

Abstract
Aim
Extracellular superoxide dismutase (ECSOD) and the cysteine/glutamate transporter (Cys)/(xCT) are tumor microenvironment (TME) redox state homeostasis regulators. Altered expression of ECSOD and xCT can lead to imbalance of the TME redox state and likely have a profound effect on cancer invasion. In the present study, we investigated whether ECSOD and xCT could be therapeutic targets for prostate cancer (PCa) invasion.
Results
Immunohistochemistry of tumor microarray PCa tissues (N=165) with high Gleason scores indicated that xCT protein expression is significantly increased while ECSOD protein expression is significantly decreased. Metastatic PCa indicated ECSOD protein expression is significantly decreased in epithelial area whereas xCT protein expression is significantly increased in stromal area. Furthermore, inhibition of extracellular O2•− by overexpression of ECSOD or alteration of the extracellular Cys/CySS ratio by knockdown of xCT protein inhibited PCa cell invasion. Simultaneous overexpression of ECSOD and knockdown xCT inhibited PCa cell invasion more than overexpression of ECSOD or knockdown of xCT alone. In the co-culturing system, simultaneous overexpression of ECSOD and knockdown of xCT in prostate stromal WPMY-1 cells inhibited PCa cell invasiveness more than overexpression of ECSOD alone. The decrease in PCa invasion correlated with increased of extracellular H2O2 levels. Notably, overexpression of catalase in TME reversed the inhibitory effect of ECSOD on cancer cell invasion.
Conclusion
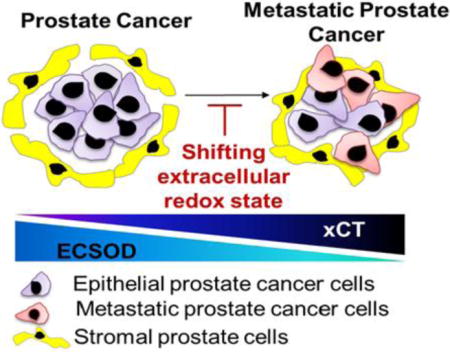
Impaired ECSOD activity and an upregulated of xCT protein expression may be clinical features of an aggressive PCa, particularly metastatic cancers and/or those with a high Gleason score. Therefore, shifting the extracellular redox state toward an oxidizing status by targeted modulation of ECSOD and xCT, in both cancer and stromal cells, may provide a greater strategy for potential therapeutic interventions of aggressive PCa.
Keywords: ECSOD, xCT, Redox state, Invasion
Graphical Abstract
Introduction
Redox state (reduction/oxidation) is the balance of reducing and oxidizing equivalents and plays important roles in several physiologic and pathologic processes. Therapies that target redox imbalances have been added to cancer regimens to target redox–related proteins that influence carcinogenesis and metastasis. Redox imbalance results from altered levels of reactive oxygen species/reactive nitrogen species (ROS/RNS) and/or antioxidant proteins. The major species of ROS/RNS include hydrogen peroxide (H2O2), hydroxyl radical (·OH), superoxide radical (O2•−), nitric oxide (NO•), and peroxynitrite (OONO•) [1]. Studies to date focused on the intracellular redox state; little is known about the redox state in the extracellular spaces (extracellular matrix and stromal cells). We previously demonstrated that an imbalance of cysteine/cystine (Cys/CySS) pools in extracellular spaces promoted an alteration of the intracellular redox state that could be monitored by the redox status of thioredoxin 1 [2–4]. The extracellular redox state acts in concert with the intracellular redox state to control influx and efflux of ROS/RNS [1, 2, 5]. Both ROS/RNS and Cys/CySS are associated with invasion, hypoxia, angiogenesis, and acidosis in the extracellular spaces of cancerous tissues [1, 6, 7].
Among redox state regulators, the cysteine/glutamate transporter (xCT) and extracellular superoxide dismutase (ECSOD) are the primarily tumor microenvironment (TME)-redox state homeostasis regulators; the former controls the redox coupling ratio (Cys/CySS) and the latter controls the prooxidant levels including O2•− and H2O2 [1]. ECSOD (SOD3) is the isoform of superoxide dismutase (SOD) that contains a signaling peptide, which directs ECSOD to the extracellular spaces and facilitates its binding to heparan sulfate proteoglycans [1, 8]. ECSOD serves as a modulator of redox homeostasis in extracellular spaces by removing plasma membranes/extracellular O2•− and producing H2O2 [1, 8, 9]. ECSOD has a longer half-life in the circulation relative to the other two SOD isoforms, copper zinc SOD and manganese SOD [10]. ECSOD expression is influenced by multiple stimuli, including angiotensin II, NO•, exercise training, and certain pathological states [8]. Several studies indicate that ECSOD has an anti-tumor effects [11], thus it is reduced in several human cancers including colon, lung, breast, thyroid, mammary, and pancreatic ductal adenocarcinoma [12–15].
On the other hand, xCT (SLC7A11) is a plasma membrane transporter of CySS from outside the cell to inside. While inside the cell, CySS is rapidly reduced to Cys by glutathione (GSH) or thioredoxin reductase 1, and is then either synthesized into GSH or exported to the extracellular spaces [16]. The constant flow of Cys from inside the cells provides a reducing extracellular microenvironment and consequently, Cys levels are low inside the cells [17]. Commonly, xCT is not universally expressed by cells; overexpression of xCT is reported for various cancers including aggressive prostate cancer (PCa) [3], colon [18], glioma [19], head and neck [20], lung [21, 22], breast [23, 24], ovarian [25, 26], liver [27], and in cancer stem-like cells [28]. Due to its ability to export glutamate in exchange for CySS, down-regulation of xCT expression markedly decreases cancer cell viability and subsequently restricts the cancer cell’s ability to switch between glucose and glutamine [16, 22].
PCa incidence is increasing significantly worldwide [29]. Metastatic PCa is the most lethal form of this disease and a better understanding of its development may lead to effective targeted therapies. The redox state in PCa cells, stromal cells, and adjacent benign tissue has not been comprehensively studied. Herein, we analyzed a large number of prostate tissue samples to determine the expression levels of the ECSOD and xCT proteins in human benign tissues and in PCa tissues of varying Gleason scores and clinical parameters. In addition, we modulated the levels of ECSOD and xCT in both PCa epithelial and stromal prostate cells to investigate the extracellular redox state upon PCa invasion. The present study demonstrates that modulation of various TME components, for both PCa and stromal prostate cells, provides an effective anti-invasive effect that may have application as therapeutic interventions.
Materials and Methods
Tumor microarray construction and immunohistochemistry staining
Paraffin-embedded tissue blocks were selectively cored from 165 PCa patients and 34 benign control patients, who underwent radical prostatectomy at the University of Kentucky, to construct a tissue microarray (TMA) through the Markey Biospecimen and Tissue Procurement Shared Resource, University of Kentucky-Lexington. Approval for use of human prostate tissue was obtained from the University of Kentucky Institutional Review Board. Tissue cores (2 mm) containing cancerous tissues, adjacent benign epithelial tissues, or human benign prostatic hyperplasia (BPH) were used for the construction of tissue microarrays with duplicate cores from each patient. There were 4 disease groups: Gleason scores ≥ 8, 7, ≤ 6, and BPH. PCa-TMA slides were deparaffinized in a 60°C oven for 1 h and then rinsed in 3 changes of xylene for 10 min each. To block endogenous peroxidases, slides were immersed in 0.3% methanol/hydrogen peroxide for 20 min and then rehydrated for 1 min in each of 100%, 95%, 75%, and 50% ethanol and then double-distilled H2O. Heat-induced epitope retrieval was performed using a digital decloaking chamber (BioCare Medical, Concord, CA) for 20 minutes at 110°C in DAKO high pH EDTA or low pH citrate antigen retrieval buffer, as indicated. Endogenous peroxidase activity was quenched using reagent from Envision+ Kits (DAKO, Glostrup Municipality, Denmark), followed by incubation with primary antibodies ECSOD (high pH, 1:100) or xCT (low pH, 1:50) overnight at 4°C in a humidified chamber. After washing, slides were incubated with Envision+ polymer-bound secondary antibody (DAKO), then visualized with 3,3′-Diaminobenzidine and lightly counterstained with hematoxylin. Slides were then dehydrated, cleared, and mounted with coverslips; followed by microscopic analysis. Negative control slides had the primary antibody replaced with rabbit or goat serum for quality control and validation of the staining. The information about PCa-TMA samples is displayed in Table 1. The assessments of ECSOD and xCT immunoreactivity were quantified with the Aperio imaging analysis system - Version 1 (Aperio Technologies, Inc., Vista, CA, USA), which can accurately measure protein expression patterns and morphometric characteristics in distinct tissue regions of interest (epithelial vs. stromal areas). The cytoplasmic algorithm was set to analyze the intensity (positive pixel count) of ECSOD and xCT in order to obtain an objective evaluation and avoid subjective interpretation (Supplementary Fig. 1). The threshold intensity for positive areas (weak, medium, and strong positive) ranged from 0–220. The threshold for strong positive intensity ranged from 0–100. The average intensities of ECSOD and xCT in the epithelial area were obtained from the strong intensity areas in each core, while the average intensities of ECSOD and xCT in the stromal area were obtained from all positive areas in each core (Fig. 1).
