

Abstract
Background
Hydrogen sulfide (H2S) is an important regulator of mitochondrial bioenergetics, but its role in regulating mitochondrial biogenesis is not well understood. Using both genetic and pharmacological approaches, we sought to determine if H2S levels directly influenced cardiac mitochondrial content.
Results
Mice deficient in the H2S-producing enzyme, cystathionine γ-lyase (CSE KO) displayed diminished cardiac mitochondrial content when compared to wild-type hearts. In contrast, mice overexpressing CSE (CSE Tg) and mice supplemented with the orally active H2S-releasing prodrug, SG-1002, displayed enhanced cardiac mitochondrial content. Additional analysis revealed that cardiac H2S levels influenced the nuclear localization and transcriptional activity of peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) with higher levels having a positive influence and lower levels having a negative influence. Studies aimed at evaluating the underlying mechanisms found that H2S required AMP-activated protein kinase (AMPK) to induce PGC1α signaling and mitochondrial biogenesis. Finally, we found that restoring H2S levels with SG-1002 in the setting of heart failure increased cardiac mitochondrial content, improved mitochondrial respiration, improved ATP production efficiency, and improved cardiac function.
Conclusions
Together, these results suggest that hydrogen sulfide is an important regulator of cardiac mitochondrial content and establishes that exogenous hydrogen sulfide can induce mitochondrial biogenesis via an AMPK-PGC1α signaling cascade.
Keywords: hydrogen sulfide, mitochondria, heart, AMPK
Graphical Abstract
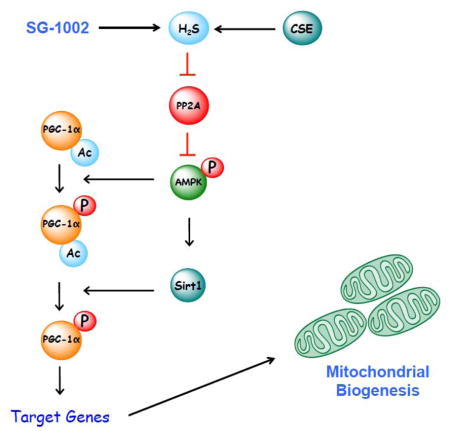
1. Introduction
Mitochondria occupy an important position as mediators of cellular homeostasis due to their role in the regulation of fuel utilization, calcium storage, intracellular signaling, and cell death [1, 2]. As such, impairments in mitochondrial function lead to the development of various disorders, such as neurodegenerative disease, cancer, aging, diabetes, and heart failure [1]. The heart is particularly susceptible to impairments in mitochondrial function given its limited regenerative capacity and persistent energy requirements [2]. As a result, the mitochondrial quality control system, consisting of mitophagy, fission and fusion, and biogenesis, is critically important in maintaining the fidelity of the heart under physiological and pathological conditions [1–3].
The mitochondrial biogenic response in the heart is tightly regulated by a complex network orchestrating both nuclear and mitochondrial genome transcription and replication [3]. This system coordinates both genomes during cardiac development and in response to physiological stimuli when there are changes in substrate availability and energetic demands [3]. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is a master regulator of mitochondrial biogenesis and energy expenditure [4, 5]. Cardiac PGC-1α is induced at birth when the heart undergoes a dramatic shift in fuel preference from relying on glucose and lactate during the fetal period to the use of fatty acids (FA) after birth [6]. PGC-1α regulates the activity of a number of transcription factors, including, peroxisome proliferator-activated receptor-α (PPARα), estrogen receptor–related α (ERRα) and nuclear respiratory factor 1 (NRF1) [5]. By regulating the transcriptional activities of these proteins, PGC-1α modulates genes involved in mitochondrial biogenesis and metabolic pathways. Mitochondrial content is significantly reduced in the failing hearts of both rodents and humans [7, 8]. Furthermore, downregulation of PGC-1α signaling has also been observed in the setting of experimental heart failure [9]. As such, understanding the mechanisms by which PGC-1α signaling is regulated in the heart could lead to the development of therapies aimed at inducing mitochondrial biogenesis and augmenting energy production in the setting of increased contractile demand [8].
Hydrogen sulfide (H2S) is a critically important physiological gaseous signaling molecule that regulates a multitude of biological processes, including angiogenesis, proliferation, redox balance, inflammation, and cell death [10]. It is produced enzymatically in all mammalian species via the actions of cysteine metabolic enzymes: cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfutransferase (3MST) [10, 11]. Although all three enzymes are expressed in the cardiovascular system, prevailing data indicates that CSE plays the foremost role in cardiovascular physiology [10]. Numerous proteins and pathways have been identified as cellular targets of H2S. However, a common cellular target for many studies aimed at understanding the biology and therapeutic potential of H2S has been the mitochondria. H2S has a dual affect on mitochondrial bioenergetics with low concentrations serving as electron donors to the electron transport chain and higher concentrations serving as inhibitors of cytochrome c oxidase [12]. H2S also influences the levels/activation of a number of proteins related to mitochondrial biogenesis (PGC1α [13, 14]; AMP-activated protein kinase (AMPK) [15, 16]; endothelial nitric oxide synthase (eNOS) [11, 17, 18]) and there is evidence that mitochondrial content is higher in brains [13] and hearts [19] treated with exogenous H2S. While these studies provided evidence for elevated mitochondrial levels in response to H2S treatment, it was not clear if the observed increase was due to a direct effect of H2S or was simply an indirect consequence of H2S altering injury. Therefore, the main goal of the current study was to address this issue by determining if H2S levels directly influence cardiac mitochondrial content under non-stressed conditions. Additionally, we sought to gain insights into the mechanisms by which H2S induces mitochondrial biogenesis in the setting of myocardial ischemia-reperfusion.
