruA or dcpA mutants, which display increased c-di-GMP levels due to loss of DcpA PDE activity [30], the intracellular concentration of c-di-GMP is not significantly elevated in the claR mutant (Fig. S2c). A non-polar deletion of the Atu1632 coding sequence immediately upstream of claR does not result in an enhancement of biofilm formation (Fig. S3).
Fig. 2.
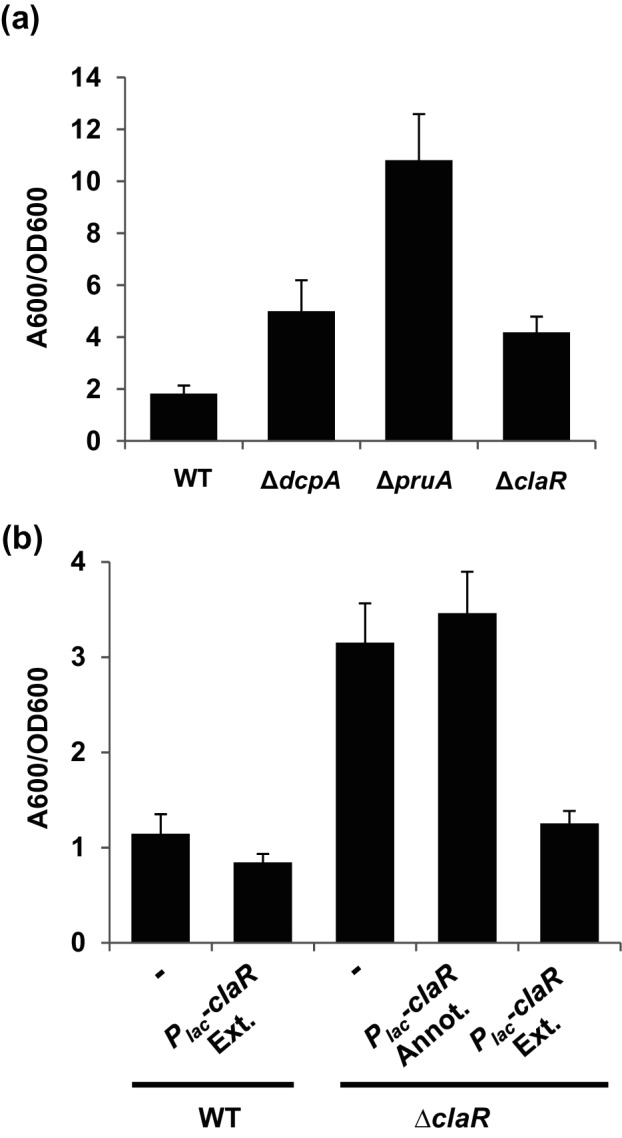
ClaR regulates A. tumefaciens biofilm formation. (a) A. tumefaciens quantitative biofilm formation on PVC coverslips after 48 h static growth at 28 °C. Adherent biomass quantified by staining with crystal violet (CV). CV absorbance quantified by absorbance at 600 nm (A600). In parallel, the OD600 of planktonic culture was determined. CV absorbance was normalized to culture growth by calculating the A600/OD600 ratio. Values are the result of two independent biological replicates consisting of three technical replicates each. Error bars: ±1 sd. (b) 48 h PVC coverslip biofilms quantified as described in Fig. 2(a). claR Annot.: 750 bp claR annotated in NCBI database. claR Ext.: annotated claR plus additional 84 bp added to 5′ end of gene. Dash represents strain without plasmid introduced. Values are the results of two independent biological replicates consisting of three technical replicates each. Error bars: ±1 sd.
Surprisingly, provision of the annotated claR coding sequence (claR-Annot.) on an IPTG-inducible, multi-copy plasmid in the ∆claR mutant did not restore biofilm formation to WT levels (Fig. 2b). Alignment of the 249 aa predicted claR coding sequence with closely related rhizobial bi-directional best-hit homologues (orthologous sequences from Agrobacterium vitis, Agrobacterium radiobacter and Rhizobium leguminosarum, Fig. S4) revealed an additional 28 N-terminal amino acid residues that are not included in the annotated A. tumefaciens claR. A longer 278-codon version of claR (claR-Ext.), including these 28 additional residues, was in fact able to complement biofilm formation of an Atu1631 mutant back down to WT levels (Fig. 2b). This indicates that removal of the N-terminal 28 aa residues of ClaR disrupts its function and/or stability. As a result, the extended 277 aa version of ClaR was used in all subsequent experiments (and the amino acid coordinates cited above are relative to this longer sequence). Overall, these results provide genetic confirmation that ClaR is a negative regulator of A. tumefaciens biofilm formation.
ClaR REC domains are structurally conserved but phosphorylation site mutants retain activity
To provide a comparison to the known secondary and tertiary structures of CheY, the structures of the ClaR REC1 and REC2 domains were predicted utilizing the PHYRE2 structure prediction algorithm (http://www.sbg.bio.ic.ac.uk/phyre2) [48]. E. coli CheY (PDB:3CHY) possesses a characteristic structure composed of a five-stranded parallel β-sheet core flanked by five α-helices (Fig. 3a). Both REC1 and REC2 are predicted to possess a similar structure, sharing the β-sheet core and the same general position of the conserved active sites mentioned above (Fig. 3b, c). However, REC1 and REC2 are predicted to possess only four α-helices, without the 5th helix (H5) found in CheY (Fig. S5a, b). In E. coli CheY, the H4-β5-H5 secondary structure element (Fig. 3a, light blue) has been shown to provide an interface for protein–protein interaction with the flagellar switch [46]. Although both REC1 and REC2 are lacking this H5 helix, they do appear to share structural conservation with each other in this region. Structural alignments of CheY with REC1 and REC2 show a relatively high degree of overall structural homology (Fig. S5a, b), with RMSD values of 0.98 and 1.11, respectively. The conserved aspartate residue is absent in the REC1 domain, with an asparagine residue at this position (N72). We therefore hypothesized that phosphorylation of the REC2 domain of ClaR might be important for its regulatory function.