Table 1.
Characteristics and clinical parameters of participants
| PCa-TMA Description | Clinical parameters | # of Cases |
|---|---|---|
| Tissues | BPH | 34 |
| PCa | 165 | |
| PCa | Gleason ≤ 6 | 33 |
| Gleason 7 | 100 | |
| Gleason ≥ 8 | 32 | |
| Clinical Stage | Stage I | 3 |
| Stage II | 86 | |
| Stage III | 47 | |
| Stage IV | 18 | |
| Unknown | 11 | |
| Metastatic PCa | Organ Confined | 141 |
| Metastatic PCa | 13 | |
| Unknown | 11 | |
| Smoking | Tobacco/cigar/others | 52 |
| Non-smoking | 56 | |
| Unknown | 57 | |
| Recurrence | Local/Distal Recurrence | 31 |
| No Recurrence | 82 | |
| Unknown | 52 | |
| PSA | < 10 ng/ml | 3 |
| 10–20 ng/ml | 4 | |
| > 20 ng/ml | 66 | |
| Age range (years) | BPH | 57–81 |
| PCa | 41–75 |
Figure 1. Quantitative analysis of ECSOD and xCT expression in PCa tissues.
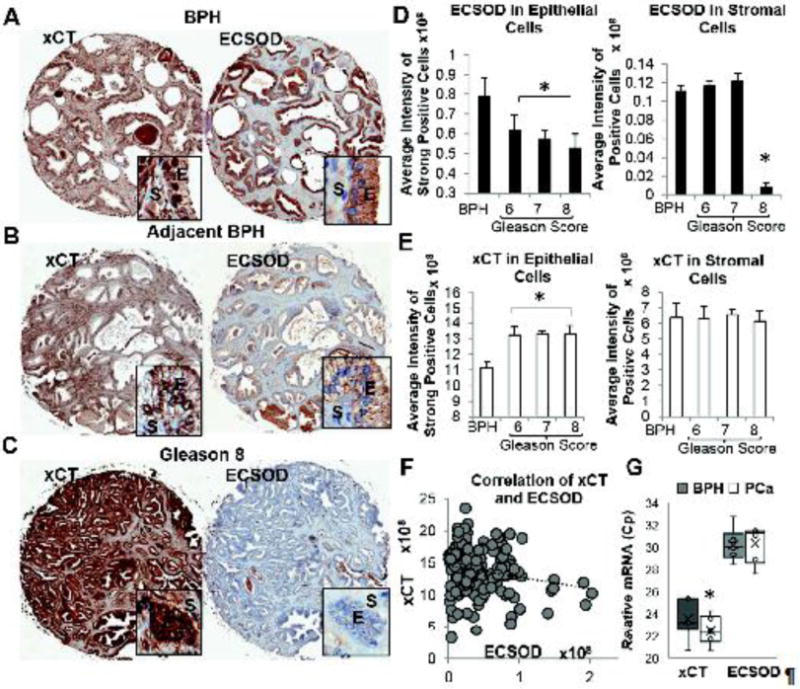
Reversible expression pattern of ECSOD and xCT on the same tissues: (A) BPH=Benign Prostatic Hyperplasia, (B) Adjacent BPH counterpart; and (C) PCa tissue (B and C are from the same patients). E=Epithelial cells, S=Stromal cells. Image magnifications: 20X for tissues and 400X for inserts. Quantitative analysis of (D) ECSOD and (E) xCT expression. ECSOD expression is significantly decreased in PCa epithelial areas while xCT expression is significantly increased in PCa epithelial areas. Intensity from strong positive area of epithelial cells was used for the analysis. (F) A negative linear correlation of ECSOD vs. xCT in PCa epithelial areas with Pearson r value = −0.198 and p-value = 0.018. Total pixel intensity of all areas (weak, medium, and strong positive areas) was used for the analysis. (G) RT-qPCR of ECSOD and xCT in prostate tissues. Relative mRNA levels of ECSOD and xCT were quantified by qRT-PCR as described in Materials and Methods. Lower Cp value indicates higher expression level. *p ≤ 0.05 when compared to BPH.
RNA extraction and real-time quantitative PCR
Total RNA was isolated from flash frozen PCa and BPH tissues (Bioserve, Beltsville, MD) using the MagNA Pure 96 System (Roche Diagnosis USA, Indianapolis, IN). RNA concentrations were determined using an RNA 6000 Nano Kit (Agilent Waldbronn, Germany). The first-strand cDNA was reverse-transcribed and then pre-amplified with the Transcriptor first strand cDNA synthesis kit and cDNA Pre-Amp Master Mix (Roche Diagnosis, USA). qPCR was performed with the LightCycler 480 Probes Master system (Roche Diagnosis, USA). All experiments were performed following manufacturers’ instructions. Actin was used as the endogenous control for normalization of gene expression. Primers (Thermo Fisher Scientific) used for qRT-PCR were as follows: SLC7A11 5′-ccatgaacggtggtgtgtt-3′ (forward) and 5′-gaccctctcgagacgcaac-3′ (reverse), SOD3 5′-ggtgcagctctcttttcagg-3′ (forward) and 5′-aacacagtagcgccagcat-3′ (reverse), β-actin 5′-ccaaccgcgagaagatga-3′ (forward) and 5′-ccagaggcgtacagggatag-3′ (reverse). Relative mRNA copy number of ECSOD and xCT were reported as crossing point-PCR-cycle (Cp).
Chemicals and reagents
All chemicals and reagents were purchased from Sigma Chemical Co., unless otherwise specified. Tissue culture supplies were from Falcon (BD Biosciences, San Jose, CA). All tissue culture reagents and Calcein-AM fluorescence dye were obtained from Thermo Fisher Scientific. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). Polycarbonated (PCF) inserts were purchased from Corning (Tewksbury MA). Matrigel was purchased from BD Biosciences. All antibodies were purchased from Santa Cruz Biotechnology, Inc., unless otherwise specified.
Cell culture and treatment
PC3 and WPMY1 cells (stromal cell) were obtained from the American Type Culture Collection. Normal prostate epithelial cells (PrEC) were obtained from Lonza Inc. (Walkersville, MA). PC3 cells were cultured in RPMI 1640 supplemented with 10% FBS and 100 mg/L kanamycin sulfate. WPMY1 cells were cultured in keratinocyte serum-free medium supplemented with 50 mg/L bovine pituitary extract, 5 μg/L recombinant epidermal growth factor, and 100 mg/L kanamycin sulfate. PrEC cells were grown in serum-free prostate epithelial growth media (Lonza/Cambrex). All cell lines were grown at 37°C in a humidified atmosphere of 95% air and 5% CO2. Various concentrations of SOD, catalase (CAT), 5-aza-2′-deoxycytidine (5-Aza-dC), or sulfasalazine (SASP) were added to the medium for various time points. For each experiment, a preliminary time course and dose-response study of these compounds was performed; only the optimal conditions were reported in the present study. Cells were authenticated using short tandem repeat profiling by ATCC.
