

Abstract
Increased mortality and diverse morbidities are globally associated with exposure to ambient air pollution (AAP), cigarette smoke (CS), and household air pollution (HAP). The AAP-CS-HAP aerosols present heterogeneous particulate matter (PM) of diverse chemical and physical characteristics. Some epidemiological models have assumed the same health hazards by PM weight for AAP, CS, and HAP regardless of the composition. While others have recognized that biological activities and toxicity will vary with components, we focus particularly on oxidation because of its major role in assay outcomes. Our review of PM assays considers misinterpretations of some chemical measures used for oxidative activity. Overall, there is low consistency across chemical and cell-based assays for oxidative and inflammatory activity. We also note gaps in understanding how much airborne PM of various sizes enter cells and organs. For CS, the body burden per cigarette may be much below current assumptions. Synergies shown for health hazards of AAP and CS suggest crosstalk in detoxification pathways mediated by AHR, NF-κB, and Nrf2. These complex genomic and biochemical interactions frustrate resolution of the toxicity of specific AAP components. We propose further strategies based on targeted gene expression based on cell-type differences.
Keywords: oxidants, nanoparticles, iron, signaling, air pollution, tobacco smoke
Graphical abstract
1. Introduction
Airborne particulate material (PM) from ambient air pollution (AAP), cigarette smoke (CS), and household air pollution (HAP) is globally associated with 15 million deaths per year (Table 1A). Synergies have also emerged for AAP and CS, which show super-additive effects (Table 1B, discussed below). The manifold components of AAP-CS-HAP aerosols present heterogeneous PM of diverse chemical and physical characteristics, including carbonaceous cores and minerals together with organic components. While carbonaceous particles may deliver exogenous toxicants, they may also effect biological responses by adsorbing endogenous compounds. Besides vehicular and industrial sources of fossil fuels, AAP often includes silicates of crustal origin [1][2].
Table 1.
Airborne PM Hazards
| A. Global premature mortality attributed to airborne PM | |
|---|---|
|
| |
| Annual excess mortality, millions (M) | |
|
| |
| Ambient air pollution (AAP) | 4.2M [5] |
|
| |
| Household air pollution (HAP) includes smoke from coal, dried dung, and wood | 4.3M [6] |
|
| |
| Cigarette smoke (CS) | |
| Direct | 6.4M [7] |
| Second-hand | 0.65M |
|
| |
| Total premature deaths from airborne toxicants | 15 M |
| B. Synergies of AAP and CS | ||
|---|---|---|
| Study | Synergy (fold-excess above additivity) | |
| Cardiovascular mortality | ACS Prevention Study II: 429,406 adults [8] | 1.1-fold excess |
| Cancer of lung | ACS Prevention Study II: 1.2 million adults [9] | 2.2-fold excess |
| Body mass index (BMI) | Southern California Children’s Health Study: 3318 children, ages 10-18 y [10] | 1.3-fold excess |
| Meta-analysis of 12 studies: 109,838 mother child pairs [11] | 1.6-fold excess | |
| Neurodegeneration cognitive decline | Health and Retirement Survey, 2004: 18,575, age ≥ 50 years [12] | 1.9-fold excess |
The sizes of aerosol PM are identified by the aerodynamic diameter in micrometers: PM10, PM2.5, and PM 0.1; the larger size classes include all smaller PM. Ambient air pollution is continually monitored by the EPA, which reports PM2.5 throughout the US by density (μg/m3 air); however, but PM composition is not regularly reported. The larger size PM10 −2.5 μ (coarse) are considered less toxic and are trapped in the upper airways; the PM0.1 and smaller (ultrafine) may be even more toxic than the PM2.5 [3][4], but is currently not monitored by the EPA. Cigarette smoke exposure is monitored by blood level of cotinine and other nicotine metabolites, and is rarely reported by size class.
Epidemiologic studies generally show robust associations of health hazards in proportion to ambient PM2.5. Besides loss of life expectancy, specific disease associations include Alzheimer’s, ischemic heart disease and stroke, lung cancer, and chronic obstructive pulmonary disease (Appendix 1). In recent U.S. Medicare data for older adults, mortality rates varied by 7.3% per 10 μg/m3 of PM2.5 over the 90% range of 6.2-15.6 μg/m3 [13]. This and a Canadian study [14] show health hazards into the lowest levels of PM2.5 [15]. Thus, for AAP as for CS, there is a no safe minimum exposure.
The highest regional North American exposures are 10-fold below some Asian cities where PM2.5 often exceeds 200 μg/m3. Some studies suggest that the American PM2.5 hazard associations for AAP extend into the many-fold higher levels of PM2.5 [16][17]. Curvilinear associations are indicated for CS. Additional complexities arise from HAP of homes and other indoor sites in the developing world often include high levels of HAP from domestic fuels of wood and dried-dung, as well as CS. Airborne dust from deserts and agricultural fields (silicates of crustal origin) is also associated with health hazards, although less clearly than urban PM2.5 [18][19][20]. The mechanisms generating these associations are poorly understood.
To our knowledge, no studies with chemical or cell assays have compared ambient PM2.5 from multiple global urban samples. Because the heterogeneous sources of PM2.5 have widely divergent activities by chemical and cell-based assays, we argue that oxidants cannot be directly estimated by the common assays. Furthermore, many publications refer to the oxidants in, or derived from cellular interaction with air pollution components as reactive oxygen species, usually abbreviated as ‘ROS’. Indeed, by using ‘ROS’, it appears that this diverse group of molecules are treated as a single entity. Unfortunately, the actual oxidant has not often been identified or assays have been used that do not accurately measure what is purported for them.
Integrated epidemiological models have considered the total impact of AAP, CS, and HAP on these diseases for global data [16,17]. The shared pathologic associations AAP and CS go from ‘lung to brain’ across humans and rodent models (Appendix 1). The convergent chronic pathologies from AAP and CS suggest biochemical and genomic processes that shared widely across populations. Less is known about cell-type specific responses to various sources of HAP.
Two epidemiological assumptions warrant inquiry. Many studies assume the same toxicity by weight for all sources of PM2.5 and that different sources of PM2.5 (fossil fuel vs cigarette) are additive, i.e. do not synergize. While these are reasonable initial assumptions, other evidence convinces us that PM2.5 by weight cannot adequately represent its toxicity, because of synergies between AAP and CS. For example, AAP and CS have supra-additive effects in adult lung cancer and childhood obesity (Table 1B). These synergies are not well explained by the existing mechanisms that remain to be experimentally studied.