Fig. 3.

Comparison of ClaR predicted secondary structures to CheY. (a) Experimentally determined crystal structure of E. coli CheY ([41], PDB ID: 3CHY). α-Helices and β-sheets coloured in green and red, respectively. Blue colouring represents predicted CheY protein–protein interaction surface. Active site residues known to be required for phosphorylation highlighted in magenta. Structure visualized using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). (b) Predicted structure of ClaR REC1 domain (residues 1–121). Structure predicted using PHYRE2 structure prediction algorithm (http://www.sbg.bio.ic.ac.uk/phyre2) [48]. Structure colouring described in Fig. 3(a). (c) Predicted structure of ClaR REC2 domain (residues 144–252). Structure predicted also using PHYRE2 structure prediction algorithm. Structure colouring described in Fig. 3(a).
The conserved aspartate phosphorylation site on the REC2 domain of ClaR (D193) was separately mutated to asparagine and alanine, both residues that would be expected to abrogate the ability of the protein to be phosphorylated. Plasmid-borne expression of both claR mutant alleles (Plac-claR D193A and Plac-claR D193N) in the ΔclaR mutant resulted in modest, yet significant, decreases in biofilm formation (Fig. 4, P-value of <0.05 by paired t-tests vs. claR). These data indicate that the predicted phosphorylation site of ClaR is not absolutely required for function. Both of the D193A and D193N ClaR alleles were able to reduce biofilm formation in the claR mutant, but not to the same extent as the wild-type copy of claR (Fig. 4, P-value of <0.05 by paired t-tests vs. ∆claR Plac-claR). This partial decrease in activity may reflect protein misfolding and perhaps turnover, rather than a strict requirement for phosphorylation.
Fig. 4.
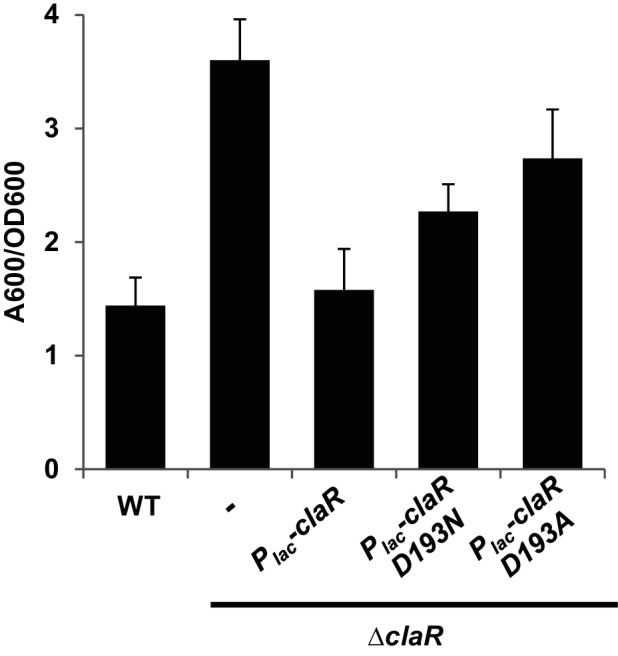
ClaR phosphorylation site not absolutely required for protein function. 48 h PVC coverslip biofilms quantified as described in Fig. 2(a). Dash represents strain without plasmid introduced. Values average three independent biological replicates consisting of three technical replicates each. Error bars: ± sd.
ClaR interactions with the A. tumefaciens pterin-dependent control pathway
Previous work from our laboratory characterized the prominent role of the PruA-PruR-DcpA signalling pathway in the control of A. tumefaciens attachment and biofilm formation [30]. DcpA has two activities related to the c-di-GMP second messenger: a diguanylate cyclase (DGC) activity that can catalyse c-di-GMP synthesis, and a phosphodiesterase (PDE) activity that drives its degradation. In laboratory culture conditions, the phosphodiesterase activity of the DcpA protein is required to restrict UPP production to those cells engaged in surface colonization, presumably through maintaining low cellular levels of c-di-GMP in free-swimming cells. The strong bias towards the dominant PDE activity of DcpA requires the pruA gene, encoding a pteridine reductase that synthesizes a derivative of the metabolite tetrahydromonapterin, and pruR, a gene that forms a bi-cistronic operon with dcpA and encodes a putative pterin-binding protein [30]. Independent null mutants in dcpA, pruA and pruR have essentially the same ECR phenotype with elevated production of UPP and hyper-adherence [30]. To test for any genetic or regulatory interaction between ClaR and this pathway, the effect of claR ectopic expression on biofilm formation and UPP production was tested in several mutant backgrounds. As observed before, plasmid-borne claR expression in the ΔclaR mutant results in a decrease of adherence back to near WT levels (Fig. 5a). Strikingly, the claR plasmid also acted to decrease attachment (Fig. 5a), Congo red staining (Fig. 5b) and c-di-GMP levels (Fig. S2c) in the dcpA mutant background compared to the plasmid-less mutant strains. Inversely, dcpA expression (WT and DGC-PDE +variants) in a claR mutant reduces biofilm formation to near-WT levels (P-value of <0.05 by paired t-tests vs. ∆claR), and this requires DcpA PDE activity (Fig. S6, P-value of <0.05 by paired t-test of DGC+PDE- DcpA vs ∆claR).