2. Materials and Methods
2.1. Animals
The following strains of mice on a C57BL/6J background were utilized in this study: (1) C57BL/6J (Jackson Labs, Bar Harbor, ME), (2) Cardiac specific cystathionase-γ-lyase transgenic (CSE Tg+), (3) Cystathionase-γ-lyase deficient (CSE KO), (4) AMPKα2 floxed (Stock#: 014142, Jackson Labs, Bar Harbor, ME), (5) αMHC-Cre transgenic (Stock#: 011038, Jackson Labs, Bar Harbor, ME), and (6) endothelial nitric oxide synthase deficient mice (eNOS KO; Stock# 002684, Jackson Labs, Bar Harbor, ME). CSE Tg+ were generated by ligating the full-length Mus musculus cystathionine γ-lyase cDNA to the murine α-myosin heavy chain promoter, followed by injection of the DNA into newly fertilized mouse embryos (FVB/n background) [20]. The mice were then backcrossed to C57BL/6J for 9 generations. Global CSE KO knockout mice were generated by replacing exon 1 (including the ATG start codon), exon 2, and exon 3 with a neomycin selection cassette [11]. The mice were then backcrossed to C57BL/6J for 9 generations. Cardiac specific AMPKα2 deficient mice (αMHC-Cre+ x AMPKfl/fl) were generated by breeding AMPKf/f mice with αMHC-Cre+ mice. In all experiments, Wild-Type (WT) littermates were used as controls. Male mice between the ages of 8–10 weeks were utilized. All experimental protocols were approved by the Institute for Animal Care and Use Committee at T3 Laboratories and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 86 -23, revised 1996), and with federal and state regulations.
2.2. Patient Samples
Left ventricular samples were procured from patients with advanced ischemic heart failure undergoing a heart transplant at Emory University in accordance with Institution Review Board protocols. Additional non-failing heart failure samples were obtained from LifeLink. All patient identifiers were removed to strictly maintain donor confidentiality and anonymity. Both sample sets included male and female patients (Supplemental Table 1).
2.3. Materials
The orally active H2S-releasing prodrug, SG-1002, was provided by Sulfagenix (Cleveland, OH). SG-1002 was administered to mice in the diet (Purina 5001; Research Diets Inc, New Brunswick, NJ) to achieve a dose of 20 mg/kg/day [11]. Mice received the diet for 4 weeks. Control mice received standard chow (Chow; Purina 5001) for the same duration. For the in vitro experiments, H2S was administered as sodium sulfide (Na2S; Sigma Aldrich).
2.4. In vitro cell culture
H9c2 cardiomyocytes were purchased from ATCC (Rockville, MD, USA). Cells were grown in ATCC-formulated Dulbecco’s Modified Eagle’s Medium (DMEM; catalog# 30-2002) with 10% fetal bovine serum (FBS). Cells were maintained in this media until 80% confluent. Cells were then maintained in DMEM with 0.5% FBS for 12 hours. Cells were then exposed to 100 μM of Na2S for 10, 20, 40, or 60 minutes. Additional groups of cells were exposed to 100 μM of Na2S for 3 consecutive days.
2.5. Cellular Fractionation and Western blot analysis
Whole cell, cytosolic, and nuclear fractions were obtained from heart homogenates as previously described [21]. Protein concentrations were measured with the DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein were loaded into lanes of Criterion™ TGX (Tris-Glycine eXtended) Stain-Free PAGE gels (BioRad). The gels were electrophoresed and activated using a ChemiDoc MP Visualization System (BioRad). The protein was then transferred to a PVDF membrane. The membranes were then imaged using a ChemiDoc MP Visualization System to obtain an assessment of proper transfer and to obtain total protein loads. The membranes were then blocked and probed with primary antibodies (Supplemental Table 2) overnight at 4 °C. Immunoblots were next processed with secondary antibodies (Cell Signaling) for 1 hour at room temperature. Immunoblots were then probed with a Super Signal West Dura kit (Thermo Fisher Scientific) to visualize signal, followed by visualization using a ChemiDoc MP Visualization System (BioRad). Data was analyzed using Image Lab (BioRad). The total protein images were used as loading controls. For each protein of interest, the portion of the protein load image corresponding to the molecular weight of the protein of interest was used as the loading control [16].
2.6. mRNA and qPCR
RNA was isolated using the RiboPure kit according to the manufacturer’s instructions (Ambion). Reverse transcription was performed in a standard fashion with QuantiTect Reverse Transcription Kit (QIAGEN) supplemented with DNase treatment. Taqman qPCR was carried out according to the manufacturer’s instructions using probe sets obtained from Thermo Fisher Scientific.
2.7. Electron Microscopy
Heart tissue was dissected along the muscle fiber while immersed in 2.5% glutaraldehyde buffered with 0.1 M sodium cacodylate (pH 7.2). Samples were stored in the fixative overnight at 4 °C. Samples were then washed with the same buffer and post-fixed in 1% buffered osmium tetroxide, dehydrated through a graded ethanol series to 100%, and embedded in Eponate 12 resin. Ultrathin sections were cut on a Leica UC6rt ultra -microtome at 70–80 nm and counter-stained with 4% aqueous uranyl acetate and 2% lead citrate. Sections were examined using a Hitachi H-7500 transmission electron microscope equipped with a Gatan BioScan CCD camera.
2.8. Citrate Synthase Activity
Cardiac citrate synthase activity was measured spectrophotometrically in homogenates [22].
2.9. Sulfhydration
Protein sulfhydration was evaluate using the modified biotin switch assay or the maleimide assay [23]. For the maleimide assay, sulfhydration was evaluated by subtracting the red fluorescent intensity of samples treated with DTT from the red fluorescent intensity of samples without DTT treatment. The values were then normalized to input and presented as a percentage. For this assay the loss of red fluorescent in the presence of DTT indicates sulfhydration.
2.10. Sulfide Measurements
Hydrogen sulfide and sulfane sulfur levels were measured in heart tissue as previously described [24]. The amount of H2S is reported as nmol/mg wet weight.
2.11. Immunoprecipitation
Heart homogenates were immunoprecipitated with an antibody to PGC-1α using the Dynabeads® Protein G Immunoprecipitation Kit according to manufacturer’s instructions. The samples were then subjected to standard Western blot techniques and the membranes probed with antibodies to phosphoserine and acetyllysine.