To approach these complex questions, Section 2 compares AAP, CS, and wood smoke for biochemical and cell responses. Section 3 evaluates chemical confounds in common assays for oxidative activity, and comments on misinterpretations of oxidant measurements that persist in the toxicology literature. Recent studies of dung smoke PM show the need to broaden the profile of inflammatory responses in other assays. Section 4 revisits the epidemiologic assumption of equal activity by PM mass, while Section 5 confronts gaps in understanding PM entry to cells and organ systems. Lastly, we propose development of assays for chemical activities and genomic responses to airborne PM with specific cell types. Our inquiry selected studies to best illustrate our main points. We hope to be fair in representing our conclusions about the uncertainties in some widely used assays, for which we suggest alternative approaches. We have not addressed the complexities of pathophysiology or attempted to review global sources. For a more detailed treatment of these complex topics, see the forthcoming monograph [1].
2. Ambient air pollution and cigarette smoke comparisons
The assumption of some epidemiological studies that PM2.5 from different sources has equivalent activity has received little evaluation. We found only one study to assay urban PM with CS and wood smoke with chemical composition by multiple assays with dose responses by Jin and colleagues [21](Table 2). PM10 were collected from two Chinese mega cities and compared with PM from a laboratory diesel engine and from the combustion of cigarette, coal, and wood. The composition of PM10 varied widely by source: up to 100-fold for water-soluble organic carbon (WSOC) and 10-fold for water-insoluble polyaromatic hydrocarbons (PAH). Among metals, iron was the most prevalent by 10-fold or more. The 20-fold higher Fe content of PM10 from Wuhan than Beijing suggests more desert dust.
Table 2.
PM10 of Chinese cities vs smoke from cigarette, fossil fuels, & wood
| A. Composition | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Beijing AAP | Wuwei AAP | Cigarette | Coal | Diesel | Wood | |
| WSOC, mgC/g PM10 | 3.9 | 1.5 | 220 | 11 | 32 | 250 |
|
| ||||||
| WIOC, μg/mL anthracene, benzo[a]pyrene, chrysene | ||||||
| 0.8 | 31 | 140 | 3.9 | 3.8 | 0.5 | |
| 0.24 | 0.56 | 7.1 | ||||
| 1.1 | 7.2 | 4.8 | 22 | 5.3 | 0.25 | |
|
| ||||||
| endotoxin (LPS), μg/g PM10 | 1.2 | 8.3 | 1.5 | 1.2 | 1.3 | 0.8 |
|
| ||||||
| Cu, μg/g PM10 | 20 | 91 | 16 | 32 | 31 | 4 |
|
| ||||||
| Fe | 800 | 17,000 | 1,200 | 2,000 | 1,700 | 100 |
|
| ||||||
| Ni | 3 | 44 | 7 | 16 | 10 | 1.5 |
|
| ||||||
| Pb | 70 | 98 | 7 | 85 | 31 | 6.3 |
|
| ||||||
| Zn | 236 | 378 | 184 | 195 | 178 | 35 |
| B. Oxidative and Cytokine Responses | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Beijing | Wuwei | Cigarette | Coal | Diesel | Wood | |
|
| ||||||
| Chemical assay | 60 | 40 | 60 | 40 | 130 | 60 |
| DTT oxidation, μmol/min μg PM10 | ||||||
|
| ||||||
| Cell assays ED50 | ||||||
| DHE, fluorescence | 500 | 500 | 750 | 550 | 550 | 1000 |
| TNFα, ng/ml media | 11 | 8 | 4 | 4 | 3.8 | 1.8 |
Cell assays for inflammatory responses and what these authors referred to as “ROS production” gave divergent responses between these samples. For TNFα release by macrophage cells, urban PM10 was >2-fold more active than the PM10 from smoke PM10 from diesel or cigarette, while wood smoke was inactive and without any dose response up to 400 μg/ml. In contrast, the cell fluorescence assay using dihydroethidium (DHE) gave up to 100% higher values for CS and wood smoke than for urban PM10; notably, the urban AAP PM10 did not induce any DHE response. The authors concluded from the reversed order that “factors affecting ROS and TNFα release were rather different”, and that TNFα also positively correlated with metals (Cu, Fe, Pb), but negatively with WSOC. The oxidation of dithiothreitol (DTT) did not correlate with TNFα induction; e.g., diesel had the most DTT activity, but the least TNFα response. (Table 2B). These divergent outcomes may be in part understood by the chemical confounds in these assays as discussed below.
The perplexing divergence in these findings are familiar to many investigators seeking the source attribution of AAP components for which the chemical and cell assays are inadequate or misleading. We respect these studies for providing both chemical composition and using multiple assays in their ‘en suite’ study of major ambient PM. We further note the need for direct chemical comparisons of CS, AAP, and wood smoke (HAP) by PM size class concurrently in the same assay.
CS may be considered less complex than AAP because it arises from immediate combustion of a single plant species directly enters the body; CS also includes multifarious soil contaminants and pesticides, as well as nicotine-specific toxicants and high levels of carbon monoxide that are atypical for AAP. Nonetheless, both AAP and CS include carbonaceous particles that derive from the incomplete combustion of organic material.
CS particle sizes and chemistry overlap with AAP: for both, the vast majority of particles is <0.2 μm [22][23] [24]. CS has thousands of trace organic components including polycyclic aromatic hydrocarbons (PAHs) and many trace metals. The 5000+ organic compounds in CS include more than 50 carcinogens recognized by the International Agency for Research on Cancer (IARC), some of which are shared with AAP from near-roadway sites (traffic associated air pollution, TRAP).
To start this inquiry, we assembled data from TRAP and CS. Figure 1 shows the distribution of several metals and water insoluble organic compounds (WIOC) in TRAP vs primary CS: For both, a group of toxic elements (arsenic, chromium, cadmium, lead) was highest in the ultrafinePM; however, the larger PM are not equally represented in these samples. The larger size classes of CS appear to contain more WISOC than in TRAP, which has implications for pathophysiology. Direct comparisons are needed using the same sampling protocol and assays for WSOC and other components by the same size classes for AAP and CS.
Figure 1.
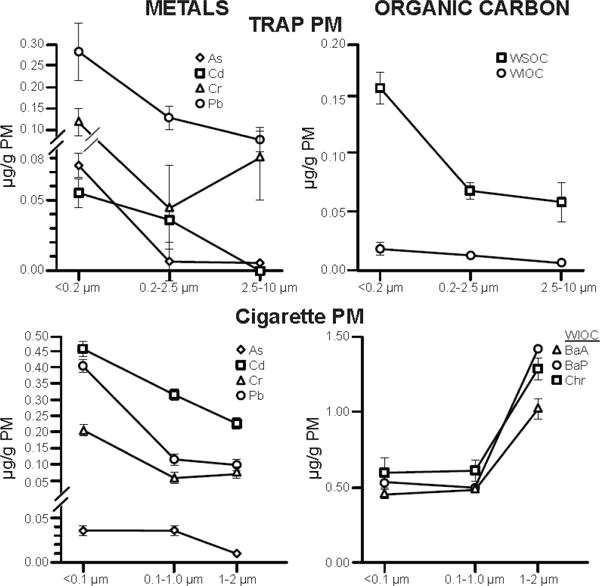
Chemical composition of traffic related air pollution (TRAP) vs cigarette smoke (CS) by PM size class. Note differences between the larger size classes of TRAP vs CS.