Fig. 5.
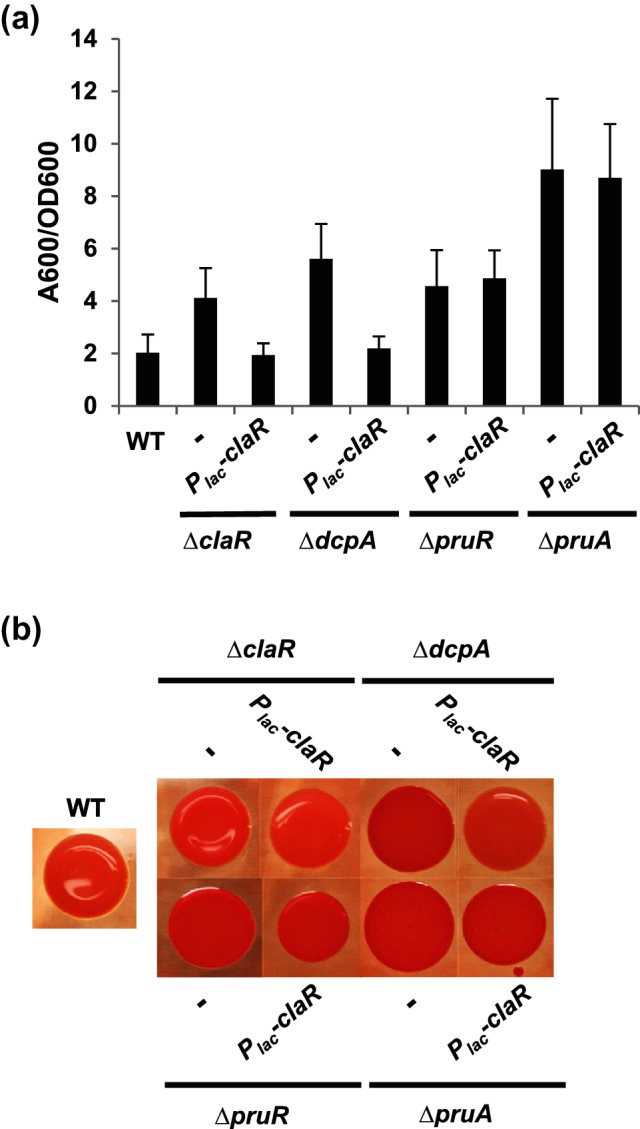
ClaR activity in various A. tumefaciens mutants. (a) 48 h PVC coverslip biofilms quantified as described in Fig. 2(a). Dash represents strain without plasmid inserted. Values average three independent biological replicates consisting of three technical replicates each. Error bars: ± sd. (b) Congo Red colony phenotypes of indicated A. tumefaciens strains. Strains were grown to mid-exponential phase, normalized to OD600 0.5 and spotted onto ATGN plates containing ~100 µg ml−1 Congo Red dye and IPTG. Photographs were taken after 48 h of growth at 28 °C. Dash represents strain lacking the introduced plasmid.
Unexpectedly, ClaR overexpression failed to significantly decrease biofilm formation or UPP production in the pruA or pruR mutant backgrounds (Fig. 5a, b, P-value of >0.05 by paired t-tests vs. pruA and pruR). We previously found that the pteridine reductase activity of PruA is abolished by mutation of the catalytic site tyrosine to alanine (Y163A), and this abrogated its activity in vitro and in vivo, as well as its ability to control biofilm formation [30]. The plasmid-borne claR allele introduced into the pruA Y163A allelic replacement mutant background was also ineffective at decreasing biofilm formation or Congo Red staining, similar to what was observed in the ΔpruA null mutant (Fig. S7a, b). These results indicate that both PruA and PruR are required for ClaR-mediated negative regulation of UPP-dependent attachment in A. tumefaciens. Furthermore, the control of ClaR activity seems to be linked specifically to PruA-catalysed pterin biosynthesis.
Discussion
In this report, we describe the genetic and phenotypic analysis of A. tumefaciens ClaR (Atu1631), initially discovered utilizing a transposon mutagenesis screen for negative regulators of UPP production [25]. We show here that ClaR is also a negative regulator of A. tumefaciens biofilm formation, and that it possesses tandem conserved CheY-like REC domains (REC1 and REC2), similar to those involved in phosphotransfer in the canonical CheY-type proteins involved in chemotaxis [38]. The REC1 domain in ClaR has an asparagine at the typical position (N72) for the phospho-accepting aspartate residue, but in contrast REC2 has an aspartate at this position (D193). Mutation of the REC2 aspartate (D193A) demonstrates that this residue is not required for ClaR-dependent regulation, suggesting that phosphorylation may not play an important role in ClaR activity. Our findings also suggest that ClaR control is integrated with the pterin-dependent PruA-PruR-DcpA pathway [30]. Although plasmid-borne expression of claR returns the elevated biofilm phenotype of a dcpA mutant back to wild-type levels, it does not do so in pruA or pruR mutants.