Adenovirus gene transduction and siRNA transfection
The adenoviral vector containing human ECSOD cDNA (AdhSOD3) and the control adenovirus with no gene insert (AdEmpty) were purchased from ViraQuest, Inc. (Iowa City, Iowa). Adenovirus containing green fluorescent protein (GFP) cDNA was used to determine transduction efficiency. Transduction efficiency of the adenoviral vector containing GFP was >85% in all cell lines as measured by fluorescence intensity of GFP in flow cytometry analysis (data not show). The adenovirus constructs and the procedure of transduction of adenoviral vectors were previously described [30]. Our published data indicate that transduction of AdhSOD3 at 300 multiplicity of infection (MOI) [30] provides optimal expression of ECSOD (at least up to 5 days after transduction). Heparin was added to the media to activate ECSOD secretion. Cells harvested at 72 h post-transduction. For siRNA, siRNA xCT (sc-76933) and scrambled (sc-36869) were purchased from Santa Cruz. siRNA transfection was performed as previously described [2]. siRNA at 40 ng/ml were used in all experiments.
In vitro invasion assay
PC3 (2.5 × 105/mL) cells with or without gene modification were cultured in serum-free RPMI 1640 before seeding onto the upper chamber. Serum-free RPMI 1640 and complete medium were added to the upper and lower chambers, respectively. After incubation for 24 h, cells that had invaded through Matrigel coated PCF membranes were detached using the cell dissociation buffer and detected using Calcein-AM fluorescence dye. Due to the close interaction between PCa epithelial and stromal cells in tissue, the effect of stromal cells on PCa invasion was investigated using a co-culturing system. Stromal cells were placed in the bottom of the chamber while PC3 cells were placed on the insert. Paracrine signaling from stromal cells induced the PC3 cells to invade from the top surface through the bottom surface of the insert (Supplementary Fig 2).
Analysis of Cys, CySS, GSH, and glutathione disulfide (GSSG)
Concentrations of Cys, CySS, GSH, and GSSG in cell culture media were determined by HPLC with fluorescence detection as described in a previous publication [2]. Briefly, media was added (1:1) to ice-cold 10% perchloric acid solution containing 0.2 M boric acid. Samples were derivatized with iodoacetic acid (IAA) and dansyl chloride. Thiols and disulfides were separated and quantified by integration of their HPLC peaks relative to the internal standard (gamma-glutamylglutamate). Extracellular redox potential of the redox couples Cys/CySS (EhCySS) and GSH/GSSG (EhGSSG) were calculated using the Nernst equation, EhCySS = −250 + 30 log [(CySS)/(Cys)2] and EhGSSG = −264 + 30 log [(GSSG)/(GSH)2], respectively.
Extracellular H2O2 and nitrite measurements
External H2O2 and nitrite levels (which are a stable and nonvolatile breakdown product of NO•) were analyzed following the manufacturer’s instructions based on the Amplex Red (ThermoFisher, Waltham, MA) and the Griess reagent system (Promega Co. Madison, WI), respectively. Media decanted from growing cells were analyzed in these experiments.
Intracellular ROS/RNS measurements
Briefly, cells were incubated with phenol red-free media containing 10 μM 2′,7′-dichlorofluorescein diacetate (H2DCFDA) or 5-(and-6)-carboxy-2′,7′-dichlorofluorescein diacetate (CDCFDA, oxidation-insensitive dye) (Thermo Fisher) for 30 min at 37°C. CDCFDA was used to normalize uptake, efflux, and ester cleavage of H2DCFDA. The data are presented as the ratio of dichlorofluorescein (DCF) to 5-(and-6)-carboxy dichlorofluorescein (CDCF) [31].
Matrix metalloproteinase (MMP) activity
Conditioned media were concentrated using an Amicon Ultra-15 filter. MMP2/9 activities (Merck Millipore, Billerica, MA) were measured in the media by following the manufacturer’s protocol.
Western blot analysis
The protocols of western blot analysis were detailed in a previous publication [32]. Proteins from crude supernatants or concentrated conditioned media were placed in each well. Proteins were identified using antibodies to ECSOD, xCT, or β-actin. The data images were analyzed using Odyssey software.
Statistics
All vitro experiments were repeated at least three separate times. One-way ANOVA or Student’s two-sample t-test was used to analyze the mean difference across groups. Multiple pairwise comparisons between groups were adjusted using LSD. Quantitative levels of ECSOD and xCT from BPH and PCa patients were calculated from the Aperio system. ECSOD and xCT immunohistochemistry (IHC) expression levels were then compared between BPH vs. PCa groups and across Gleason groups using two sample t-test, one-way ANOVA, and post hoc test. Correlation between ECSOD with xCT expressions from PCa patients and with prostate specific antigen levels and age were calculated using Pearson’s correlation coefficient while univariate comparison of IHC with clinical features; smoking history, metastasis, recurrence, and survival, was performed using two group and multiple group comparisons. We then utilized total intensity positive pixels of ECSOD and xCT from PCa patients versus BPH subjects to calculate the Youden’s index [33] in order to dichotomize ECSOD and xCT into two levels (low vs. high expression levels). Overall survival was estimated using Kaplan Meier curves between high vs. low ECSOD and xCT levels and compared using the generalized Wilcoxon test. Statistical analyses were performed with SPSS 14 software (SPSS, Inc., Chicago, IL, USA) and SAS 9.4 (Cary, NC). A two-sided p-value of less than 0.05 was considered significant.
Results
The extracellular redox state is progressively altered as PCa becomes more aggressive with a decrease in ECSOD and an increase in xCT protein expression
In order to understand the role of ECSOD and xCT during PCa progression, we analyzed the expression levels of ECSOD and xCT in prostate epithelial and stromal cells of BPH and PCa tissues of varying Gleason scores. As displayed in Table 1, a total of 165 PCa tissues and 34 BPH tissues were used in construction of a PCa-tumor microarray (PCa-TMA). Among PCa samples, stage II disease (52%) and Gleason score 7 (60%) are the predominant types (Table 1). Local recurrence was exhibited in 19% of patients, while 18% of patients experienced distant metastatic cancer. The average age of PCa patients was 58 and 32% of PCa patients had a smoking history. In general, staining intensity for both ECSOD and xCT was stronger in epithelial cells than stromal cells. Remarkably, a high Gleason score demonstrated intense staining for xCT and a weak staining for ECSOD in PCa when compared to adjacent BPH from the same patients (Fig. 1A–C). Quantitative analysis of the staining intensity of ECSOD and xCT expression using the Aperio system are presented in Fig. 1D–E. High grade (Gleason score ≥ 8), intermediate grade (Gleason score 7), and low grade (Gleason score 6) PCa tissues exhibited significantly higher levels of xCT but lower levels of ECSOD in epithelial area compared to adjacent BPH tissues. The decrease of ECSOD in epithelial areas was significant across all Gleason scores. PCa with Gleason score ≥ 8 demonstrated a greater decrease in ECSOD in epithelial areas compared to samples with lower Gleason scores of 7 and 6. Interestingly, ECSOD expression in stromal areas was significantly decreased only in PCa with Gleason score ≥ 8. In contrast, xCT expression was significantly increased in PCa epithelial areas but not in the stromal areas. Concomitant down-regulation of ECSOD and up-regulation of xCT expressions was also observed in PCa with stages II, II, and IV (Supplementary Fig. 3). Pearson correlation confirms the reverse correlation; as ECSOD level is decreased while xCT level is increased in each sample (Fig. 1F).