PAH, polycyclic aromatic hydrocarbons; BaA, benz(a)anthrocene; BaP, benzo(a)pyrene; Chr, chrysene; WSOC, water-soluble organic carbon; WIOC, water-insoluble OC.
TRAP PM: From Los Angeles County, 2013-2014; WSOC and WIOC are averaged across sampling sites from Fig 2 of Hu et al. [25]; metal levels obtained by Dr. Arian Saffari from data of [26][27]. WIOC includes PAHs, n-alkanes, hopanes, and steranes, WSOC includes n-alkanoic acids, resin acids. These data resemble Tokyo samples which had 2-fold greater PAH densities in ultrafine PM than fine PM [28].
Cigarette PM: From 3R4F reference cigarette; PM per cigarette, 10.5 mg. Unimodal distribution (mean 0.26 μm, range 0.02-5.9). Tobacco-specific nitrosamines and heavy metals are enriched in smaller PM; heavy metals, larger; nicotine evenly distributed. Redrawn from [29]. The size distribution confirms Li et al [30].
The next section examines in detail chemical assays for PM activity that are widely used. Our discussion aims to illustrate mechanisms and confounds with examples, and does not attempt to comprehensive review the extensive literature on diverse sources of PM with different chemistries.
3. Assays for ambient particles
A decade ago, Künzli et al. [31] provided a thorough comparison of the chemistry and biological responses to samples from multiple sites, “PM oxidative activity varied significantly among European sampling sites. Correlations between oxidative activity and all other characteristics of PM were low, both within centers (temporal correlation) and across communities (annual mean). Thus, no single surrogate measure of PM redox activity could be identified. Therefore, it is important to define the appropriate methods to determine oxidative activity of PM.” Attempts to find such an appropriate method have continued. Three prominent and commonly used chemical assays are discussed next, followed by cell-based assays.
3a. Chemical assays
To evaluate the potential toxicity of PM2.5 with varying composition, many studies use chemical assays for the oxidative activity of PM2.5. Widely used assay substrates include dithiothreitol (DTT) and ascorbate acid (AA)-glutathione (GSH), which we discuss in sequence.
Dithiothreitol
Findings for DTT are often reported as units of reactive oxygen species (ROS). However, as stated earlier this is not chemically valid because ‘“ROS” is not a single molecular species. In fact, the DTT and other assays are sensitive to diverse agents: some are true free radicals with unpaired electrons (superoxide O2 − and hydroxyl HO•). Additionally, these assays detect other redox active agents that are not free radicals (hydrogen peroxide, ozone, singlet oxygen, and peroxynitrite). Each differs markedly in physical, chemical, and biochemical properties. Contrarily, the putative values given for ROS by the DTT assay actually represent the non-specific production of undefined oxidation products. Thus, most reports using DTT cannot be considered as either quantitative or qualitative for the oxidants in the sample. Conclusions from the DTT assay do not serve well to predict APP-associated cell response (inflammatory and/or anti-inflammatory) and cell damage. Furthermore, the heterogeneity and variability of AAP frustrates simple interpretation of these measurements as end-points for oxidative activity.
The DTT assay as initially reported assumed that quinones mediated DTT oxidation by catalysis [32]. While thermodynamically feasible, this reaction scheme cannot account for the DTT oxidation observed in the assay (see Appendix 2). We suggest a more plausible mechanism.
Quinones mediate many AAP and CS oxidative mechanisms, and occur in the water soluble organic fraction (WSOC) of both TRAP [33] and CS tar [34][35][36]. Quinones are defined as aromatic ring compounds with two carbonyl groups at various locations. During combustion, quinols (the reduced form of quinones) are oxidized to semi-quinone free radicals which can propagate oxidation by redox re-cycling [36]; Reactions 1-5 below.
As described 80 years ago, quinones (1,2- or 1,4) readily combine with thiols in a Michael addition reaction [37]. 1,4-naphthoquionone (NQ), is a component of AAP [33]. Here we show it forms an adduct with thiols (RSH), such as cysteine or GSH in Reaction 1:
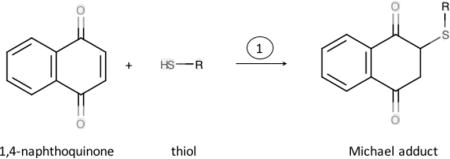
The reaction of GSH with quinones is a potential contributor to GSH depletion observed in interactions of AAP with lung-lining fluid components [31]. In contrast, because DTT has two thiol groups in close proximity that readily form a stable six membered ring when oxidized, DTT reacts in a concerted reaction with NQ in which the NQ is reduced (Reaction 2). NQ then rearranges to form the 1,4-naphthoquinol (Reaction 3).
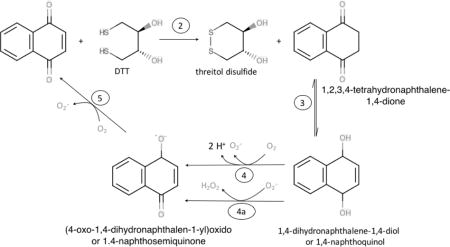
Quinols can then react either with O2 (Reaction 4) to generate superoxide (O2·−) or with O2·− (Reaction 4a) to generate hydrogen peroxide (H2O2). In either case, a semiquinone is formed, which is a free radical. The semiquinone then is restored to the original quinone by reacting with another O2 (Reaction 5). The key point is that quinones can cycle though oxidation and reduction, but catalyzing DTT oxidation without being consumed. The O2·− generated could also react with DTT to produce the thiyl radical, as reported [32]:
| (6) |
The thiyl radical will then react with another molecule of O2, thus accounting for
| (7) |
the observed acceleration of DTT consumption by O2. Consistent with recycling of the quinone, 10-fold more DTT can be consumed than the quinone that is lost. The loss is most likely through adduct formation with one of the thiols of DTT in Reaction 1.
Transition metals (M, below), the most abundant of which is iron, catalyze the oxidation of DTT even more easily than quinones because of their larger reduction potential and reaction rates.
| (8) |
| (7) |
| (9) |
| (10) |
| (11) |
The hydroxyl radical generated in Reaction 11 (Fenton chemistry) will oxidize rapidly oxidize DTT to sulfinic and sulfonic acid forms.
In sum, the widely used DTT oxidative assays are confounded by facts that both the quinone and the metal reactions are neither completely catalytic, nor first order reactions driven by substrate concentration.
Ascorbate acid (AA)-glutathione (GSH)
The oxidative activity of PM2.5 in AAP is also often assayed by AA and GSH, the normal reductants in lung fluids [31]. We focus on relationships of oxidative activity to metal content.