CheY domain-containing proteins in A. tumefaciens
There are multiple A. tumefaciens proteins that contain CheY-like REC domains. The chemotaxis locus in A. tumefaciens and related rhizobia differs somewhat from the well-studied E. coli model system, with the presence of two CheY homologues (CheY1/CheY2) while lacking a CheZ phosphatase [49]. Both CheY1 and CheY2 contribute to chemotaxis, but cheY2 mutants exhibit the most severe defect in chemotactic motility [50]. Based primarily on studies in Sinorhizobium meliloti, CheY2 is thought to act as the main mediator of chemotaxis [51]. In contrast, CheY1 is hypothesized to act as a phosphate sink, in effect fulfilling the role of CheZ in modulating the sensitivity of the chemotactic pathway [52]. Several lines of evidence suggest that ClaR is not a component of the chemotaxis pathway. As we demonstrate above, a ∆claR mutant does not display a motility defect (Fig. S2a). In addition, the components of the canonical chemotaxis pathway have been characterized in A. tumefaciens and they are shared with other members of the Rhizobiaciae [50–52]. Therefore, we predict that ClaR possesses a separate and distinct regulatory output.
In A. tumefaciens, there are other examples of proteins that possess CheY-like REC domains but are not directly involved in chemotaxis. One of the best described is PleD (also called CelR). Homologues of PleD contain dual REC domains coupled to a c-di-GMP-synthesizing diguanylate cyclase (DGC) domain, the first of which was characterized as an important cell cycle regulator in Caulobacter crescentus [53]. The DGC activity of PleD in C. crescentus is controlled by phosphorylation [54, 55], predominantly via the DivJ kinase and PleC phosphatase [56]. In A. tumefaciens, high levels of pleD expression result in elevation of intracellular c-di-GMP levels with a concomitant increase in UPP production, and this requires the PleD DGC catalytic GGDEF motif [25]. PleD has also been demonstrated to play a role in the regulation of A. tumefaciens cellulose production, biofilm formation and virulence [25, 57]. In addition to its role in attachment and polysaccharide production, PleD is thought to influence A. tumefaciens division and development [58].
Phosphorylation and ClaR function
We have found that the aspartate residue on ClaR REC2 is not absolutely required for negative regulation of attachment and biofilm formation, suggesting that phosphorylation is not a critical aspect of ClaR activation (although the D193 mutants are somewhat less effective than wild-type claR, an observation perhaps due to defects in protein folding or altered stability). This observation does not exclude the possibility that ClaR is phosphorylated by an as yet unidentified kinase, but rather that phosphorylation of this residue is not absolutely required for ClaR-dependent regulation of attachment and biofilm formation. In structural studies, phosphorylation of CheY-like REC domains did not cause major conformational rearrangements [59], and there remains some uncertainty about the exact mechanism of how phosphorylation-mediated REC domain activation leads to the formation of productive protein–protein or inter-domain interactions [47, 59, 60]. It is additionally a possibility that ClaR has structural flexibility that allows interaction with a downstream partner(s) in the absence of phosphorylation.
Alternatively, phosphorylation via an as yet unidentified kinase might enhance or stabilize these interactions to a degree that is undetectable by our phenotypic assays. It is therefore possible that ClaR phosphorylation potentiates the negative regulation of UPP production and biofilm formation by ClaR. A related phenomenon is observed with PleD in C. crescentus, with the non-phosphorylated protein possessing low DGC activity that is further stimulated by DivJ-mediated phosphorylation [54]. Future work probing for possible ClaR interaction partners, with particular focus on its phosphorylation state and the members of the PruA-PruR-DcpA pathway, will shed light on the molecular requirements for ClaR-dependent signalling.
Receiver domain-containing proteins can have distinct cellular roles depending on the phosphorylation state of the protein [61]. One example is B. subtilis DegU, with the phosphorylated form of the protein activating genes involved in biofilm formation and the unphosphorylated form promoting genetic competence [62, 63]. Phosphorylation-independent dimerization and activity has been observed for a variety of response regulators [61]. In some instances, a conserved non-phosphorylated aspartate residue is required for dimerization and full activity [64]. This may be the case with ClaR, with the modest decrease in activity for the D193A claR allele due to the lack of proper intermolecular interactions.
Che-protein control of non-chemotaxis phenotypes
Despite ClaR having a pronounced regulatory effect on UPP production and biofilm formation, motility is not affected in a claR mutant (Fig. S2a). This observation is unsurprising, as there are several examples in the literature of CheY-like proteins controlling non-motility phenotypes. The fact that half of bacterial genomes that contain che loci encode multiple cheY homologues, with many genomes containing greater than four predicted chemosensory systems, further reinforces this concept [65]. Chemosensory systems have been functionally classified by the regulation of three categories of bacterial phenotypes [66]: flagellar motility, Type IV-pili mediated motility and alternative cellular functions (ACF).
ACF phenotypes regulated by chemosensory systems are diverse and found in a variety of bacteria. Myxococcus xanthus encodes eight distinct chemosensory systems that include several prominent examples of ACF systems. The Che3 system plays no role in motility but predominantly controls entry into development, specifically promoting fruiting body formation [67]. Developmental gene expression is also controlled by Che3 activity. Another M. xanthus Che-type system, the Dif (Che2) chemosensory system, has been shown to regulate both social gliding motility and subsequent fruiting body formation [68]. DifD, a CheY homologue, negatively regulates fibril polysaccharide production via an unknown mechanism.