2.12. AMPK Activity
The activity of AMPK was measured in homogenates prepared from heart tissue. The samples were first immunoprecipitated with a specific anti- AMPKα2 antibody (abcam). An aliquot of the immunoprecipitated samples were incubated in a reaction buffer containing 12.5 mM Tris-HCl (pH 7.5), 2.5 mM β-glycerophosphate, 1 mM dithiothreitol, 0.05 mM Na3VO4, 5 mM MgCl2, 0.050 mM ATP, and 0.2 mM of SAMS (AMPK synthetic substrate peptide). The rate of ADP formed from the incorporation of ATP in the synthetic peptide was then measured with the ADP-Glo Kinase Assay kit (Promega) according to the manufacturer’s instructions. Activity was expressed as ADP generated (in picomoles) per minute per milligram of protein.
2.13. Sirt1 Activity
The activity of cardiac Sirt1 was evaluated using the SIRT1 Activity Assay (catalog# ab156065, abcam).
2.14. PP2A Activity
The activity of cardiac PP2A was evaluated using the PP2A Immunoprecipitation Phosphatase Assay Kit (catalog# 12-313, Millipore Sigma).
2.15. Nitric Oxide Metabolite Analysis
Nitrite levels were quantified by ion chromatography (ENO20 Analyzer, Eicom). Tissue nitrosothiol compounds were quantified by using group-specific reductive denitrosation by iodine-iodide with subsequent detection of the liberated NO by using gas-phase chemiluminescence [25].
2.16. Adenosine Metabolite Analysis
Adenosine metabolites were evaluated in cardiac tissue using the ATP/ADP/AMP Assay Kit (Cat #: A-125, Biomedical Research Service Center, University at Buffalo-SUNY).
2.17. Mitochondria Respiration and ATP
Cardiac fibers were isolated and permeabilized wtih saponin as previously described [26]. Respiration was monitored using a Clark-type oxygen electrode (Hansatech Instruments, Amesbury, MA) in the presence of pyruvate or palmitoyl-L-carnitine. To evaluate ATP synthesis, aliquots were taken from the respiration chamber over a 1-minute period after the addition of ADP. ATP was then quantified with a bioluminescence assay using an ATP determination kit (A-22066; Molecular Probes, Eugene, OR). The rate of ATP synthesis was then normalized to the oxygen consumption rates measured over the time the aliquots were collected to obtain a measure of ATP synthesis efficiency (ATP/Oxygen ratio). This measurement reflects the ratio of state 3 (ADP stimulated respiration) ATP synthesis rates to state 3 oxygen consumption. A higher value indicates better efficiency.
2.18. Myocardial Ischemia-Reperfusion Protocol and Myocardial Injury Assessment
Mice were subjected to surgical ligation of the left coronary artery followed by reperfusion for 2 weeks. Echocardiography was performed as previously described [19].
2.19. Statistics
All data are expressed as mean ± SEM. The data was first evaluated for normal distribution using the D’Agostina and Pearson omnibus normality test. Subsequent, statistical significance was evaluated as follows: (1) unpaired Student t-test for comparison between 2 means; (2) a 1-way ANOVA with a Tukey test as the posthoc analysis for comparison among 3 or more groups; and (3) a 2-way ANOVA with a Bonferroni test as the posthoc analysis for comparison among the means from groups of WT and AMPK KO mice and WT and eNOS KO mice. For the echocardiography data, a 2-way repeated measures ANOVA with a Bonferroni test as the posthoc analysis was used. The following comparisons were made separately: (1) baseline vs. post-baseline measurements for each group, (2) differences between each groups baseline measurements, and (3) differences between each groups post-baseline measurements. The p-value for these evaluations was adjusted by applying the Bonferroni correction for multiple comparisons. A value of p < 0.05 denoted statistical significance and p-values were two-sided. All statistical analysis was performed using Prism 5 (GraphPad Software Inc).
3. Results
3.1. Endogenous H2S Influences Cardiac Mitochondrial Content
We sought to determine if there was a direct relationship between cardiac H2S levels and mitochondrial content. First, we used a genetic model to address this question. We collected heart samples from mice with significantly elevated levels of H2S (CSE Tg+) and from mice with attenuated levels of H2S (CSE KO) (Supplemental Figure 1). Analysis revealed that the hearts of CSE Tg+ mice displayed an increase in mitochondrial DNA levels and citrate synthase activity (Figure 1A–B). In contrast, both were decreased in the hearts of CSE KO mice. Together these findings suggest that H2S levels influence cardiac mitochondrial content. This was further confirmed by analysis of electron microscopy images of cardiac ventricles from CSE Tg+ and CSE KO mice demonstrating an increase and decrease in the number of mitochondria per field, respectively (Figure 1C–D).
Figure 1.

(A) Ratio of mitochondrial DNA to nuclear DNA. (B) Citrate synthase activity. (C) Representative electron microscopy images of mitochondria. Scale bar equals 3 μm. (D) Number of mitochondria per field of view. (E) Summary of mitochondria area and perimeter measurements (μm2) and (F) percentage of mitochondria in a given field that fell into three size categories based on area: <0.6 μm2, 0.6 μm2 – 1.0 μm2, and >1.0 μm2. (G) Basal and maximum (state 3) oxygen consumption rates for permeabilized myocardial fibers in the presence of pyruvate and (H) palmitoyl-L-carnitine. (I) Efficiency of ATP synthesis [ATP produced per oxygen consumed (ATP/O)] in permeabilized myocardial fibers. All samples were collected from hearts of Wild-Type, cystathionase-γ-lyase transgenic (CSE Tg+), and CSE deficient (CSE KO) mice. Values are means ±SEM. *p<0.05, **p<0.01, and ***p<0.001 vs. Wild-type.