Next, we evaluated ECSOD and xCT mRNA expressions in specimens obtained from PCa’s patients with Gleason score ≥ 8 (N=4) compared to BPH (N=4). The relative mRNA expression levels of ECSOD and xCT in BPH and PCa are shown in Fig. 1G indicating that xCT levels were significantly increased in PCa patients (lower Cp indicates higher level of mRNA) while ECSOD levels were slightly decreased. Oncomine data analysis revealed the similar expression trends for ECSOD and xCT for all three prostate data sets (Grasso, Taylor, and Yu) with one data set reaching statistical significance (Supplementary Fig. 4).
Analysis of ECSOD and xCT expression with clinical outcome revealed that ECSOD expression was significantly decreased, especially in the epithelial area of metastatic PCa; in contrast, xCT protein expression was significantly increased in the stromal area (Fig. 2A). Furthermore, ECSOD expression in epithelial area significantly decreased in recurrence PCa but not xCT expression (Fig. 2B). xCT expression slightly increased in PCa patients who resist to radiation treatment (Supplementary Fig. 5). More importantly, Kaplan-Meier survival curve of high expression of xCT (cut-off at 1.4 × 109) is associated with low survival rate of PCa patients after diagnosis based on the generalized Wilxoson test, whereas low expression of ECSOD was not (cut-off at 7.5 × 106) (Fig. 2C). Data from Figs. 1 and 2 suggest that the redox state at the cell surface may be modified due to changes in ECSOD and xCT levels. Low ECSOD levels with a Gleason score ≥ 8 and metastasis PCa indicates that significant redox imbalance precedes malignancy.
Figure 2. Correlation of ECSOD and xCT expression with PCa clinical parameters.
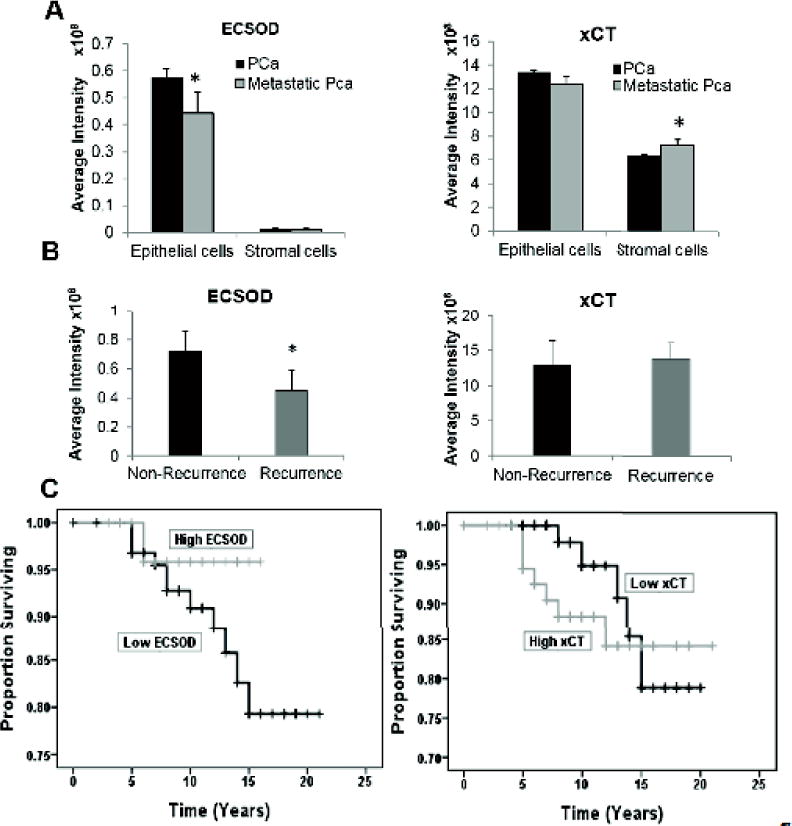
(A) Expression levels of ECSOD and xCT in epithelial and stromal areas of metastatic PCa. (B) Expression levels of ECSOD and xCT in epithelial area of recurrence PCa. (C) Kaplan-Meier curves of PCa patients from University of Kentucky’s hospital who have high vs. low expression of ECSOD (generalized Wilcoxon p-value = 0.5) or xCT (generalized Wilcoxon p-value =0.068). *p ≤ 0.05 when compared to PCa.
Simultaneously increasing ECSOD and inhibiting xCT expression suppresses PCa cell invasion, alters extracellular H2O2 production, and shifts intra- and extracellular redox states toward the oxidizing state in PCa cells
We confirmed that the extracellular redox states in PCa cell lines mirrored data obtained from the human PCa tissues. As demonstrated in Fig. 3A, PC3 cells exhibited significantly higher levels of xCT and significantly lower levels of ECSOD compared with normal human prostate epithelial cells (PrEC), a finding consistent with those from PCa tissues. Down-regulation of ECSOD expression was suggested to be from methylation of the ECSOD promoter [3]. We treated PrEC and PC3 cells with 5 μM 5-Aza-dC, a DNA methylation inhibitor, for 96 h and found that 5-Aza-dC restored ECSOD expression levels when compared with non-treatment group (Fig. 3B). In addition, it is well known that expression of xCT is regulated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2) in response to oxidative stress [24]. Herein, western blot analysis confirmed that up-regulation of xCT in PC3 cells is correlated with the up-regulation of Nrf2 expression (Fig. 3C). Next, we addressed the role of ECSOD and xCT in PCa cell invasion by treating PC3 cells with either (1) 100 µM SASP, a xCT inhibitor, (2) 100 Units SOD (non-permeable purified), or (3) 100 µM SASP plus 100 Units SOD in the upper chamber for 18 hs. As shown in Fig. 3D, combined treatment of SASP and non-permeable purified SOD in the media significantly decreased PCa invasion more effectively than addition of SOD or SASP alone. Due to the possibility of non-specific inhibition of SASP and the labile property of SOD, we further confirmed the result by overexpressing ECSOD using an adenovirus contain cDNA for SOD3 gene (AdhSOD3) and knockdown of xCT using siRNA directed against the SLC7A11 gene (siRNA xCT). As shown in Fig. 3E, transduction of AdhSOD3 at 300 MOI for 48 h resulted in an approximate 3-fold increase in ECSOD inside and outside the cells, while transfection of xCT (40 nM/ML) siRNA resulted in a decrease in xCT expression by 80%. Interestingly, we observed a slight decrease in xCT expression after overexpression of ECSOD and a slight increase of ECSOD after inhibition of xCT. As shown in Fig. 3F, overexpression of ECSOD or knockdown of xCT significantly decreased PC3 cell invasion. As expected, concurrent overexpression of ECSOD and knockdown of xCT inhibited PC3 cell invasion to a greater extent than overexpression of ECSOD or knockdown of xCT alone.
Figure 3. Overexpression of ECSOD and knockdown of xCT suppresses the invasive ability of aggressive PC3.