Many studies show associations of oxidative activity with the iron content of AAP. Positive correlations of AA and GSH oxidation with PM2.5 iron content were reported for Paris [38]; for 8 of 9 sites in the Los Angeles Basin [39] and for a Dutch train station [40]. In contrast, PM2.5 from the Barcelona Metro showed negative correlations of iron levels with oxidative activity (Table 3) [41]. The electrified Barcelona Metro PM2.5 are notably rich in metals from mechanical abrasion of metal surfaces and of brake linings, but have minor contribution from fossil fuels. When iron or other transition metals fail to cause DTT or AA oxidation, we suggest this is due to their location within the particle or their chelation which prevents their redox cycling. Studies discussed below illustrate this for surface iron in silica particles.
Table 3.
Correlation of metals vs oxidative activity in assays of biological reductants
| Ascorbic acid assay (AA) | Glutathione assay (GSH) | |
|---|---|---|
| Antimony | 0.27 | 0.05, p<0.01 |
| Copper | 0.29 | 0.64, p< 0.01 |
| Iron | −0.56, p <0.01 | −0.14 |
| Nickel | −0.05 | −0.12 |
| Lead | 0.40, p<0.05 | 0.34, p<0.05 |
| Zinc | 0.05 | 0.21 |
Adapted from Moreno et al [39]. Entries without P values were not significant.
For iron to participate in cell responses to particles, it must be accessible to interact. Thus, the iron must be on the surface rather than buried within the structure. It also must be chemically reactive. Thus, metallic iron (no charge) or iron that is complexed in such a way as to make it chemically inert, will not participate in reactions. Indeed, iron being part of the composition of the internal structure or inert, is largely the reason its content is so poorly correlated with biological responses. When iron participates in biological responses to particles, it is usually through initiation of lipid peroxidation. In reactions 10 and 11 above, iron cycles between the ferric (3+) and ferrous (2+) state while generating hydroxyl radicals that then initiate lipid peroxidation. But, we also note that AA can reduce iron from ferric to ferrous and may thereby act, seemingly counterintuitively, as a pro-oxidant. This pro-oxidant effect of AA and other reductants that is due to their reduction of iron has been known since the 1960s [42].
We note that some correlations would be considered anomalous and misleading by biochemists. Oxidative activity with AA and GSH was positively correlated with antimony and lead in these studies (Table 3) and by [38]. However, neither antimony nor lead can directly oxidize glutathione, although these metals are bound ionically by GSH and other thiols (RSH) and may thereby facilitate their oxidation. Nickel and zinc did not correlate with oxidative activity. This is reasonable because under physiolologic conditions, these metals also may, under some conditions, facilitate but cannot oxidize ascorbate, glutathione, or other organic compounds. The potential in vivo toxicity of nickel, zinc, and other trace metals is indisputable, but cannot be evaluated by these and other in vitro chemical assays.
3b Cell assays
Cells will react with both quinones and metals both at the cell plasma membrane and internally if taken up, with differences by cell type. Cell types also vary in reactivity depending on handling of metals and whether quinones are reduced and/or conjugated. Cell response to PM also differ in production of O2 − and H2O2 by activating NADPH oxidases, which mediate signaling pathways (Section 3e below). Next, we discuss cell- based assays for oxidative activity for three widely used markers: DCF, DHE, and MTT.
Dichlorofluorescein (DCF)
The substrate 2′,7′-dichlorofluorescin diacetate (DCFH-DA) is hydrolyzed within cells to DCFH that, in turn, may be oxidized to the fluorophore 2′,7′-DCF. The assay described for H2O2 originally required the presence of a peroxidase catalyst [43]. Mistakenly, the assay began to be used for H2O2 and other oxidants without regard for the need for catalysis. Rigorous studies showed that dye oxidation was primarily due to iron-catalyzed reaction with H2O2. [44][45][46]. Moreover, DCFH is oxidized by diverse agents that lack specificity for H2O2 or other reactive species [47]. The DCF assay may be even more problematic because the DCF radical generates superoxide and H2O2 from spontaneous free radical chain reactions [48]. By neglecting this established chemistry, erroneous conclusions continue. For example, one study employed microfluidic electrophoresis with fluorescence of DCF enhanced by plasmon-resonance, from which it was calculated that each puff of CS had 8 pmol of “ROS” [49]. Despite the elegant plasmon technology for directly assaying ambient CS, the DCF response was not defined chemically for any of the oxidants in the assay. As described above, calculation of ROS per mole has no chemical meaning. Furthermore, because of chain reactions intrinsic to DCFH chemistry, the fluorescence DCF lacks chemical specificity.
Dihydroethidium (DHE) can be a specific assay for superoxide
The substrate hydroethidine is oxidized to 2-hydroxyethidium by superoxide, but also by many other agents. To identify superoxide requires separation of the products by HPLC [50]. Thus, DHE is superior to DCF for superoxide if properly conducted. However, superoxide formed in the media away from the cell surface would dismute to oxygen and H2O2 (see Reaction X), which could affect cells. Regardless, formation of superoxide is dependent on particle surface reactions and will not correlate with bulk measurements of metals, quinones or other particle components.
Ferricytochrome c
The spectrophotometric measurement of SOD-inhibitable reduction of ferricytochrome c remains among the simplest real-time assays [51]. However, PM may interfere with the assay by physically blocking ferricytochrome c, a 12 kDalton protein, from accessing the site of O2·− production. Alternatively, the smaller substrate nitro-blue tetrazolium can be used with appropriate controls [52].
MTT
The reduction of MTT (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to its purple formazan product gives a measure of mitochondrial activity. However, large decreases of the MTT substrate do not necessarily indicate cell death or irreversible cell stress: depending on the toxicant, cell death may be overestimated [53] or underestimated [54].
Reliable assays for cell death include release of the easily measured lactate dehydrogenase (LDH) and the uptake of propidium iodide which binds to DNA, [55]. Cell damage may also be assessed by cell exclusion of the vital dyes, Trypan Blue and Erythrosine, or binding of fluorescently tagged Annexin V. While all of these assays may indicate cell damage by particles, none of them can show that the damage is specifically due to oxidative stress. Although airborne PM may include agents that potentially cause oxidative damage, it difficult to prove causality of one agent among a suite of candidates in a complex composition.
Lastly we note two major studies which used expanded arrays of multiple chemical and cell responses to PM2.5, but reached the same frustrating conclusions as did Kunzli et al. [31]. Crobeddu et al. [38] included DTT, AA, and GSH; a plasmid scission assay, and DCFH; expression of two antioxidant enzymes and pro-inflammatory IL-6. They also measured a large group of metals and multiple PAH species. They concluded that AA and GSH oxidation correlated best with both DCFH oxidation and expression of selected genes. However, Visentin et al. (2016)[54]?? did not find correlations of DTT and ascorbate assays; contrarily, the content of zinc was highly correlated with DTT oxidation, which makes sense to biochemists because Zn is not oxidizable under these conditions (Section 3, Table 2). The state-of-the-art for source apportionment of disease risk assessment for based on these assays looks more like the randomness of a Jackson Pollack painting than the orderliness of a Piet Mondrian.