Alterations in mitochondrial content can arise from biogenesis or from fusion and fission. Our data suggest biogenesis. A series of experiments were, therefore, undertaken to determine the contribution of fusion-fission to the altered mitochondrial content observed in the hearts of CSE Tg+ and CSE KO mice (Supplemental Figure 2). First, the expression of proteins involved in mitochondrial fusion and fission were evaluated. The expression of the fusion proteins, mitofusin-1 (Mfn1), mitofusin-2 (Mfn2), and Opa-1, as well as the fission protein, fission-1 (Fis1) were not altered in the hearts of CSE Tg+ or CSE KO mice. Second by electron microscopy, we observed no differences in the area or perimeter of the mitochondria in the hearts of each strain (Figure 1E). Finally, further analysis of mitochondrial fusion-fission was achieved by calculating the percentage of mitochondria in a given field that fell into three size categories based on area: <0.6 μm2, 0.6 μm2 – 1.0 μm2, and >1.0 μm2 [27]. In support of the overall area and perimeter calculations, this analysis also revealed no differences in the hearts of each strain (Figure 1F). Overall these results suggest that H2S levels influence cardiac mitochondrial content via biogenesis.
To characterize the mitochondrial functional phenotype of hearts from Wild -type, CSE Tg+ and CSE KO mice in a manner that would detect potential differences in mitochondrial volume density or function, respiration (oxygen consumption) experiments were performed on saponin-permeabilized myocardial fibers. This technique allowed for the selective use of different metabolic substrates to define the maximal respiratory capacity of specific mitochondrial oxidative pathways [26]. Experiments were performed with LV fibers using pyruvate and palmitoyl-carnitine. For these experiments, basal respiration was assessed followed by maximal ADP-stimulated state 3 respiration. LV fibers from CSE Tg+ mice did not display any changes in state 3 respiration rates in the presence of pyruvate (Figure 1G). They did however display increased state 3 respiration rates in the presence of palmitoyl-carnitine (Figure 1H). In contrast, LV fibers from CSE KO mice displayed increased state 3 respiration rates in the presence of pyruvate and decreased rates in the presence of palmitoyl-carnitine. Additionally, maximal rates of ATP synthesis from ADP were normalized to state 3 respiration rates to determine the efficacy of ATP synthesis in the presence of each substrate (Figure 1I). LV fibers from CSE Tg+ mice displayed enhanced ATP/oxygen ratios in the presence of palmitoyl-carnitine. In contrast, fibers from CSE KO mice displayed lower ATP/oxygen ratios in the presence of both pyruvate and palmitoyl-carnitine.
3.2. Exogenous H2S Induces Cardiac Mitochondrial Biogenesis
Next, we sought to determine if exogenous H2S could influence cardiac mitochondrial content. For these studies, we administered SG-1002 (20 mg/kg/day) in the chow for 4 weeks. SG-1002 is an H2S prodrug that we have previously shown to exert cardioprotective effects [11, 16]. Initial studies confirmed that the administration of SG-1002 significantly increased cardiac H2S levels (Supplemental Figure 1). We then examined if the increase in H2S influenced cardiac mitochondrial content. Analysis revealed that SG-1002 increased mitochondrial DNA levels, citrate synthase activity, and the number of mitochondria per field of electron microscopy images (Figure 2A–D). Further analysis confirmed that SG-1002 did not influence mitochondrial fusion-fission (Supplemental Figure 2 and Figure 2E–F). Finally, SG-1002 did not alter the state 3 respiration rates of LV fibers in the presence of pyruvate (Figure 2G). It did however, improve the state 3 respiration rates and ATP/oxygen ratios of LV fibers in the presence of palmitoyl-carnitine (Figure 2H–I).
Figure 2.

(A) Ratio of mitochondrial DNA to nuclear DNA. (B) Citrate synthase activity. (C) Representative electron microscopy images of mitochondria. Scale bar equals 3 μm. (D) Number of mitochondria per field of view. (E) Summary of mitochondria area and perimeter measurements (μm2) and (F) percentage of mitochondria in a given field that fell into three size categories based on area: <0.6 μm2, 0.6 μm2 – 1.0 μm2, and >1.0 μm2. (G) Basal and maximum (state 3) oxygen consumption rates for permeabilized myocardial fibers in the presence of pyruvate and (H) palmitoyl-L-carnitine. (I) Efficiency of ATP synthesis [ATP produced per oxygen consumed (ATP/O)] in permeabilized myocardial fibers. All samples were collected from hearts of mice administered standard diet (Chow) or diet supplemented with SG-1002 (SG-1002; 20mg/kg/day) for 4 weeks. Values are means ±SEM. *p<0.05, **p<0.01, and ***p<0.001 vs. Chow.
3.3. H2S Levels Influence PGC-1α
Given that PGC-1α is a master regulator of mitochondrial biogenesis [4, 5], we sought to determine if H2S influenced the expression or activation of PGC-1α. Initial analysis revealed that the whole cell expression of PGC-1α was not altered in the hearts of CSE Tg+ or CSE KO mice (Figure 3A–B). However, the cytosolic levels of PGC-1α were significantly lower in the hearts of CSE Tg+ mice (Figure 3A–B). Correspondingly, the nuclear levels of PGC-1α were significantly higher in the hearts of CSE Tg+ mice. In contrast, the cytosolic levels of PGC-1α were unaltered in the hearts of CSE KO mice. However, the nuclear levels of PGC-1α were significantly lower in the hearts of CSE KO mice (Figure 3A–B). In agreement with these changes, the hearts of CSE Tg+ mice displayed elevated expression of a number of PGC-1α target genes, whereas the expression of the same genes were lower in the hearts of CSE KO mice (Figure 3D). Similarly, SG-1002 did not alter the whole cell expression of PGC-1α, but it did decrease the cytosolic levels of PGC-1α, increase the nuclear expression of PGC-1α, and increase the expression of PGC-1α target genes (Figure 3D–F). Together, this data suggests that cardiac H2S levels influence the nuclear localization and transcriptional activity of PGC-1α.
Figure 3.