Rhodospirillum centenum, a photosynthetic member of the Alphaproteobacteria, contains three chemosensory loci [66]. The first locus (Che1) contains the canonical chemotaxis system, while the second (Che2) encodes a cytosolic pathway that regulates swarming motility through the synthesis of both polar and lateral flagella [69]. The third locus (Che3) is not associated with motility but instead plays a role in R. centenum cyst development [70].
Chemosensory systems can also play an important role in c-di-GMP metabolism and the regulation of bacterial attachment. WspR, which regulates attachment phenotypes in Pseudomonas aeruginosa [71, 72], is a CheY homologue that is encoded as part of a predicted seven-gene chemosensory locus. WspR contains a REC domain coupled to a GGDEF motif, and its diguanylate cyclase activity is significantly increased upon phosphorylation of the REC domain [72]. Phosphorylation of WspR, which only occurs upon surface contact, also induces localization of the protein into distinct cytoplasmic clusters [73, 74]. The mechanism of signal transduction coupling surface contact to a protein phosphorylation cascade is currently unknown.
ClaR interaction with PruA-PruR-DcpA regulatory pathway
Our previous work revealed that the pteridine reductase PruA and the putative pterin-binding protein PruR were required to bias the activity of the DcpA protein towards a PDE-dominant state. Until the current study, we considered this to be specifically through DcpA. Our findings with ClaR suggest that this prior DcpA-specific model is oversimplified. Plasmid-borne expression of claR can effectively reverse the elevated adherence and ECR phenotypes of a dcpA deletion mutant. The increased c-di-GMP pools in a dcpA mutant are reduced by elevated ClaR levels (Fig. S2c, P-value of <0.05 by paired t-tests of ∆dcpA Plac-claR vs. ∆dcpA), indicating that ClaR possibly interacts with members of the c-di-GMP regulon in A. tumefaciens besides DcpA. We also cannot exclude the possibility that ClaR might also interact directly with biosynthetic proteins for UPP and cellulose to modulate their activity, analogous to regulatory interactions between CheY and the flagellar motor complex [40].
We were, however, surprised to find that the effect of the plasmid-borne claR gene absolutely requires the presence of both PruA and PruR for its regulatory activity, as shown by the epistatic effect of pruA and pruR mutations in strains expressing the plasmid-borne claR gene (Fig. 5). Specifically, ClaR requires PruA enzymatic activity for proper function, as the claR plasmid is likewise ineffective in a mutant background that is abrogated for PruA-catalysed pterin synthesis (Fig. S7). These findings are intriguing and functionally separate the impact of claR, as well as pruA and pruR, from DcpA activity. Models in which ClaR interacts with DcpA to exert its activity are refuted by the robust phenotypic impact of plasmid-borne expression of claR in the ΔdcpA mutant. The simplest model is that, in addition to the impact of pterins on DcpA activity, in parallel, ClaR activity is also pterin-responsive, and thus also requires PruA and PruR (Fig. 6). Dual pterin-dependent adherence regulation via ClaR and DcpA would explain the observation that the ΔpruA mutant displays significantly stronger adherence stimulation than the ΔdcpA mutant (although the pruR mutant does not). Our recent findings suggest that PruR resides in the periplasm (Feirer and Fuqua, unpublished data), and thus it seems likely that PruR cannot exert its effect directly, and perhaps functions to impact ClaR activity through another transmembrane receptor distinct from DcpA.
Fig. 6.

Model of ClaR regulation. The tentative model of ClaR regulation is based on data described in the figures and text, and on previously published work [30]. ClaR requires PruA and PruR, but not DcpA, to exert negative regulation of A. tumefaciens biofilm formation. The biosynthesis of the dominant PruA-dependent monapterin, 2′OMet-H4MPt, is depicted, with the compound represented as a double-ringed structure with a short acyl tail. The physical association of the monapterin species with PruR in the periplasm has not been proven, but is supported by recent work. Solid arrows are enzymatic reactions and dashed arrows are putative regulatory interactions. The ClaR dual REC domain structure with the conserved D residue is shown. Synthesis of c-di-GMP is indicated for GTP by DcpA (thin arrow) and by the other active DGCs in A. tumefaciens (double-stalked arrow) that all contribute to cellular pools of the signal. The bold arrow for c-di-GMP breakdown by DcpA is meant to show that this activity dominates under standard conditions. The Wzy-type complex is diagrammatically shown for the presumptive UPP biosynthetic machinery.
Funding information
This project was supported by National Institutes of Health (NIH) grants GM080546 and GM120337 (C. F.) and GM109259 (C. M. W.). N. F. was funded on the Indiana University Genetics, Molecular and Cellular Sciences NIH Training Grant T32-GM007757.
Acknowledgements
We thank the MSU Mass Spectrometry Facility for assistance with the c-di-GMP measurements.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
Footnotes
Abbreviations: 2’OMet-H4MPt, 2’-O-Methyl- tetrahydromonapterin; ATGN, Agrobacterium tumefaciens minimal medium with glucose and ammonium sulfate; c-di-GMP, cyclic diguanylate monophosphate; CR, Congo red; CV, Crystal violet; DGC, diguanylate cyclase; LC, liquid chromatography; MS, mass spectrometry; OD, optical density; PDE, phosphodiesterase; REC, receiver domain; SOE, splicing by overlapping extension; UPP, unipolar polysaccharide; WT, wild type.
Two supplementary tables and seven supplementary figures are available with the online Supplementary Material.