3c. Ambient free radicals
Could persisting free-radicals in ambient particles also be factors in these assays? We observed free radical signals from re-aerosolized ultrafinePM collected a month earlier and stored in frozen aqueous suspension (from co-author Nicos Petasis) [57]. These EPR spectra are consistent with carbon-based free radicals in the graphitelike particles [33]. The long-term stability of free radicals in TRAP suggests that these radicals are ‘buried’ within the particle matrix and thus inaccessible for reaction.
Long-lived carbon-based free radicals also occur in coal dust [35]. Because the half-life of CS-tar derived free radicals in solution is short (seconds) [35], it is likely that the soluble free radicals are rapidly quenched by ascorbate and other antioxidants that are enriched in the lung surfactant fluids [58]. Although CS tar can stimulate production of hydroxyl radicals (HO•) in chemical and cell assays, this activity critically depends upon the presence of iron and other metals. If the hydroxyl radical HO• is produced in fluids covering cells, its effect would be minimal because its reactivity would be quenched by ascorbate and glutathione, together with lipids and proteins in the fluids lining the airways and alveoli, even before any contact with cell surfaces. HO• reacts promiscuously with neighboring molecules with near 100% efficiency, causing in vivo half-lifes of nanoseconds [59]. Because iron and other transition metals are essential to HO• formation, this radical can only be involved when the metal is in direct proximity. Thus, most if not all free radicals in ambient PM measured in these assays cannot not contribute to oxidative signals in cells. In contrast, H2O2 formed in the media may last long enough to interact at the cell membrane or enter cells to mediate redox signaling [60].
3d. Particle content: solubility and accessibility
Another major issue is the solubility and accessibility of prospective toxicants in PM to the assay, as shown for iron in silica PM. Iron has well documented roles in pro-inflammatory responses and injury to cells [61]. In macrophage responses to natural silica PM, Forman and colleagues showed that the surface iron was essential for induction of TNFα and IL1β [62]. For a 100 nm spherical particle composed of a uniform iron-containing material, only 6% of the iron would be on the surface. Silica PM from which iron was leached by acid treatment failed to induce IL-1β and TNFα in macrophages, indicating that an iron-particle-cell interaction was essential for these cytokine effects. As further evidence, adding an equal amount of iron to that on the particle surfaces (0.5 nM) in the absence of particles did not alter expression of IL1β or TNFα. Thus, iron chemistry and availability may vary considerably dependent on whether it is bound to carbonaceous particles, or present as multinucleate ferric hydroxide on the surface of silicate PM, or in a nano-chunk of steel.
The accessibility of iron and other metals may underlie assay complexities shown for PM from five Canadian industrial sites including smelters and petrochemicals [63]. This study is exemplary for its detailed dose responses in cell assays by source and PM size class, but did not include chemical assays. PM size classes (<0.1 to >10 μm) were characterized for metals and polycyclic aromatic hydrocarbons (PAH) in assays of cytotoxicity and cytokine responses with cell lines representing macrophages (J774A.1) and lung epithelia (A549, type II alveolar cells). As expected, endotoxin levels strongly correlated with cytokine release, which gives an important validation. Smaller PM varied in metal content between sampling sites more than PM >2.5 μm, especially for water-soluble iron and other metals. For all PM classes, cytokine release correlated weakly with iron and other metals, as well as with PAH. In dose responses, PM0.1 had lower cytotoxicity and cytokine (IL6) release than larger PM, contrary to many reports [39].
In considering, the role of metals in CS toxicity, iron is often discounted as a major contributor [64]. This is counter-productive, as iron is the most prevalent metal in combustion products from coal, fly ash, wood, diesel exhaust, and AAP. Iron is also enriched in the humic-like substances (HLS) of CS PM0.2 [22][65][66]. The HLS occur globally in AAP [67][68]. The iron bound to HLS largely comes from the microbial degradation residues found in soil, peat, and coal [69]. Its complex composition includes aromatic rings and aliphatic chains with multiple hydroxyl and carboxyl moieties that can bind metals, particularly iron. The association of HLS with oxidant generation could be a key to toxicity of combustants from fossil fuels, forest fires and cigarettes [70]. In rat lung, HLS from CS caused deposits of iron with inflammatory responses that resembled iron-rich foci in smokers’ lungs [65][71]. Long-term deposits of HLS are associated with sequestered iron and focal disruption of iron homeostasis [22].
Ghio and colleagues [69] also showed that the production of the inflammatory mediator LTB4 by macrophages could be stimulated by iron on silica in the presence of ascorbate, which is present in the airway and alveolar fluids. While iron is the most studied transition metal for oxidative stress, other transition metals including copper, manganese catalyze the same reactions, which raises the same issues regarding bulk content versus surface availability.
3e. Signaling pathways for gene expression
Most toxicology assays have examined a small number of oxidative or inflammatory markers. The value of a broader genomic-based approach is illustrated in pioneering studies of smoke from burning dried dung (DS) [72][73]. Dung is common household fuel of impoverished households of Asia and Africa, with undefined contributions to the mortality associated with household air pollution (Table 1). Based on their size of 0.57 + 0.15 μm, DS particles are predicted to deposit in bronchioles, with limited penetration to alveoli. Accordingly, these experimenters chose to model cell responses with human small airway epithelial cells (SAEC), obtained from adult donors with no smoking history. In a novel air-liquid culture model, SAEC cells were exposed to ambient DS at 1000 μg/m3 density for 60 minutes to simulate household cooking times. Surprisingly, after 24 hours, only two cytokines, IL8 and GM-CSF responded with several-fold increase, while there was no response by IL6, TNFα or more than 30 other cytokines, or of NF-κB, a key inflammatory transcription factor. Cell type may be underlie the lack of NFκB activation, because epithelial cells are unresponsive to TLR2 agonists [74]. Nonetheless, DS induced Cox-2 via the JNK-AP-1 and aryl-hydrocarbon receptor (AhR) pathways, which also mediate inflammatory responses to urban AAP. DS from six herbivores including foregut and hindgut fermenters all activated AhR and AP-1. Notably, DS impaired the interferon response to a viral analog (poly I:C), suggesting a molecular mechanism for the lung infections associated with household air pollution. These findings indicate that cell responses to DS may have major differences from the smokes of fossil fuel and wood, which were not tested in parallel. These pathways are summarized in Fig. 3.
Figure 3.