(A) Representative immunoblots and (B) analysis of the whole cell, cytosolic and nuclear expression of PGC1α. (C) Relative gene expression of PGC1α target genes associated with mitochondrial biogenesis (tfam, cox4i7, cox1, atp5b). Samples were collected from Wild-Type, CSE Tg+, and CSE KO mice. Values are means ±SEM. *p<0.05 and **p<0.01 vs. Wild-type. (D) Representative immunoblots and (E) analysis of the whole cell, cytosolic and nuclear expression of PGC1α. (F) Relative gene expression of PGC1α target genes associated with mitochondrial biogenesis (tfam, cox4i7, cox1, atp5b). Samples were collected from mice administered Chow or diet supplemented with SG-1002. Values are means ±SEM. *p<0.05 vs. Chow.
3.4. H2S Does Not Induce Mitochondrial Biogenesis via eNOS/NO
We next turned our attention to the mechanism(s) by which H2S induces PGC-1α-signaling. Specifically, we focused on three proteins known to mediate mitochondrial biogenesis through PGC-1α; eNOS/NO [28, 29]; AMPK; and Sirtuin 1 (Sirt1) [5]. Given that the cardioprotective actions of H2S have been reported to be mediated, in part, via eNOS [18], we first asked if H2S-induced mitochondrial biogenesis was channeled through eNOS/NO. SG-1002 increased the phosphorylation of eNOSSer1177 (activation site) and increased the cardiac levels of nitrite and nitrosothiols (RXNO) (Supplemental Figure 3). To determine if the elevated levels of NO contributed to H2S-induced mitochondrial biogenesis, wild-type (WT) and eNOS KO mice were administered SG-1002 for 4 weeks. SG-1002 increased mitochondrial DNA levels and citrate synthase activity in both strains of mice (Supplemental Figure 3), suggesting that eNOS/NO was not responsible for H2S-induced mitochondrial biogenesis.
3.5. H2S Induces Mitochondrial Biogenesis via AMPK
The transcriptional activity of PGC-1α is regulated by post-translational modifications. For instance, AMPK regulates the transcriptional activity of PGC -1α via phosphorylation [30], whereas Sirt1 regulates its transcriptional activity via deacetylation [5]. Evidence suggests that AMPK both activates Sirt1 and cooperates with it in enhancing the ability of PGC-1α to stimulate mitochondrial biogenesis [31]. So, the next series of experiments sought to determine if H2S-induced mitochondrial biogenesis was channeled through AMPK and Sirt1. SG-1002 increased the phosphorylation and activity of AMPK (Figure 4A–C). No changes were observed in the expression of Sirt1, but SG-1002 did increase its activity (Figure 4D–F). In agreement with these changes, SG-1002 increased the serine phosphorylation of PGC-1α and decreased its acetylation. To determine if AMPK was directly responsible for H2S induced PGC-1α-signaling and mitochondrial biogenesis, αMHC-Cre+ x AMPKfl/fl mice were given SG-1002 for 4 weeks. The deficiency of AMPK did not affect the expression of PGC-1α or Sirt1 (Figure 5A–C). However, SG-1002 failed to increase the serine phosphorylation of PGC-1α, failed to increase Sirt1 activity, and failed to decrease the acetylation of PGC-1α (Figure 5D–H). Importantly, SG-1002 was unable to increase the expression of PGC-1α target genes and mitochondrial DNA levels in the hearts of αMHC-Cre+ x AMPKfl/fl mice (Figure 5I). Together this data suggests that H2S induced mitochondrial biogenesis via an AMPK-Sirt1-PGC-1α signaling cascade.
Figure 4.

(A–B) Representative immunoblots and analysis of phosphorylated AMPK and total AMPK. (C) AMPK activity. (D–E) Representative immunoblots and analysis of Sirt1. (F) Sirt1 activity. (G) Representative immunoblots and analysis from immunoprecipitation experiments examining the (H) serine phosphorylation and (I) acetylation status of PGC1α. All samples were collected from hearts of mice administered standard diet (Chow) or diet supplemented with SG-1002 (SG-1002; 20mg/kg/day) for 4 weeks. Values are means ±SEM. *p<0.05 and **p<0.01 vs. Chow.
Figure 5.

(A) Representative immunoblots and analysis of (B) PGC1α and (C) Sirt1. (D) Representative immunoblots and analysis from immunoprecipitation experiments examining the serine phosphorylation status of PGC1α. (E) Sirt1 activity. (F) Representative immunoblots and analysis from immunoprecipitation experiments examining the acetylation status of PGC1α. (G) Relative gene expression of tfam, cox417, and mitochondria DNA. All samples were collected from hearts of Wild-type (αMHC-Cre+) and αMHC-Cre+ x AMPK floxed (αMHC-Cre+ x AMPKfl/fl) mice administered standard diet (Chow) or diet supplemented with SG-1002 (SG; 20mg/kg/day) for 4 weeks. Values are means ±SEM. *p<0.05, **p<0.01, and ***p<0.001 vs. WT Chow.
3.6. H2S Regulates AMPK by Inhibiting PP2A
We next addressed how H2S regulates AMPK. The phosphorylation of AMPK is regulated through a balance between the actions of upstream kinases and phosphatases [32]. In the heart, the liver kinase B1 (LKB1) is the major upstream AMPK kinase. SG-1002 did not alter the phosphorylation (activation) of LKB1 (Figure 6A–B). Alterations in the phosphorylation of LKB1 are not always necessary for the induction of AMPK phosphorylation, as changes in AMP and/or ADP levels can influence the confirmation of AMPK, thus allowing for LKB1 to have better access to threonine 172 [32]. The levels of ATP, ADP and AMP were not altered in hearts of mice administered SG-1002 (Figure 6C–D). Together this data indicates that SG-1002 does not influence the phosphorylation of AMPK via the action of LKB1. We then turned our attention to protein phosphatase 2A (PP2A). PP2A is a serine/threonine protein phosphatase consisting of scaffolding (PP2Aa), regulatory (PP2Ab), and catalytic (PP2Ac) subunits [33]. Analysis revealed that SG-1002 did not alter the protein expression of any of the PP2A subunits (Figure 6E–F). H2S modifies cysteine residues in proteins through the formation of a persulfide (-SSH) bond by a process termed sulfhydration or persulfidation [34]. Although several proteins have been found to be sulfhydrated, a key finding in recent years was the discovery that the sulfhydration of protein tyrosine phosphatase 1B (PTP1B) inhibits its activity [35]. We, therefore, asked if PP2A was sulfhydrated and if so could this affect its activity. Initial studies revealed that SG-1002 increased the sulfhydration of cardiac proteins (Supplemental Figure 4). Subsequent analysis using both the modified biotin switch assay and the maleimide assay revealed that SG-1002 increased the sulfhydration of PP2Aa, PP2Ab, and PP2Ac (Figure 6G–H and Supplemental Figure 4C–H). This was associated with a decrease in the activity of PP2A (Figure 6I), suggesting that H2S regulates the phosphorylation of AMPK via the inhibition of PP2A.