Edited by: I. J. Oresnik and G. H. Thomas
References
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Flemming HC, Wingender J. The biofilm matrix. Nat Rev Microbiol. 2010;8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 3.Mckew BA, Taylor JD, Mcgenity TJ, Underwood GJ. Resistance and resilience of benthic biofilm communities from a temperate saltmarsh to desiccation and rewetting. ISME J. 2011;5:30–41. doi: 10.1038/ismej.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderl JN, Franklin MJ, Stewart PS. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob Agents Chemother. 2000;44:1818–1824. doi: 10.1128/AAC.44.7.1818-1824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mah TF, O'Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 6.Houry A, Gohar M, Deschamps J, Tischenko E, Aymerich S, et al. Bacterial swimmers that infiltrate and take over the biofilm matrix. Proc Natl Acad Sci USA. 2012;109:13088–13093. doi: 10.1073/pnas.1200791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks LR, Reddinger RM, Hakansson AP. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. MBio. 2012;3:e00200-12. doi: 10.1128/mBio.00200-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marks LR, Mashburn-Warren L, Federle MJ, Hakansson AP. Streptococcus pyogenes biofilm growth in vitro and in vivo and its role in colonization, virulence, and genetic exchange. J Infect Dis. 2014;210:25–34. doi: 10.1093/infdis/jiu058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.del Pozo JL, Patel R. The challenge of treating biofilm-associated bacterial infections. Clin Pharmacol Ther. 2007;82:204–209. doi: 10.1038/sj.clpt.6100247. [DOI] [PubMed] [Google Scholar]
- 10.van Larebeke N, Engler G, Holsters M, van den Elsacker S, Zaenen I, et al. Large plasmid in Agrobacterium tumefaciens essential for crown gall-inducing ability. Nature. 1974;252:169–170. doi: 10.1038/252169a0. [DOI] [PubMed] [Google Scholar]
- 11.Watson B, Currier TC, Gordon MP, Chilton MD, Nester EW. Plasmid required for virulence of Agrobacterium tumefaciens. J Bacteriol. 1975;123:255–264. doi: 10.1128/jb.123.1.255-264.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escobar MA, Dandekar AM. Agrobacterium tumefaciens as an agent of disease. Trends Plant Sci. 2003;8:380–386. doi: 10.1016/S1360-1385(03)00162-6. [DOI] [PubMed] [Google Scholar]
- 13.Ramey BE, Matthysse AG, Fuqua C. The FNR-type transcriptional regulator SinR controls maturation of Agrobacterium tumefaciens biofilms. Mol Microbiol. 2004;52:1495–1511. doi: 10.1111/j.1365-2958.2004.04079.x. [DOI] [PubMed] [Google Scholar]
- 14.Abarca-Grau AM, Penyalver R, López MM, Marco-Noales E. Pathogenic and non-pathogenic Agrobacterium tumefaciens, A. rhizogenes and A. vitis strains form biofilms on abiotic as well as on root surfaces. Plant Pathol. 2011;60:416–425. doi: 10.1111/j.1365-3059.2010.02385.x. [DOI] [Google Scholar]
- 15.Heindl JE, Wang Y, Heckel BC, Mohari B, Feirer N, et al. Mechanisms and regulation of surface interactions and biofilm formation in Agrobacterium. Front Plant Sci. 2014;5:176. doi: 10.3389/fpls.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomlinson AD, Fuqua C. Mechanisms and regulation of polar surface attachment in Agrobacterium tumefaciens. Curr Opin Microbiol. 2009;12:708–714. doi: 10.1016/j.mib.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li G, Brown PJ, Tang JX, Xu J, Quardokus EM, et al. Surface contact stimulates the just-in-time deployment of bacterial adhesins. Mol Microbiol. 2012;83:41–51. doi: 10.1111/j.1365-2958.2011.07909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J, Kim J, Danhorn T, Merritt PM, Fuqua C. Phosphorus limitation increases attachment in Agrobacterium tumefaciens and reveals a conditional functional redundancy in adhesin biosynthesis. Res Microbiol. 2012;163:674–684. doi: 10.1016/j.resmic.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthysse AG, Holmes KV, Gurlitz RH. Elaboration of cellulose fibrils by Agrobacterium tumefaciens during attachment to carrot cells. J Bacteriol. 1981;145:583–595. doi: 10.1128/jb.145.1.583-595.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthysse AG, Marry M, Krall L, Kaye M, Ramey BE, et al. The effect of cellulose overproduction on binding and biofilm formation on roots by Agrobacterium tumefaciens. Mol Plant Microbe Interact. 2005;18:1002–1010. doi: 10.1094/MPMI-18-1002. [DOI] [PubMed] [Google Scholar]
- 21.Tomlinson AD, Ramey-Hartung B, Day TW, Merritt PM, Fuqua C. Agrobacterium tumefaciens ExoR represses succinoglycan biosynthesis and is required for biofilm formation and motility. Microbiology. 2010;156:2670–2681. doi: 10.1099/mic.0.039032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heckel BC, Tomlinson AD, Morton ER, Choi JH, Fuqua C. Agrobacterium tumefaciens exoR controls acid response genes and impacts exopolysaccharide synthesis, horizontal gene transfer, and virulence gene expression. J Bacteriol. 2014;196:3221–3233. doi: 10.1128/JB.01751-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heindl JE, Hibbing ME, Xu J, Natarajan R, Buechlein AM, et al. Discrete responses to limitation for Iron and Manganese in Agrobacterium tumefaciens: influence on attachment and biofilm formation. J Bacteriol. 2015;198:816–829. doi: 10.1128/JB.00668-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merritt PM, Danhorn T, Fuqua C. Motility and chemotaxis in Agrobacterium tumefaciens surface attachment and biofilm formation. J Bacteriol. 2007;189:8005–8014. doi: 10.1128/JB.00566-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Kim J, Koestler BJ, Choi JH, Waters CM, et al. Genetic analysis of Agrobacterium tumefaciens unipolar polysaccharide production reveals complex integrated control of the motile-to-sessile switch. Mol Microbiol. 2013;89:929–948. doi: 10.1111/mmi.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schirmer T, Jenal U. Structural and mechanistic determinants of c-di-GMP signalling. Nat Rev Microbiol. 2009;7:724–735. doi: 10.1038/nrmicro2203. [DOI] [PubMed] [Google Scholar]