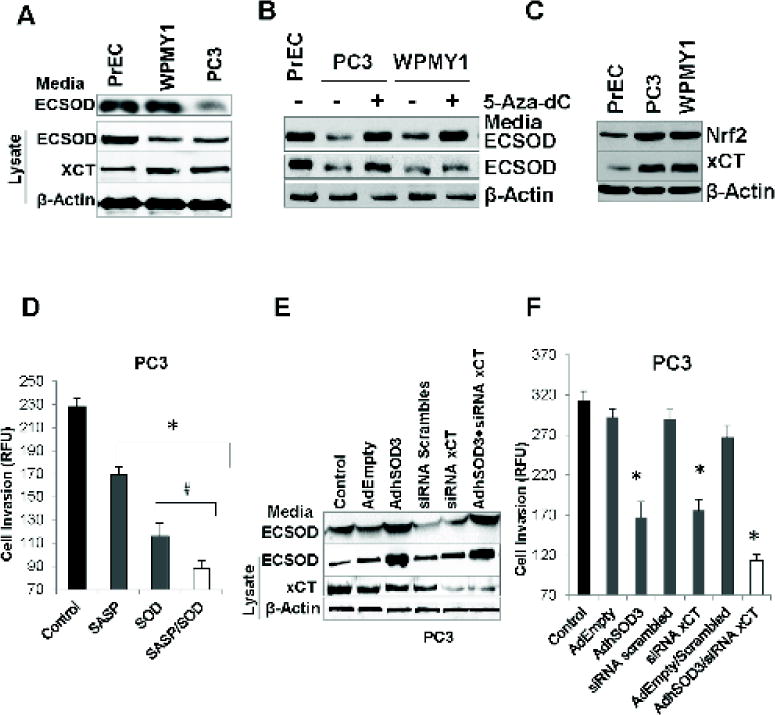
(A) Western blots display comparatively lower ECSOD (~31 kDa) and greater xCT (~55 kDa) expressions in PrEC, PC3, and WPMY1 cell lines. (B). Western blots indicate an increase of ECSOD expression in PC3 and WPMY1 cells after treatment with the DNA methylation inhibitor, 5-Aza-dC, for 96 h. (C) Western blot shows an up-regulation of Nrf2 and xCT expression in PC3 and WPMY1 cells relative to the PrEC cells. (D) Cell invasive ability of PC3 after treatment with SOD and the xCT inhibitor (SASP) for 24 h. (E) Western blot indicates an overexpression of ECSOD in the media and in cell lysates with AdhSOD3 (300 MOI) treatment and a down-regulation of xCT protein by siRNA (siRNA xCT). (F) Cell invasive ability of PC3 cells after overexpression of ECSOD in the media and cells, and with concurrent inhibition of xCT expression. AdEmpty; adenovirus empty vector. 300 MOI was used for AdEmpty and AdhSOD3. *p-value ≤ 0.05 compared with the control.
Since overexpression of ECSOD combined with inhibition of xCT may contribute to alteration of the TME redox state, we measured H2O2, NO•, and redox status in the media. Increasing ECSOD alone increased extracellular H2O2 and nitrite levels as well as intracellular ROS level. Similarly, xCT knockdown alone increased extracellular H2O2 and intracellular ROS levels. Expectedly, simultaneously increasing ECSOD and knockdown of xCT expression increased extracellular H2O2 and nitrite levels (Fig. 4A–B) as well intracellular ROS/RNS levels (Fig. 4C). The changes in extracellular and intracellular redox states are greater in the group with overexpression of ECSOD combined with knockdown of xCT when compared with overexpressing ECSOD alone or xCT knockdown alone. Furthermore, the extracellular redox potential of the redox couples Cys/CySS (EhCySS) exhibited an increase in redox potential value from −154 ± −17.5 mV to −102 ± −9.8 mV after concurrent overexpression of ECSOD and knockdown of xCT (Fig. 4D). The increase in redox potential value indicates an oxidized redox state when compared to the control condition. Transduction of AdEmpty, transfection of siRNA scrambled, and combination of both did not significantly changed cell invasion or redox state. This data indicates that overexpression of ECSOD and inhibition of xCT push the TME and intracellular redox state toward the oxidized state and inhibit PCa invasion.
Figure 4. Overexpression of ECSOD and knock-down of xCT in PC3 cells shift intra-and extracellular redox states to oxidizing status.
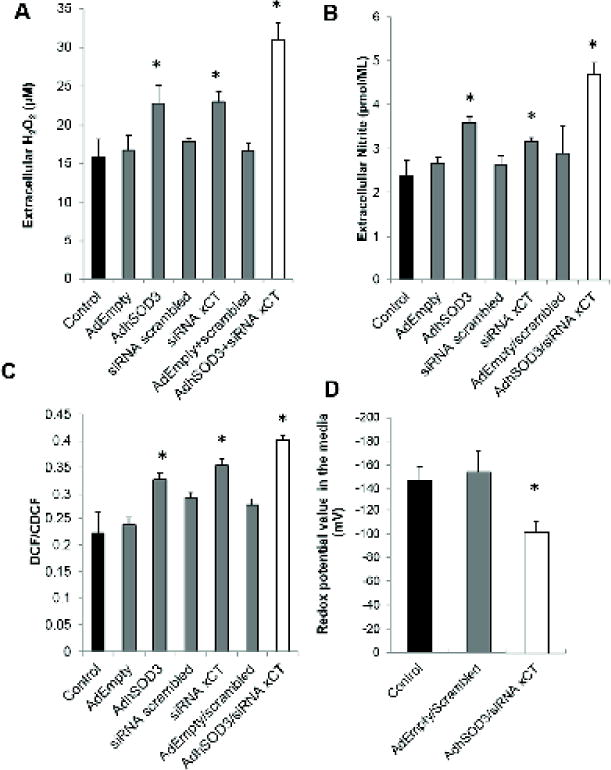
(A) H2O2 level in the media using the Amplex Red assay. (B) Measurement of nitrite level in the media using Griess reaction assay. (C) Measurement of Intracellular ROS/RNS using H2DCFDA. (D) Redox potential values in the media based on Nernst equation calculation of Cys and CySS. *p-value ≤ 0.05 compared with the control.
Increasing ECSOD and inhibiting xCT in stromal cells significantly inhibited PC3 invasion in the co-culturing system
Since there was a significant decrease in ECSOD levels in the stromal area of PCa with a Gleason score ≥ 8 and a significant increase in xCT levels in the stromal area of metastatic PCa (Fig. 1D), we investigated whether ECSOD and xCT in stromal cells support PCa cell invasion. As with the epithelial cells, immortalized prostate stromal cells, WPMY1, exhibited significantly higher levels of xCT and significantly lower levels of ECSOD compared to PrEC cells (Fig. 3A, lysates). Similarly, treatment with 5-Aza-dC induced expression of ECSOD in WPMY1 cells (Fig. 3B) and Nrf2 expression is up-regulated in WPMY1 cells (Fig. 3C).
To address the role of stromal redox state, ECSOD was overexpressed and/or xCT was knock-downed in WPMY1 cells. Western blot analysis (Fig. 5A) demonstrated an approximate two-fold increase in ECSOD levels in the spent media and cell lysates of WPMY1 cells after transduction with AdhSOD3. As expected, transfection of WPMY1 cells with siRNA directed against xCT inhibited xCT expression by approximately 70%. Similar to PC3, we observed a decrease in xCT expression after overexpression of ECSOD, while inhibition of xCT slightly increased ECSOD expression in the media. As shown in Fig. 5B, co-culture of PC3 cells with WPMY1 cells increased the ability of PC3 cells to invade through the in-vitro invasion system when compared to PC3 cells with no WPMY1 cells in the lower chamber. Notably, we observed a significant decrease in PC3 cell invasion after overexpressing ECSOD or knockdown of xCT in WPMY1 cells. However, simultaneous overexpression of ECSOD and knockdown of xCT significantly inhibited PC3 invasion, greater than with ECSOD overexpression or knockdown of xCT alone. These data confirm that ECSOD and xCT work in concert to regulate PCa invasion; altering one of them alone provides a suboptimal inhibitory effect. Next, we measured extracellular H2O2 and found that overexpression of ECSOD combined with knockdown of xCT in WPMY1 cells increased extracellular H2O2 levels more than that observed by overexpressing ECSOD alone or xCT knockdown alone. Correlatively, the redox potential in the conditioned media of WPMY1 cells overexpressing ECSOD combined with knockdown of xCT group significantly increased (Fig. 5C–D). Collectively, these changes in redox state parameters contribute to an increase in the extracellular oxidized state.
Figure 5. Concomitant overexpression of ECSOD and knockdown of xCT in prostate cancer stromal cells (WPMY1) significantly inhibits PC3 invasion and alters the redox state in the TME.