Figure 6.

(A–B) Representative immunoblots and analysis of phosphorylated LKB1 and total LKB1. (C) Cardiac levels of ATP, ADP, and AMP. (D) Ratio of AMP to ATP. (E–F) Representative immunoblots and analysis of PP2A subunits. (G–H) Representative immunoblots and analysis from the modified biotin switch assay examining the sulfhydration status of PP2A subunits. (I) PP2A activity. All samples were collected from hearts of mice administered standard diet (Chow) or diet supplemented with SG-1002 (SG-1002; 20mg/kg/day) for 4 weeks. Values are means ±SEM. *p<0.05 and ***p<0.001 vs. Chow.
Subsequent experiments sought to evaluate the temporal nature of this regulation. Beginning 10 minutes after exposing H9c2 cells to Na2S (100 μM) the activity of PP2A was reduced when compared to untreated cells (Supplemental Figure 5). This decrease continued for 60 minutes with a gradual return to untreated levels. Concurrently, the phosphorylation levels of AMPK followed the opposite trend. Further analysis, revealed that exposing cells to Na2S for 3 consecutive days resulted in a decrease in the activity of PP2A accompanied by an increase in AMPK phosphorylation. Together these studies indicate that the regulation of this pathway by H2S can be acute and also maintained chronically.
3.7. H2S Levels and Mitochondria Content are Reduced in Response to Heart Failure
There is evidence for impaired mitochondrial biogenesis from animal models of heart failure and from studies utilizing human heart failure samples [7, 36]. Additionally, studies report lower circulating H2S levels in human heart failure patients [37]. However, an association between cardiac H2S levels and cardiac mitochondrial content has not been explored. Here, we were able to obtain left ventricular samples from end-stage heart failure patients at the time of transplant. Analysis revealed diminished H2S levels (Figure 7A–B), as well as lower levels of PGC-1α target genes (Figure 7C) and mitochondrial DNA levels (Figure 7D). Similar results in regards to H2S levels and mitochondrial content were also found in samples from mice collected at 2 weeks following myocardial ischemia and reperfusion (Figure 8A–C). We, therefore, sought to determine if restoring H2S levels could influence the mitochondrial content of the ischemic heart via the proposed PP2A-AMPK signaling cascade. Previously, we found that H2S therapy increased mitochondrial content and function in hearts subjected to myocardial ischemia [19, 20]. However, in these studies H2S was administered at the time of reperfusion resulting in a reduction in infarct size. As such, the observed improvements in mitochondria content and function could have been due to the initial reduction in injury. We, therefore, decided to delay the timing of treatment here. Mice subjected to myocardial ischemia and reperfusion were administered normal diet or diet supplemented with SG-1002 beginning 24 hours after reperfusion. The mice were then followed for 2 weeks. Our analysis revealed that restoring H2S levels via the dietary supplementation of SG-1002 increased the sulfhydration of cardiac proteins and increased the sulfhydration of PP2Ab and PP2Ac (Figure 8A–B and Supplemental Figure 6A–B). This was associated with a decrease in the activity of PP2A, an increase in the phosphorylation of AMPK, an increase in the gene expression of PGC-1α target genes, an increase in cardiac mitochondrial content, improved mitochondrial respiration, improved ATP production efficiency, and improved cardiac dilatation and function (Figure 8C–H and Supplemental Figure 6C–H).
Figure 7.

Cardiac levels of (A) free H2S and (B) sulfane sulfur. (C) Relative gene expression of atp5b, mt-co1, and tfam. (D) Ratio of mitochondrial DNA to nuclear DNA. Left ventricular samples were collected from non-failing and end stage heart failure patients. Number in bars represent sample sizes. Values are means ±SEM. *p<0.05 and ***p<0.001 vs. Non-Failing.
Figure 8.

Cardiac levels of (A) free H2S and (B) sulfane sulfur. (C) Ratio of mitochondrial DNA to nuclear DNA. (D) Basal and maximum (state 3) oxygen consumption rates for permeabilized myocardial fibers in the presence of palmitoyl-L-carnitine. (E) Efficiency of ATP synthesis [ATP produced per oxygen consumed (ATP/O)] in permeabilized myocardial fibers. Samples were collected from hearts of mice subjected to 60 minutes of ischemia and 2 weeks of reperfusion. Values are means ±SEM. *p<0.05, **p<0.01, and ***p<0.001 vs. Sham. (F) Left ventricular end-diastolic diameter (LVEDD), LV end-systolic diameter (LVESD), and LV Ejection Fraction measured in groups of mice using echocardiography images 2 weeks following myocardial ischemia and reperfusion (Post). Values are means ±SEM. ***p<0.001 vs. Baseline.