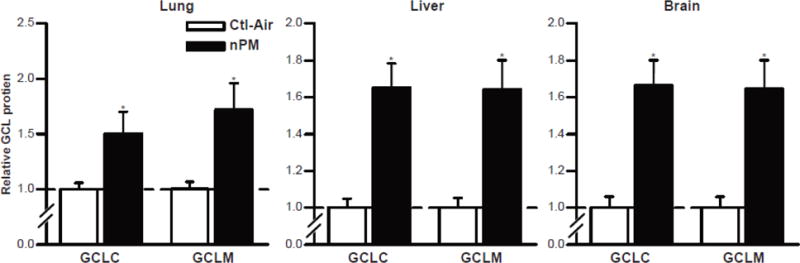
Induction of Nrf2-dependent phase II detoxifying enzymes in young mice exposed chronically to defined levels of nPM, a water-soluble subfraction of ultrafinePM [135]. The graph shows protein levels of glutamate-cysteine ligase (GCL) subunits GCLC (catalytic) and GCLM (modifier), induced to 50-60% in all three organs.
Together with data of [21] (Table 2), these findings suggest that smoke PM from wood and dung has less inflammatory activity than from AAP and CS in cell assays, and that these chemical assays did not strongly predict cell responses. Further direct comparisons of dung and wood smoke with PM2.5 from CS and AAP are needed.
NF-κB is recognized is a key regulatory of inflammatory responses to AAP in vivo, shown in rodent models [75]. Most of inhaled PM initially disperse onto trachealbronchial epithelial cells and alveolar macrophages, which receive the largest per cell dose. Activation of NF-κB in response to AAP in macrophages and epithelial cells [76][77][78] stimulated transcription of TNFα, IL-1β, and other pro-inflammatory cytokines.
Using the model of natural silica from which surface iron was removed and replenished, we showed that iron-dependent lipid peroxidation at the surface of the human THP-1 macrophages resulted in with lipid raft disruption [62]. This disruption led to activation of the phosphatidylcholine specific phospholipase C (PC-PLC) pathway that, in turn activated NF-κB, with following induction of TNFα and IL1β. Air pollution PM-stimulated production of cytokines by human airway epithelial cells is also metal-dependent [78]. NF-κB activation by TNFα through the PC-PLC pathway was also proposed for human airway epithelium [79][80], but remains untested. The PC-PLC pathway was activated by other agents in epithelial cells in several studies [81][82][83]. Nonetheless, the PC-PLC pathway is not considered as a classical pathway to NF-κB activation by oxidants.
We recently compared glial genomic responses of ultrafinePM with LPS, a widely used model for endotoxin-induced inflammation, by whole genome microarray [84]. Both agents induced TNFα and NFκB through the shared TLR4 pathway. These responses were verified in the hippocampus of mice chronically exposed to ultrafinePM. Because these ultrafinePM had minimal LPS activity, the activation of TLR4 suggests additional signaling pathways for AAP components.
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), is a master regulator of transcriptional responses to stresses from electrophiles including the oxidants in, or generated by, exposure to PM from both air pollution and cigarettes (Figure 2). Nrf2 activation is mediates increased transcription of groups of antioxidant enzymes, of detoxification enzymes that catabolize xenobiotic compounds, proteasome subunit expression [85].
Figure 2.
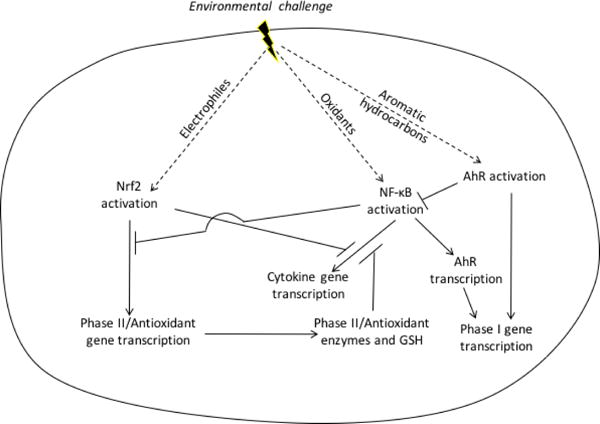
Schema of Nrf2, NFκB, AhR responses to airborne toxicants. AAP, DS, and CS induce antioxidant enzymes through electrophilic Nrf2 activation and NF-κB activation through oxidative signaling. Nrf2 activation is inhibited by c-Myc and Bach1, while NFκB activation is opposed by Nrf2-dependent antioxidant enzymes and glutathione. Also, there is non-redox dependent crosstalk by which NFκB inhibits Nrf2-dependent signaling and Nrf2 inhibits NF-κB-dependent transcription. This cross-talk includes antagonistic binding to DNA and interference with transcription through interaction with co-activators and repressors. AhR also forms a heterodimer with RelB, which enables cross-over regulation of NF-κB regulated genes.
Nrf2 is continuously synthesized and then rapidly destroyed through interaction with Keap1. These dynamics facilitate its rapid degradation by the ubiquitin-proteasome pathway. In a simplified view of these mechanisms, modification by alkylation or oxidation of specific cysteines in Keap1 inhibits Nrf2 degradation, thereby allowing newly synthesized Nrf2 to reach the nucleus. Electrophiles, including quinones in nPM [33] and acrolein [86] and benzene-derived quinones [87] in tobacco smoke, modify Keap1 and thereby activate Nrf2. H2O2 is also an activator of Nrf2 [88]. H2O2 can be generated by activation of NADPH oxidases or from redox cycling of quinones, a common pathway for PM from AAP and CS.
Responses to air pollution PM as three tiers proposed by Nel and colleagues [89] a decade ago extended the concept of a balance between induced antioxidant defenses and oxidative stress resulting from inflammation to the realm of signaling involving Nrf2 for antioxidant defense and NF-κB for inflammation. However, recent research shows that these two signaling systems are not dichotomous and interact at multiple levels. For example, activation of NF-κB by PM can result in activation of Nrf2 signaling several hours later [90].
The Nrf2 pathway also mediates body-wide responses to AAP. In our mouse model, chronic exposure to ultrafinePM, induced robust Nrf2-dependent responses in lung, liver and brain of young mice, specifically for glutamate-cysteine ligase (GCL) which is one of the classic phase II xenobiotic detoxifying enzymes (Figure 2). Surprisingly, the size of GCL induction (50-60%) was equally large in brain (cerebellum) as in lung and liver, We discuss this puzzle further in Section 5.
CS extract also induced Nrf2 in epithelial cells more rapidly and at lower levels than HO-1 and other components of the phase 2 detoxifying pathway [91] The oxidative stress associated with CS extract was alleviated by repression of an Nrf2 inhibitor KEAP1 [92]. Systemic and local cell responses to direct contact with inhaled particles are discussed below.