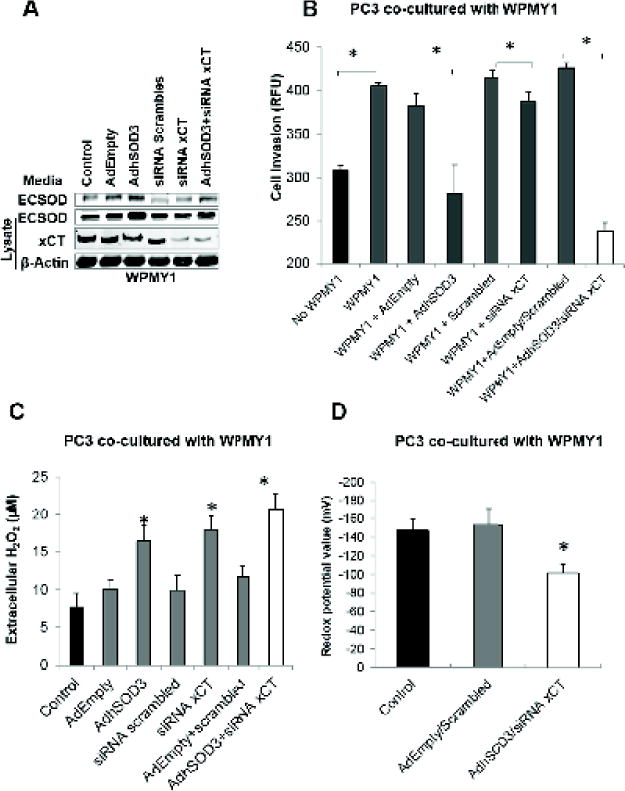
(A) Western blot shows overexpression of ECSOD in the media and in stromal cells by AdhSOD3 (300 MOI) and concomitant down-regulation of the xCT protein by siRNA. (B) Cell invasive ability of PC3 cells that was co-cultured with WPMY1 cells or WPMY1 cells with overexpression of ECSOD and knockdown of xCT. *p-value ≤ 0.05. (C) H2O2 level and (D) redox state in the media of the co-culturing system in WPMY1 cells with overexpression of ECSOD and concomitant knockdown of xCT. *p-value ≤ 0.05 compared with the control.
Treatment with CAT significantly promoted PC3 cell invasion and increased MMP activities
To confirm the role of an oxidized TME and extracellular H2O2 in PC3 cell invasion, we added 10,000 Units of CAT to the media during cell invasion. As expected, treatment with CAT significantly increased invasion of PC3 cells that overexpress ECSOD, or with xCT knockdown, and the combination of ECSOD overexpression and xCT knockdown (Fig. 6A). Correspondingly, CAT treatment increased MMP2/9 activities (Fig. 6B) as well as decreased extracellular H2O2 and nitrite levels (Supplementary Fig. 6A–B). Transduction of AdEmpty, transfection of siRNA scrambled, and a combination of both did not significantly changed cell invasion ability and MMP activities. These data imply that CAT reverses the anti-invasive effect of ECSOD on cancer cells and promotes the pro-invasive effect of xCT.
Figure 6. Removal of extracellular H2O2 by Catalase promotes PC3 cell invasion and increases MMP activities.
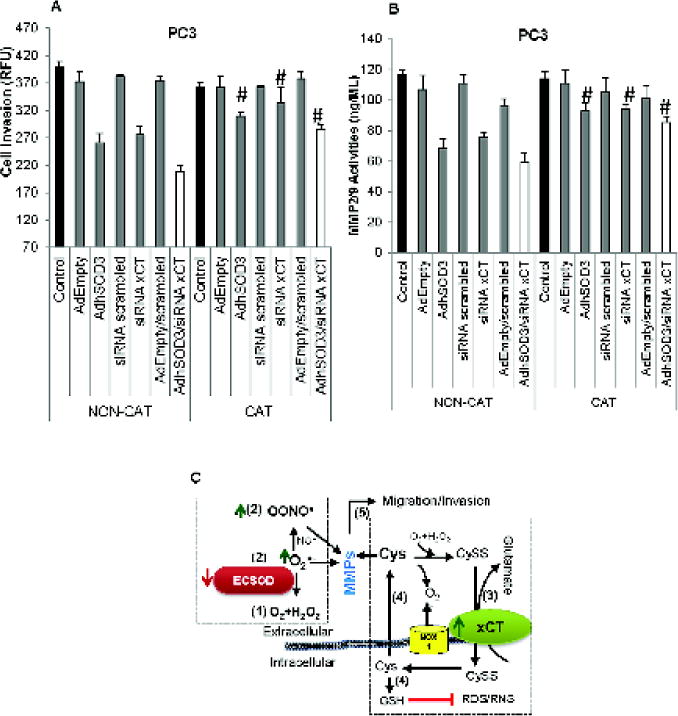
(A) Inhibition of PC3 cell invasion by overexpression of ECSOD and inhibition of xCT expression was reversed by treatment with 10,000 Units of CAT. (B) Inhibition of MMP2/9 activities by overexpression of ECSOD and inhibition of xCT expression was reversed by treatment with 10,000 Units of CAT. #p-value ≤ 0.05 when compared with Non-Catalase treatment. (C) Proposed mechanism by which ECSOD and xCT regulate PCa invasion. A decrease in ECSOD (1) results in an elevation of extracellular O2•− and OONO• (2), while an increase in xCT (3) results in an imbalance of extracellular thiol couples, e.g. an increased level of extracellular Cys (4) and a decreased level of extracellular CySS. These changes in the extracellular redox state are likely to have a profound effect on the activation of migration/invasion-related proteins (e.g., MMPs) (5), resulting in migration/invasion/metastasis of PCa. (Green arrows = increased levels, Red arrow = decreased levels).
Discussion
PCa invasion and metastasis is a complex multistep cascade that leads to secondary site deposits in distant organs. Several in-vitro studies showed a relationship between the intracellular redox state and cancer cell metastasis. Nevertheless, the interpretation of these results from cell lysates is uncertain because the cell is compartmentalized into subcellular organelles and the redox state of each compartment is significantly distinctive [34]. Using the Aperio imaging analysis system, we are the first to show that ECSOD expression is significantly decreased in epithelial and stromal areas of high Gleason PCa. The molecular mechanisms governing ECSOD expression have not been elucidated in detail. The NCI’s Genomic Data Commons database [35] revealed that 5 of 12 PCa studies reveal a deletion of SOD3 gene (Supplementary Fig. 7). Since both mRNA and protein ECSOD levels are decreased in PCa, the inhibition likely occurs at the transcriptional stage. We previously demonstrated that treatment of PC3 cells with a DNA methyltransferase inhibitor; 5-Aza-Dc increases ECSOD expression [3]. This suggests that aberrant cytosine methylation of the ECSOD promoter may contribute to the loss of ECSOD expression in PCa tissues and cells [13, 14]. The single nucleotide polymorphism, rs699473, of SOD in the serum is reportedly associated with high-grade PCa [36]. Similarly, ten-eleven translocation, a dioxygenase of 5-methylcytosine that plays a central role in DNA demethylation processes, is abundantly expressed in lung, breast, skin fibroblasts, and prostate cancers [37]. These results are consistent with the expression pattern of ECSOD and its DNA methylation status in PCa.
Next, we demonstrated that xCT protein expression is significantly increased in PCa epithelial tissues, which corroborates other studies [38–40]. In fact, both xCT mRNA and the SLC7A11 gene expressions are significantly increased in PCa tissues (Supplementary Fig. 7). We previously showed that redox-responsive Nrf2 is up regulated in high-grade PCa tissues [3, 24]. We further found that the oxidized form of peroxiredoxin and 8-hydroxy-2′-doxyguanosine are significantly increased in the matching PCa-TMA (unpublished data). Accordingly, these data suggest that an increase in xCT protein expression in aggressive PCa is due to oxidative stress activation of Nfr2. It is worth noting that, a direct interaction between ECSOD and xCT remains inconclusive. Interestingly, a slight decrease in xCT expression was observed when ECSOD was overexpressed. We propose that increasing ECSOD suppresses Nrf2 activity, which subsequently down-regulates xCT expression and alters the level of extracellular Cys and homocysteine (cysteine intermediate).