4. Discussion
The transcriptional activity of PGC-1α is regulated by a number of stimuli, exemplifying the range of settings in which mitochondrial biogenesis is induced. The mechanisms governing this regulation have been extensively studied and have been determined to vary between tissues and different settings [5]. AMPK, a member of the metabolite-sensing protein kinase family, is activated in response to alterations in cellular energy levels [32]. Activation of AMPK acts to maintain cellular energy stores, switching on catabolic pathways that produce ATP, while switching off anabolic pathways that consume ATP [38]. AMPK also induces mitochondrial biogenesis via the activation of PGC-1α. Specifically, AMPK regulates PGC-1α directly through serine phosphorylation, as well as indirectly via the activation of Sirt1, which leads to the deacetylation of PGC-1α [5]. This coordinated signaling cascade, whereby the phosphorylation of PGC-1α by AMPK is required for the subsequent Sirt1-mediated deacetylation, demonstrates the complex and specific nature by which PGC-1α is regulated by AMPK [29]. Here, we found that increasing cardiac H2S levels through the dietary supplementation of SG-1002 led to the activation of AMPK and Sirt1 resulting in the phosphorylation and deacetylation of PGC-1α. More importantly, we found that SG-1002 failed to alter PGC-1α, induce PGC-1α target genes, and induce mitochondrial biogenesis in the hearts of AMPKα2 deficient mice – indicating that H2S induces mitochondrial biogenesis via AMPK. Our data also provides some insights into how H2S regulates AMPK. The activation of AMPK is mediated through different mechanisms involving the allosteric regulation of AMPK subunits through changes in adenosine metabolites, activation by LKB1, and changes in the activity of PP2A [32]. Here, we found that H2S did not change the levels of adenosine metabolites nor alter the phosphorylation of LKB1. Rather, our evidence indicates that H2S affects AMPK by decreasing the activity of the serine/threonine protein phosphatase, PP2A.
Recently, H2S was shown to inhibit the activity of PTP1B via sulfhydration [35]. Sulfhydration or persulfidation is a post-translational modification by which H2S alters cysteine residues in proteins through the formation of a persulfide (-SSH) bond [34]. Studies indicate that protein sulfhydration is a major event by which H2S elicits cellular signaling. For instance, the antiapoptotic actions of NFkB are dependent on H2S sulfhydrating its p65 subunit [23]. Also, H2S induces endothelial cell and smooth muscle cell hyperpolarization and vasorelaxation by sulfhydrating ATP-sensitive potassium channels [39]. Here, we found that all three PP2A subunits displayed enhanced sulfhydration in the hearts of mice supplemented with SG-1002. Since this modification was accompanied by a decrease in activity, it can be suggested that like for PTP1B, sulfhydration of PP2A imparts inactivation. Further studies are warranted to elucidate the specific cysteine residues that are modified on each subunit and to determine how each affects the activity of PP2A.
As noted, H2S is a known regulator of cellular bioenergetics via its actions on mitochondrial function. For instance, H2S acts as a stimulator of mitochondrial bioenergetics through its ability to donate electrons to the mitochondrial electron transport chain [12, 40]. This action serves a physiological role in the maintenance of mitochondrial electron transport, as well as complementing and balancing the bioenergetic role of Krebs cycle-derived electron donors [12]. In contrast, H2S is a potent and reversible inhibitor of mitochondrial function via its regulation of cytochrome c oxidase (complex IV of the mitochondrial electron transport chain) [41]. Paradoxically, this action contributes to the cardioprotective effects of exogenous H2S, as inhibition of mitochondrial respiration during the early stages of reperfusion injury limits the generation of reactive oxygen species, which ultimately preserves mitochondrial function [18–20]. In addition, H2S targets several cellular pathways that influence mitochondrial function [42]. PGC-1α not only regulates mitochondrial biogenesis, but also regulates energy expenditure in the heart. Specifically, PGC-1α is essential for the maintenance of maximal, efficient cardiac mitochondrial fatty acid oxidation and ATP synthesis [26]. Consistent with our findings regarding the induction of PGC-1α signaling, we observed that augmenting cardiac H2S levels increased the maximal capacity for mitochondrial fatty acid β-oxidation and ATP synthesis, whereas lower H2S levels had the opposite effect.
PGC-1α-mediated mitochondrial biogenesis in brown fat and endothelial cells is in part regulated by eNOS and NO [28, 29]. Recent studies indicate that H2S not only augments NO bioavailability and signaling [11, 17], but provides protection against cardiac injury in an eNOS-dependent manner [15, 18]. Based on this evidence, we speculated that H2S induced mitochondrial biogenesis by activating eNOS and increasing NO levels. Indeed, we found that H2S therapy increased the phosphorylation of eNOS (activation site) and increased cardiac NO levels. However, H2S therapy did not induce mitochondrial biogenesis in an eNOS-dependent manner, as evidenced by a significant increase in mitochondrial content in the hearts of eNOS KO mice given SG-1002. This suggests that although eNOS/NO is important for H2S signaling in certain situations, it is not necessary for the induction of mitochondrial biogenesis in the naïve heart. With that being said, given the complexity by which PGC-1α signaling is regulated, we cannot rule out the possibility that under certain conditions or in different tissues eNOS/NO contributes to H2S-mediated mitochondrial biogenesis in some way. For instance, in the setting of ischemic injury, the induction of eNOS/NO could contribute to a pro-survival environment, which could indirectly aid in the promotion of mitochondrial biogenesis. Additionally, NO also modifies cysteine residues in a manner similar to sulfhydration; a process termed nitrosylation. While similar in some aspects to sulfhydration, nitrosylation appears to diminish cysteine reactivity, whereas sulfhydration seems to enhance it [43]. There also is some overlap in the proteins targeted by NO and H2S. However, it is not clear if NO and H2S target the same cysteine residues in these proteins or if they target distinct cysteine residues. Further it is not clear if they work together or oppose each other to regulate the function of proteins. Finally, it is not clear if there is a balance of sulfhydration and nitrosylation needed for proper function. Therefore, we cannot rule out the possibility that under certain conditions both NO and H2S target PP2A at the same or different cysteine residues to regulate its activity. Further, under certain conditions it is also possible that H2S induces the nitrosylation of PP2A via its actions on eNOS. Therefore, future studies aimed at identifying the specific residues in PP2A modified by H2S and NO are needed to fully understand if/how these signaling molecules work together to regulate of this protein.