- 27.Römling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sondermann H, Shikuma NJ, Yildiz FH. You've come a long way: c-di-GMP signaling. Curr Opin Microbiol. 2012;15:140–146. doi: 10.1016/j.mib.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amikam D, Benziman M. Cyclic diguanylic acid and cellulose synthesis in Agrobacterium tumefaciens. J Bacteriol. 1989;171:6649–6655. doi: 10.1128/jb.171.12.6649-6655.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feirer N, Xu J, Allen KD, Koestler BJ, Bruger EL, et al. A pterin-dependent signaling pathway regulates a dual-function diguanylate cyclase-phosphodiesterase controlling surface attachment in Agrobacterium tumefaciens. MBio. 2015;6:e00156-15. doi: 10.1128/mBio.00156-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howie AJ, Brewer DB. Optical properties of amyloid stained by Congo red: history and mechanisms. Micron. 2009;40:285–301. doi: 10.1016/j.micron.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Morton ER, Fuqua C. Genetic manipulation of Agrobacterium. Curr Protoc Microbiol. 2012;Chapter 3:Unit 3D 2. doi: 10.1002/9780471729259.mc03d02s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tempé J, Petit A, Holsters M, Montagu M, Schell J. Thermosensitive step associated with transfer of the Ti plasmid during conjugation: possible relation to transformation in crown gall. Proc Natl Acad Sci USA. 1977;74:2848–2849. doi: 10.1073/pnas.74.7.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan SR, Gaines J, Roop RM, Farrand SK. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol. 2008;74:5053–5062. doi: 10.1128/AEM.01098-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morton ER, Fuqua C. Phenotypic analyses of Agrobacterium. Curr Protoc Microbiol. 2012;Chapter 3:Unit 3D. doi: 10.1002/9780471729259.mc03d03s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massie JP, Reynolds EL, Koestler BJ, Cong JP, Agostoni M, et al. Quantification of high-specificity cyclic diguanylate signaling. Proc Natl Acad Sci USA. 2012;109:12746–12751. doi: 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 38.Wadhams GH, Armitage JP. Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol. 2004;5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Swanson RV, Simon MI, Weis RM. The response regulators CheB and CheY exhibit competitive binding to the kinase CheA. Biochemistry. 1995;34:14626–14636. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- 40.Welch M, Oosawa K, Aizawa S, Eisenbach M. Phosphorylation-dependent binding of a signal molecule to the flagellar switch of bacteria. Proc Natl Acad Sci USA. 1993;90:8787–8791. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7-A resolution. J Biol Chem. 1991;266:15511–15519. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- 42.Stock AM, Mottonen JM, Stock JB, Schutt CE. Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature. 1989;337:745–749. doi: 10.1038/337745a0. [DOI] [PubMed] [Google Scholar]
- 43.Volz K. Structural conservation in the CheY superfamily. Biochemistry. 1993;32:11741–11753. doi: 10.1021/bi00095a001. [DOI] [PubMed] [Google Scholar]
- 44.Sanders DA, Gillece-Castro BL, Stock AM, Burlingame AL, Koshland DE. Identification of the site of phosphorylation of the chemotaxis response regulator protein, CheY. J Biol Chem. 1989;264:21770–21778. [PubMed] [Google Scholar]
- 45.Lukat GS, Stock AM, Stock JB. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry. 1990;29:5436–5442. doi: 10.1021/bi00475a004. [DOI] [PubMed] [Google Scholar]
- 46.Lee SY, Cho HS, Pelton JG, Yan D, Henderson RK, et al. Crystal structure of an activated response regulator bound to its target. Nat Struct Biol. 2001;8:789–794. doi: 10.1038/nsb0901-789. [DOI] [PubMed] [Google Scholar]
- 47.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, et al. Crystal structure of activated CheY. Comparison with other activated receiver domains. J Biol Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 48.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright EL, Deakin WJ, Shaw CH. A chemotaxis cluster from Agrobacterium tumefaciens. Gene. 1998;220:83–89. doi: 10.1016/S0378-1119(98)00438-7. [DOI] [PubMed] [Google Scholar]
- 50.Harighi B. Role of CheY1 and CheY2 in the chemotaxis of A. tumefaciens toward acetosyringone. Curr Microbiol. 2008;56:547–552. doi: 10.1007/s00284-008-9120-1. [DOI] [PubMed] [Google Scholar]
- 51.Sourjik V, Schmitt R. Different roles of CheY1 and CheY2 in the chemotaxis of Rhizobium meliloti. Mol Microbiol. 1996;22:427–436. doi: 10.1046/j.1365-2958.1996.1291489.x. [DOI] [PubMed] [Google Scholar]