To investigate potential therapeutic applications, we altered the expression of ECSOD and xCT. We found that xCT is a pro-invasive protein while ECSOD is an anti-invasive protein; the dual actions of increasing ECSOD and decreasing xCT work in concert to regulate PCa cell invasion. The decrease in PCa invasion correlated with 1) an increase in extracellular H2O2 and nitrite (a by-product of NO•); 2) a change in the extracellular redox state toward the oxidizing state; and 3) inhibition of MMP activities. Treatment with extracellular CAT reversed the anti-invasive effect of ECSOD and shifted the TME toward a reducing status. It is conceivable that extracellular H2O2 and the oxidized redox state negatively mediate prostate cell invasion [41, 42]. Of note, inhibition of cancer cell invasion by H2O2 is controversial, potentially due to differences in cancer cell types, microenvironmental proteins, diffusion distances, and sub-cytotoxic concentrations [43].
In addition to extracellular H2O2, we suggest that NO• may act as a negative mediator in the regulation of PCa invasion, whereas O2•- may increase PCa invasion. Our previous work indicated that the NO• donor, SNAP, inhibits PC3’s invasiveness while the O2•- donors, xanthine/xanthine oxidase and a high extracellular Cys/CySS ratio, increase PC3’s invasive capacity [2, 32]. Consistent with our data, systemic depletion of extracellular CySS with cyst(e)inase enzyme mediates ROS production, depletes intracellular GSH, induces cell cycle arrest, and suppresses tumor growth of both prostate and breast cancer xenografts [44]. These results suggest that induction of extracellular O2•− and Cys/CySS increase PCa invasiveness while H2O2 and NO• decrease PCa cell invasion [2, 30].
Although the precise mechanism of ECSOD and xCT on MMP activities is not fully elucidated, we previously demonstrated that induction of O2•− or Cys/CySS levels enhance MMP2 activity [30]. Since overexpression of ECSOD and inhibition of xCT expression inhibited MMP2/9 activities and co-treatment with CAT reversed the inhibitory effect. Thus, O2•−, H2O2, and Cys/CySS could act as a redox switch at thiol coordinated Zn2+-MMP active sites to directly amplify MMP activities [45].
In addition to therapeutic targets, expressions of ECSOD and xCT have potential as clinical markers for prognosis and monitoring response to cancer treatment. Loss of ECSOD expression has been associated with decreased long-term survival in patients with pancreatic ductal adenocarcinoma [46], while overexpression of xCT has been linked to the resistance to anti-cancer drugs [25]. Clinical studies by noninvasive PET imaging in assessing xCT activity in tumors are underway [47], while Balsalazide, an xCT inhibitor derivative, is already being used to prevent or reduce symptoms of acute radiation-induced proctosigmoiditis in patients undergoing radiation therapy for PCa [48]. These findings highlight the important roles of ECSOD and xCT as therapeutic targets and markers for treatment and diagnosis of aggressive cancers.
Information on the paracrine signaling of ROS/RNS and Cys/CySS between cancer and stromal cells is currently limited. It was reported that extracellular H2O2 from cancer-associated fibroblasts promoted invasiveness in epithelial skin cancer cells by activation of the ERK and JNK pathways [43]. In contrast, thyroid tumor stromal ECSOD inhibited cancer cell migration by regulating IL1- as well as MCP1 inflammatory cytokines [49]. Our results indicated that overexpression of ECSOD and downregulation of xCT in stromal cells significantly inhibits PCa cell invasion in a co-culturing system. The data implies that extracellular O2•−, H2O2, and the Cys paracrine signals promote PC3 cell invasion. Due to its half-life in aqueous phase, diffusion permeability, and highly reactive properties, extracellular H2O2 is an ideal molecular candidate as a paracrine signaling.
To gain a greater understanding of extracellular redox state and PCa metastasis, confirming the protection effect of ECSOD and xCT in a pre-clinical model is clearly warranted. However, the limitations of currently available PCa metastasis models are a problem that must be addressed. Bone is the major site of metastasis in PCa. Transgenic spontaneous PCa models rarely result in bone metastases and orthotopic xenograft models are invasive, predominantly to adjacent lymph nodes. Furthermore, direct injection of PCa cells into bone results in growth of PCa cells at the point of injection only. Thus, development of relevant animal models for PCa metastasis should be a future goal for the research community. Nevertheless, our data on human prostate cancer tissues and co-culturing systems in vitro strongly suggest that shifting the extracellular redox state toward an oxidizing status by targeted modulation of ECSOD and xCT, in both cancer and stromal cells, provides a better strategy for potential therapeutic of invasive PCa.
Pro-oxidants are a double-edged sword; they can be beneficial or harmful to cancer cells. Hence, increasing intra- and extracellular pro-oxidant levels beyond the antioxidant capacity of tumor cells by shifting the TME redox state provides a logical approach for treating metastatic PCa. Our current study suggests that extracellular H2O2 and the oxidized redox state are negative mediators of PCa cell invasion. Overexpression of ECSOD and knockdown of xCT alter the extracellular and intracellular redox states and subsequently inhibit invasion of PCa cells. Targeting ECSOD and xCT in both epithelial and stromal cells may provide greater inhibition of PCa invasion than targeting these proteins in epithelial cells alone. Accordingly, modulation of various TME components as a network would provide a greater strategy for potential therapeutic interventions of aggressive PCa.
Supplementary Material
Highlights.
Alteration of the extracellular redox state is a clinical feature of aggressive prostate cancers.
ECSOD is down-regulated in high Gleason score and metastatic prostate cancers.
xCT is up-regulated in high Gleason score and metastatic prostate cancers.
Simultaneously targeting ECSOD and xCT decreases prostate cancer cell invasion.
Acknowledgments
This work was supported mainly by NIH grants CA188792 to Dr. Chaiswing. Our research utilized the service facilities of the Biospecimen Procurement and Translational Pathology Shared Resource Facility, the Flow Cytometer Shared Resource Facility, and the Biostatistics and Bioinformatics Shared Resource Facility, which were funded by a Markey Cancer Center support grant (P30 CA177558). The authors thank 1) Dr. Dean Jones and Dr. Bill Yang of Emory University, GA, who provided technical support for measurements of thiols in the media, and 2) Drs. Chi Wang and Liu Jinpeng, the Markey Biostatistics and Bioinformatics Shared Resource facility, who provided assistance with Oncomine data analysis. We would like to dedicate this article to the memory of Dr. Terry D. Oberley. His dedication to the field of free radical biology and medicine was an inspiration to all of us.