There is a complex interaction between Nrf2 and NF-κB signaling. At one level, the gene products of the two pathways affect the expression of the other. NFκB can be activated by endogenously produced H2O2 [93]. Nrf2-regulated enzymes, and production of GSH, and thioredoxin, and NADPH, which all contribute to the elimination of intracellular H2O2, should also affect the H2O2-dependent activation of NFκB even when cells are not stimulated [94]. Thus, increases in antioxidant genes through Nrf2 activation by AAP, CS, or dung smoke should attenuate pro-inflammatory gene induction. There are also direct interactions between the NFκB and Nrf2, with reciprocal negative feedback loops that inhibit transcription dependent upon the other [95]. At a third level, the proteins produced by NFκB activation further stimulate the activation of Nrf2. Most recently, we used iron-coated silica nPM to investigate the second and third levels [90]. Within the first six hours after nPM exposure of macrophages, pro-inflammatory cytokines were induced by NFκB activation, but without increase in the mRNAs of four Nrf2-dependent genes. However, at 18 hours post exposure, the four Nrf2-dependent mRNAs were all elevated through an NFκB-dependent mechanism Interestingly, one Nrf2-dependent gene, glutamate cysteine ligase catalytic subunit, was depressed at 6 hours in an NFκB-dependent manner.
The aryl-hydrocarbon receptor (AhR) pathway catabolizes polycyclic aromatic hydrocarbons (PAH) and is a major mediator of responses to airborne toxicants from AAP, CS, and HAP. The AhR is a ligand-activated transcription factor residing in a cytoplasmic complex with HSP90 and other chaperones. After ligation, AhR binds to xenobiotic response elements of de-toxifying genes, as well as genes mediating other functions. The broad pleiotropies of AhR extend to inflammatory cytokines, cell proliferation, apoptosis, and stem cells. The NFκB module also mediates CS-induced apoptosis through the AhR [96]. The AhR protein can also dimerize with RelB, with cross-over regulation of NF-κB regulated genes [97].
CS induced AhR and NF-κB, in vitro and in vivo [98]. Dung smoke (DS) also induced the AhR, but without induction of NFκB in both studies, discussed above. This is the first indication that DS and CS could activate different subcellular pathways. CS also alters DNA methylation (DNAme) of the AhRR (aryl hydrocarbon repressor) [99], a tumor suppressor gene for xenobiotics that we suggest also could influence vulnerability to air pollution.
4. The epidemiological quandary: how to evaluate different sources of PM
Lung cancer, known for strong CS associations, is also linked to AAP with increased risk of 8% per 10 μg/m3 PM2.5 [100][101 ][102]. These findings led the International Agency for Research on Cancer in 2013 to classify both outdoor air pollution and airborne PM as carcinogenic. Synergies of AP and CS for lung cancer are shown in a benchmark analysis from the Cancer Prevention Study II [9] (Table 1B): The combined mortality from lung cancer for CS with high PM2.5 exposure was 2-fold over simple additive effects of high PM2.5 or CS, and accounted for 14% of lung cancer mortality. While lifetime CS exposure can be reasonably estimated, the total PM2.5 exposure is less certain. The authors commented “Potential biological mechanisms for greater-than-additive effect remain unclear”. No animal model studies have included both AAP/TRAP and CS (Appendix 1).
Beyond lung cancer, AAP-CS synergies have not been widely considered in epidemiological studies. A baseline for further studies is the integrative dose-response model [16][17], which includes these diverse sources. Lung cancer shows linear increases across a 10,000-fold range of PM2.5, while ischemic heart disease reaches a plateau at much lower levels. These studies assumed an inhaled dose of PM2.5 of 12 mg per cigarette, equivalent to a daily intake of ambient PM2.5 at 667 μg/m3. However, the assumed equivalent toxicity by weight for all-source PM2.5 is challenged by a little noted gap between initial nicotine content and resulting blood levels.
Using careful pharmokinetics, Benowitz and Jacob [103] showed that 1 mg of nicotine per cigarette reaches the circulation, which is 90% less than the 8-9 mg of nicotine in the unsmoked cigarette Because nicotine is evenly distributed across particle sizes [29] and is soluble, we suggest that this 10% also approximates CS delivery of soluble toxicants associated with CS PM2.5. Thus, the effective dose of PM2.5 per cigarette would be 10% of the initial 12 mg of PM2.5 per cigarette, or 1.2 mg. This deduction still allows conclusions from many studies that lungs retain the majority (60-90%) of particles inhaled by smokers [104]. However, none of these studies considered the solubility of potential toxicants, many of which are water soluble, e.g. metals and oxidized PAH and other carbon compounds [26]. These adjustments of delivered PM2.5 would recalculate these data with a left-ward shift by about 10-fold for values above 10,000. This recalculation would not alter the main conclusion that the IHD risk reaches a plateau, while lung cancer increases linearly with at higher levels of PM2.5 exposure.
Second-hand smoke (SHS) also interacted with AAP in two studies of childhood obesity. In the Children’s Health Study of Los Angeles, the body mass index (BMI) was higher in adolescents exposed to SHS, with further increases in those living near roadways [10](Table 1B). The net impact was 3.0 BMI units above those with low PM2.5 and no SHS exposure by age 18, and scaled with the number of household smokers. Similarly, children of the National Health and Nutrition Survey (NHANES) also showed super-additive synergy of SHS and traffic exposure for obesity [105]. Urinary PAH was used as a measure of exposure to fossil fuel exhaust, with correction by serum cotinine, a marker for SHS. Compared with the lowest exposures, children with high SHS and PAH exposure had 30-fold more obesity. The strongest PAH-obesity associations were with naphthalene metabolites (2-benzene rings. Consistent with these findings, German adolescents [106] and adults [23] showed strong association of insulin resistance with PM2.5. The recognition of AAP as an endocrine disruptor for insulin resistance [107] must now be expanded to include these synergies with CS.
As an approach to synergies of airborne toxicants, urban PM from a Canadian city (EHC-93) were eluted by sonication from filters followed by centrifugation and passage through 0.2 μ filters, yielding water soluble (WS) and water insoluble fractions (WIS) [108]. Protein changes and cytoxicity of A459 lung cells and in vivo exposures marked differed in response between the WIS and WS, including a group of inflammatory proteins that were increased by WS and increased by WIS. In vivo installation of the WS fraction caused mild lung injury in rats, whereas WIS was less toxic, unlike in vitro cell responses. It was concluded that the response to the combination of the two fractions was the result of both synergistic and antagonistic interactions. However, the delivery of water-soluble agents to cells in this method cannot reflect the much higher local concentrations of an agent as it diffuses from particles on the cell surface or as it taken up by endocytosis into cells.
5. How do particles enter the cell and our body?
We briefly discuss major gaps in how inhaled PM enters cells, and impacts organs systemically in adults and during development.