- 52.Sourjik V, Schmitt R. Phosphotransfer between CheA, CheY1, and CheY2 in the chemotaxis signal transduction chain of Rhizobium meliloti. Biochemistry. 1998;37:2327–2335. doi: 10.1021/bi972330a. [DOI] [PubMed] [Google Scholar]
- 53.Aldridge P, Jenal U. Cell cycle-dependent degradation of a flagellar motor component requires a novel-type response regulator. Mol Microbiol. 1999;32:379–391. doi: 10.1046/j.1365-2958.1999.01358.x. [DOI] [PubMed] [Google Scholar]
- 54.Paul R, Weiser S, Amiot NC, Chan C, Schirmer T, et al. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 2004;18:715–727. doi: 10.1101/gad.289504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan C, Paul R, Samoray D, Amiot NC, Giese B, et al. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci USA. 2004;101:17084–17089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curtis PD, Brun YV. Getting in the loop: regulation of development in Caulobacter crescentus. Microbiol Mol Biol Rev. 2010;74:13–41. doi: 10.1128/MMBR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barnhart DM, Su S, Baccaro BE, Banta LM, Farrand SK. CelR, an ortholog of the diguanylate cyclase PleD of Caulobacter, regulates cellulose synthesis in Agrobacterium tumefaciens. Appl Environ Microbiol. 2013;79:7188–7202. doi: 10.1128/AEM.02148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim J, Heindl JE, Fuqua C. Coordination of division and development influences complex multicellular behavior in Agrobacterium tumefaciens. PLoS One. 2013;8:e56682. doi: 10.1371/journal.pone.0056682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho HS, Lee SY, Yan D, Pan X, Parkinson JS, et al. NMR structure of activated CheY. J Mol Biol. 2000;297:543–551. doi: 10.1006/jmbi.2000.3595. [DOI] [PubMed] [Google Scholar]
- 60.Villali J, Pontiggia F, Clarkson MW, Hagan MF, Kern D. Evidence against the "Y-T coupling" mechanism of activation in the response regulator NtrC. J Mol Biol. 2014;426:1554–1567. doi: 10.1016/j.jmb.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desai SK, Kenney LJ. To approximately P or Not to approximately P? Non-canonical activation by two-component response regulators. Mol Microbiol. 2017;103:203–213. doi: 10.1111/mmi.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dahl MK, Msadek T, Kunst F, Rapoport G. The phosphorylation state of the DegU response regulator acts as a molecular switch allowing either degradative enzyme synthesis or expression of genetic competence in Bacillus subtilis. J Biol Chem. 1992;267:14509–14514. [PubMed] [Google Scholar]
- 63.Kobayashi K. Gradual activation of the response regulator DegU controls serial expression of genes for flagellum formation and biofilm formation in Bacillus subtilis. Mol Microbiol. 2007;66:395–409. doi: 10.1111/j.1365-2958.2007.05923.x. [DOI] [PubMed] [Google Scholar]
- 64.Lin W, Wang Y, Han X, Zhang Z, Wang C, et al. Atypical OmpR/PhoB subfamily response regulator GlnR of actinomycetes functions as a homodimer, stabilized by the unphosphorylated conserved Asp-focused charge interactions. J Biol Chem. 2014;289:15413–15425. doi: 10.1074/jbc.M113.543504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wuichet K, Zhulin IB. Origins and diversification of a complex signal transduction system in prokaryotes. Sci Signal. 2010;3:ra50. doi: 10.1126/scisignal.2000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He K, Bauer CE. Chemosensory signaling systems that control bacterial survival. Trends Microbiol. 2014;22:389–398. doi: 10.1016/j.tim.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kirby JR, Zusman DR. Chemosensory regulation of developmental gene expression in Myxococcus xanthus. Proc Natl Acad Sci USA. 2003;100:2008–2013. doi: 10.1073/pnas.0330944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Black WP, Yang Z. Myxococcus xanthus chemotaxis homologs DifD and DifG negatively regulate fibril polysaccharide production. J Bacteriol. 2004;186:1001–1008. doi: 10.1128/JB.186.4.1001-1008.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berleman JE, Bauer CE. A che-like signal transduction cascade involved in controlling flagella biosynthesis in Rhodospirillum centenum. Mol Microbiol. 2005;55:1390–1402. doi: 10.1111/j.1365-2958.2005.04489.x. [DOI] [PubMed] [Google Scholar]
- 70.Berleman JE, Bauer CE. Involvement of a Che-like signal transduction cascade in regulating cyst cell development in Rhodospirillum centenum. Mol Microbiol. 2005;56:1457–1466. doi: 10.1111/j.1365-2958.2005.04646.x. [DOI] [PubMed] [Google Scholar]
- 71.D'Argenio DA, Calfee MW, Rainey PB, Pesci EC. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol. 2002;184:6481–6489. doi: 10.1128/JB.184.23.6481-6489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci USA. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Güvener ZT, Harwood CS. Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol Microbiol. 2007;66:1459–1473. doi: 10.1111/j.1365-2958.2007.06008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huangyutitham V, Güvener ZT, Harwood CS. Subcellular clustering of the phosphorylated WspR response regulator protein stimulates its diguanylate cyclase activity. MBio. 2013;4:e00242-13. doi: 10.1128/mBio.00242-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.