Abbreviations
- H2DCFDA
2′ 7′-dichlorofluorescein diacetate
- 5-Aza-dC
5-aza-2-deoxycytidine
- CDCFDA
5-(and-6)-carboxy-2′ 7′-dichlorofluorescein diacetate
- AdhSOD3
adenoviral vector containing human ECSOD cDNA
- GFP
adenovirus containing green fluorescent protein
- AdEmpty
adenovirus with no gene inserted
- BPH
benign prostatic hyperplasia
- CAT
catalase
- Cys
cysteine
- xCT
cysteine/glutamate transporter
- CySS
cystine
- ECSOD
extracellular superoxide dismutase
- FBS
fetal bovine serum
- GSH
glutathione
- GSSG
glutathione disulfide
- H2O2
hydrogen peroxide
- ·OH
hydroxyl radical
- IHC
immunohistochemistry
- MOI
multiplicity of infection
- NO•
nitric oxide
- OONO•
peroxynitrite
- PrEC
normal human prostate epithelial cells
- Nrf2
nuclear factor (erythroid-derived 2)-Like 2
- PCF
polycarbonated
- PCa
prostate cancer
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SASP
sulfasalazine
- SOD
superoxide dismutase
- O2•−
superoxide radical
- TMA
tissue microarray
- TME
tumor microenvironment
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chaiswing L, Oberley TD. Extracellular/microenvironmental redox state. Antioxid Redox Signal. 2010;13(4):449–65. doi: 10.1089/ars.2009.3020. [DOI] [PubMed] [Google Scholar]
- 2.Chaiswing L, et al. Regulation of prostate cancer cell invasion by modulation of extra- and intracellular redox balance. Free Radic Biol Med. 2012;52(2):452–61. doi: 10.1016/j.freeradbiomed.2011.10.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaiswing L, Zhong W, Oberley TD. Increasing discordant antioxidant protein levels and enzymatic activities contribute to increasing redox imbalance observed during human prostate cancer progression. Free Radic Biol Med. 2014;67:342–52. doi: 10.1016/j.freeradbiomed.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shan W, et al. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic Biol Med. 2010;49(12):2078–87. doi: 10.1016/j.freeradbiomed.2010.10.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee R. Redox outside the box: linking extracellular redox remodeling with intracellular redox metabolism. J Biol Chem. 2012;287(7):4397–402. doi: 10.1074/jbc.R111.287995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaiswing L, Zhong W, Oberley TD. Increasing discordant antioxidant protein levels and enzymatic activities contribute to increasing redox imbalance observed during human prostate cancer progression. Free Radic Biol Med. 2013;67C:342–352. doi: 10.1016/j.freeradbiomed.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Policastro LL, et al. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal. 2013;19(8):854–95. doi: 10.1089/ars.2011.4367. [DOI] [PubMed] [Google Scholar]
- 8.Qin Z, et al. Extracellular superoxide dismutase (ecSOD) in vascular biology: an update on exogenous gene transfer and endogenous regulators of ecSOD. Transl Res. 2008;151(2):68–78. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest. 1996;75(5):617–36. [PubMed] [Google Scholar]
- 10.Karlsson K, et al. Pharmacokinetics of extracellular-superoxide dismutase in the vascular system. Free Radic Biol Med. 1993;14(2):185–90. doi: 10.1016/0891-5849(93)90009-j. [DOI] [PubMed] [Google Scholar]
- 11.Laukkanen MO. Extracellular Superoxide Dismutase: Growth Promoter or Tumor Suppressor? Oxid Med Cell Longev. 2016;2016:3612589. doi: 10.1155/2016/3612589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, et al. Proteomic analysis of minute amount of colonic biopsies by enteroscopy sampling. Biochem Biophys Res Commun. 2016;476(4):286–92. doi: 10.1016/j.bbrc.2016.05.114. [DOI] [PubMed] [Google Scholar]
- 13.Teoh-Fitzgerald ML, et al. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol Cancer Res. 2012;10(1):40–51. doi: 10.1158/1541-7786.MCR-11-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teoh-Fitzgerald ML, et al. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene. 2014;33(3):358–68. doi: 10.1038/onc.2012.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teoh ML, et al. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res. 2009;69(15):6355–63. doi: 10.1158/0008-5472.CAN-09-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savaskan NE, Eyupoglu IY. xCT modulation in gliomas: relevance to energy metabolism and tumor microenvironment normalization. Ann Anat. 2010;192(5):309–13. doi: 10.1016/j.aanat.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Lewerenz J, Maher P, Methner A. Regulation of xCT expression and system x (c) (-) function in neuronal cells. Amino Acids. 2012;42(1):171–9. doi: 10.1007/s00726-011-0862-x. [DOI] [PubMed] [Google Scholar]
- 18.Ma MZ, et al. Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015;368(1):88–96. doi: 10.1016/j.canlet.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 19.Patel SA, et al. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology. 2004;46(2):273–84. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Zhang P, et al. xCT expression modulates cisplatin resistance in Tca8113 tongue carcinoma cells. Oncol Lett. 2016;12(1):307–314. doi: 10.3892/ol.2016.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyer SS, et al. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2009;296(1):L37–45. doi: 10.1152/ajplung.90401.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yae T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. doi: 10.1038/ncomms1892. [DOI] [PubMed] [Google Scholar]
- 23.Baek S, et al. Exploratory clinical trial of (4S)-4-(3-[18F]fluoropropyl)-L-glutamate for imaging xC- transporter using positron emission tomography in patients with non-small cell lung or breast cancer. Clin Cancer Res. 2012;18(19):5427–37. doi: 10.1158/1078-0432.CCR-12-0214. [DOI] [PubMed] [Google Scholar]
- 24.Habib E, et al. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015;5:33–42. doi: 10.1016/j.redox.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, et al. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65(16):7446–54. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- 26.Okuno S, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer. 2003;88(6):951–6. doi: 10.1038/sj.bjc.6600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinoshita H, et al. Cystine/glutamic acid transporter is a novel marker for predicting poor survival in patients with hepatocellular carcinoma. Oncol Rep. 2013;29(2):685–9. doi: 10.3892/or.2012.2162. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida GJ. The heterogeneity of cancer stem-like cells at the invasive front. Cancer Cell Int. 2017;17:23. doi: 10.1186/s12935-017-0393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 30.Chaiswing L, et al. Extracellular redox state regulates features associated with prostate cancer cell invasion. Cancer Res. 2008;68(14):5820–6. doi: 10.1158/0008-5472.CAN-08-0162. [DOI] [PubMed] [Google Scholar]
- 31.Chaiswing L, et al. Characterization of redox state of two human prostate carcinoma cell lines with different degrees of aggressiveness. Free Radic Biol Med. 2007;43(2):202–15. doi: 10.1016/j.freeradbiomed.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 32.Chaiswing L, Zhong W, Oberley TD. Distinct redox profiles of selected human prostate carcinoma cell lines: implications for rational design of redox therapy. Cancers (Basel) 2011;3(3):3557–84. doi: 10.3390/cancers3033557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Budczies J, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One. 2012;7(12):e51862. doi: 10.1371/journal.pone.0051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones DP, Sies H. The Redox Code. Antioxid Redox Signal. 2015;23(9):734–46. doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nordstrom T, et al. Associations between circulating carotenoids, genomic instability and the risk of high-grade prostate cancer. Prostate. 2016;76(4):339–48. doi: 10.1002/pros.23125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamiya T, et al. Ten-eleven translocation 1 functions as a mediator of SOD3 expression in human lung cancer A549 cells. Free Radic Res. 2017;51(3):329–336. doi: 10.1080/10715762.2017.1313415. [DOI] [PubMed] [Google Scholar]
- 38.Dun Y, et al. Expression of the cystine-glutamate exchanger (xc-) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324(2):189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-) : cystine supplier and beyond. Amino Acids. 2012;42(1):231–46. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 40.Doxsee DW, et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate. 2007;67(2):162–71. doi: 10.1002/pros.20508. [DOI] [PubMed] [Google Scholar]
- 41.Bohm B, et al. Extracellular localization of catalase is associated with the transformed state of malignant cells. Biol Chem. 2015;396(12):1339–56. doi: 10.1515/hsz-2014-0234. [DOI] [PubMed] [Google Scholar]
- 42.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan JS, et al. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017;8(1):e2562. doi: 10.1038/cddis.2016.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cramer SL, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23(1):120–127. doi: 10.1038/nm.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274(31):21491–4. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 46.O’Leary BR, et al. Loss of SOD3 (EcSOD) Expression Promotes an Aggressive Phenotype in Human Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2015;21(7):1741–51. doi: 10.1158/1078-0432.CCR-14-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baek S, et al. (4S)-4-(3-18F-fluoropropyl)-L-glutamate for imaging of xC transporter activity in hepatocellular carcinoma using PET: preclinical and exploratory clinical studies. J Nucl Med. 2013;54(1):117–23. doi: 10.2967/jnumed.112.108704. [DOI] [PubMed] [Google Scholar]
- 48.Jahraus CD, et al. Prevention of acute radiation-induced proctosigmoiditis by balsalazide: a randomized, double-blind, placebo controlled trial in prostate cancer patients. Int J Radiat Oncol Biol Phys. 2005;63(5):1483–7. doi: 10.1016/j.ijrobp.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 49.Parascandolo A, et al. Extracellular Superoxide Dismutase Expression in Papillary Thyroid Cancer Mesenchymal Stem/Stromal Cells Modulates Cancer Cell Growth and Migration. Sci Rep. 2017;7:41416. doi: 10.1038/srep41416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.