The AMPK-PGC-1α signaling cascade has been extensively studied in the context of cardiac metabolism [32]. So, the novel aspects of this study rest not in the determination that AMPK-PGC-1α signaling leads to mitochondrial biogenesis, but in the evidence that endogenous and exogenous H2S influence this pathway. More so, the finding that a decrease in endogenous H2S levels led to an impairment in AMPK-PGC-1α signaling becomes important when considering the evidence that H2S levels are decreased in pathological conditions – i.e. heart failure (Figure 7 and [37]) – that also present with reduced mitochondrial content [7, 36]. Based on this evidence it can be suggested that endogenous H2S levels not only play an important role in maintaining the mitochondrial content of the heart, but that a reduction in endogenous H2S levels contributes to the pathophysiology of heart failure through a disruption in mitochondrial biogenesis. This idea is supported by our findings that restoring H2S levels with SG-1002 increased mitochondrial content, improved ATP production, and attenuated LV dysfunction in a murine model of ischemia-reperfusion injury. Our study has, therefore, identified an important regulatory mechanism in the mitochondrial biogenesis pathway that if corrected by restoring H2S levels could protect the failing heart and ultimately reduce mortality and morbidity associated with heart failure. Future studies aimed at determining if H2S therapy coupled with other pharmacological agents that target the AMPK-PGC-1α signaling pathway are warranted to fully test this postulate.
Supplementary Material
Highlights.
Hydrogen Sulfide levels influence cardiac mitochondrial content
Hydrogen Sulfide induces mitochondrial biogenesis in an AMPK-dependent manner
Hydrogen Sulfide activates AMPK via the sulfhydration and inhibition of PP2A
Acknowledgments
This work was funded by grants from the American Heart Association (15POST25610016 and 16GRNT31190016 to J.W.C) and the National Institutes of Health (NIH) (1R01DK115213-01 and 5R01HL098481-05 to J.W.C. and 1R01HL092141, 1R01HL093579, 1U24HL094373, and 1P20HL113452 to D.J.L.). This work was also supported by funding from the Carlyle Fraser Heart Center of Emory University Hospital Midtown to J.W.C. and an operation grant from the Canadian Institutes of Health Research to R.W.
Footnotes
6. Disclosures
SG-1002 was provided by Sulfagenix, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Literature Cited
- 1.Andres AM, Stotland A, Queliconi BB, Gottlieb RA. A time to reap, a time to sow: mitophagy and biogenesis in cardiac pathophysiology. J Mol Cell Cardiol. 2015;78:62–72. doi: 10.1016/j.yjmcc.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, 2nd, Kitsis RN, et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ Res. 2016;118:1960–91. doi: 10.1161/RES.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorn GW, 2nd, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981–91. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–21. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S–90. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leone TC, Kelly DP. Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harb Symp Quant Biol. 2011;76:175–82. doi: 10.1101/sqb.2011.76.011965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541–8. doi: 10.1161/CIRCRESAHA.109.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2013;61:599–610. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faerber G, Barreto-Perreia F, Schoepe M, Gilsbach R, Schrepper A, Schwarzer M, et al. Induction of heart failure by minimally invasive aortic constriction in mice: reduced peroxisome proliferator-activated receptor gamma coactivator levels and mitochondrial dysfunction. J Thorac Cardiovasc Surg. 2011;141:492–500. e1. doi: 10.1016/j.jtcvs.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson CK, Calvert JW. Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol Res. 2010;62:289–97. doi: 10.1016/j.phrs.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, et al. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–27. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 13.Pan H, Xie X, Chen D, Zhang J, Zhou Y, Yang G. Protective and biogenesis effects of sodium hydrosulfide on brain mitochondria after cardiac arrest and resuscitation. Eur J Pharmacol. 2014;741:74–82. doi: 10.1016/j.ejphar.2014.07.037. [DOI] [PubMed] [Google Scholar]
- 14.Untereiner AA, Fu M, Modis K, Wang R, Ju Y, Wu L. Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide. 2016;58:67–76. doi: 10.1016/j.niox.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, et al. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120:888–96. doi: 10.1161/CIRCULATIONAHA.108.833491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barr LA, Shimizu Y, Lambert JP, Nicholson CK, Calvert JW. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. 2015;46:145–56. doi: 10.1016/j.niox.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polhemus D, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, et al. Hydrogen Sulfide Attenuates Cardiac Dysfunction Following Heart Failure via Induction of Angiogenesis. Circ Heart Fail. 2013 doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci U S A. 2014;111:3182–7. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, et al. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–9. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia -reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–5. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–74. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mo L, Wang Y, Geary L, Corey C, Alef MJ, Beer-Stolz D, et al. Nitrite activates AMP kinase to stimulate mitochondrial biogenesis independent of soluble guanylate cyclase. Free Radic Biol Med. 2012;53:1440–50. doi: 10.1016/j.freeradbiomed.2012.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, et al. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol Cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholson CK, Lambert JP, Molkentin JD, Sadoshima J, Calvert JW. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler Thromb Vasc Biol. 2013;33:744–51. doi: 10.1161/ATVBAHA.112.300484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calvert JW, Condit ME, Aragon JP, Nicholson CK, Moody BF, Hood RL, et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circ Res. 2011;108:1448–58. doi: 10.1161/CIRCRESAHA.111.241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, et al. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol. 2008;295:H185–96. doi: 10.1152/ajpheart.00081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nature medicine. 2011;17:71–8. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- 28.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–9. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 29.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A. 2004;101:16507–12. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan W, Zhang H, Liu P, Wang H, Liu J, Gao C, et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1alpha signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol. 2013;108:329. doi: 10.1007/s00395-013-0329-1. [DOI] [PubMed] [Google Scholar]
- 32.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circulation research. 2012;111:800–14. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lubbers ER, Mohler PJ. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J Mol Cell Cardiol. 2016;101:127–33. doi: 10.1016/j.yjmcc.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul BD, Snyder SH. H(2)S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 35.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–91. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polhemus DJ, Calvert JW, Butler J, Lefer DJ. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica (Cairo) 2014;2014:768607. doi: 10.1155/2014/768607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–68. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci U S A. 2012;109:2943–8. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem J. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Modis K, Panopoulos P, Coletta C, Papapetropoulos A, Szabo C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem Pharmacol. 2013;86:1311–9. doi: 10.1016/j.bcp.2013.08.064. [DOI] [PubMed] [Google Scholar]
- 43.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.