5a. Cell entry
PM may enter cells by endocytosis, in which part of the plasma membrane surrounds the particle and then buds off into the cytosol [109]. Four pathways are described for endocytosis of ultrafinePM; caveolar-mediated endocytosis dependent upon clathrin, phagocytosis, macropinocytosis, and pinocytosis [110]. PM components could leak from the endosomes. However, because endosomal membranes are similar to the plasma membrane from which they originate, only those materials that enter from the extracellular fluid would likely escape the endosome into the cytosol. This route could enable delivery of soluble components of AAP into the cytosols, as proposed for drug delivery by nanoparticles [111]. Zinc and other metal ions from AAP may escape from endosomes, particularly if they fuse with lysosomes in which the acidity may solubilize them [112]. It seems unlikely that particles larger than small molecules can escape endosomes to enter mitochondria or nuclei.
PM entering a cell from the apical surface can form an endosome, which may cross the cell to the basolateral surface and fuse with the plasma membrane thereby releasing a particle into the extracellular fluid on the basolateral side. This trans-tissue transport can also work in reverse. Translocation across tissues depends on both the physico-chemical properties of the PM and the mechanism of endosome formation [110][113]. Independently of endocytosis, cell signaling processes may be initiated by interactions at the plasma membrane, including receptor binding in macrophages [114] or disruption of membrane lipid rafts [62]. Thus, cell internalization PM is not required for toxicity.
5b: Inhalation
Inhalation is our main exposure to airborne pollutants. While lungs receive the bulk of PM2.5, some is ingested after mucociliary transport into the throat, discussed below. There is definitive evidence that some particles enter the brain, but we lack consensus on the extent of ‘nose-to-brain’ transport by human olfactory neurons [115]. Humans breathe mostly by nose, with increasing mouth air intake during exertion. In contrast, rodents are ‘obligate nose breathers’ [116]. For humans and rodents, some of the nasal PM hits olfactory nerve endings, which potentially transport them directly into the forebrain. In mice, we showed inflammatory responses of the olfactory neuroepithelium and olfactory bulb within 24 hours [117].
‘Nose-to-brain’ passage of inhaled ultrafine PM was shown in classic studies with radio-tracers by Oberdorster, Elder, and colleagues. Their initial study exposed rats to aerosolized nano-scale [13C]-graphite PM (37nm ± 1.7 dia.) [118]. In several brain regions, the [13C] peaked at day 1, with modest if any decline by day 7, whereas lung [13C] sharply declined after day 1. Notably, levels of [13C] in olfactory bulb (OB) approximated that of cerebellum, which is several synapses from the olfactory input. Similarly, manganese oxide PM of 30 nm yielded the same levels in the frontal cortex and cerebellum after 6 and 12 days [119]. This equivalence suggests a systemic input of [13C] throughout the brain.
The importance of PM size for neuronal transport was shown by larger Mn of 1.9 μm, which restricted Mn elevations to the OB, without elevation in cerebellum [120]. A possible anatomical basis for size exclusion is the small diameter of olfactory neurons, which for smaller mammals, are among the smallest diameter brain neurons. In the most detailed analysis, guinea pig olfactory neurons had 0.28μm mean axon diameter with range 0.2-0.45 pm [121]. We have not found diameter measurements for human olfactory neurons. Calduron-Garciduenas et al. [122] described “abundant particulate material in the cytoplasm” of OB neurons from a Mexico City teenager, but did not give PM size estimates.
Moving into the respiratory tract, the PM contacts the epithelial cells lining our airways, nose and throat to lung. The ‘mucocilary escalator’ conveys particles, particularly the coarse PM, into the throat where they are swallowed. The portion of inhaled PM that reaches the gut is not well-defined. Gut delivery of PM is also used in some studies [123][124]. Fine and ultrafine PM from CS reach deep lung alveoli with high efficiency, 50-90% [116][104]. Deeper in the lung at alveolar surfaces, particles are phagocytosed by macrophages, with increasing efficiency <0.2 μm [125]. Ambient PM inhalability measured by radioactive tracers was similar for rodents and humans up to 5 μm [126]. For artificial aerosols of 2.5 μm, children had less efficient nasal filtering than adults, suggesting greater alveolar deposition [127]. This age-difference is relevant to interactions of household smoking with freeway exposure of that increase obesity risk of children [10].
Particle translocation from lung to blood is very inefficient. In human and rodents, 1-25% of inhaled artificial ultrafinePM reaching the lung was detected in blood cells [125]. Nonetheless, the above tracer studies definitively show that some PM reaches the brain from the lung, as well as from the nose. The fetal brain is also adversely impacted by vehicular exhaust PM as shown by experimental studies from three labs [128][129].
For inhaled PM to act directly on the fetus requires passage across at least three barriers: I, maternal air-lung interface into the maternal circulation; II, phagocytosis by circulating macrophages and by the liver; III, the placenta. Placental transfer of artificial PM depends on particle size and chemistry [130].
Human placentas can transport nanoscalePM into the fetal side of circulation [131][132], which is mediated by endocytosis at placental villi [132]. This research was motivated by concerns about the largely unregulated nanoparticles in cosmetics, and as food additives, baby food to beer. We anticipate adverse synergies of cosmetic and food nanoPM additives with air pollution in brain development.
System-wide cell reactions suggest a ‘lung-to-brain” route [133]. Serum from ozone exposed rats increased production of TNFα and H2O2 when added to cultured microglia [133]. Because ozone is quenched immediately on contact with respiratory tract fluids, we anticipate that these ‘ozone effects’ represent chemically modified blood lipids or proteins, or from innate immune responses. The inhaled PM may cause oxidative damage to circulating blood cells, proteins, and lipids, as well as inducing innate immune responses.
The time course of inflammatory response to PM0.2 in a mouse model also suggest systemic factors. We did not see increased TNFα or other inflammatory responses in the cerebral cortex or cerebellum until after 3 weeks of exposure to PM0.2, when the increases were as large in the olfactory epithelium as in the remotely connected cerebellum [134]. Recall from above that the radioactively-labelled graphite also yielded same levels in cerebellum as forebrain [118]. Moreover, we found that Nrf2 induction and other genomic detoxification responses to chronic PM0.2 were as large in cerebellum as in lung or liver (Fig.2). Because the lung traps most of the inhaled particles, it is hard to see how the liver and brain could have equal responses, without considering systemic down-stream effects from the lung adducts formed in the lung, as postulated for ozone [133].
6. Recommendations for alternative approaches
The prediction of epidemiological effects from lab assays remains problematic, as illustrated in examples of weak correlations and divergent outcomes in Sections 2 and 3. Readers may share our consternation, and wonder if any assays are more meaningful. In principle, free radicals can be detected by spin-trapping in cells exposed to AAP [136] or CS [36] [40]. However, spin-trapping requires electron paramagnetic spectrometers that are not commonly available. High concentrations of spin probes that that do not interfere with metabolism can be used to assess by spin-trapping for semi-quantitative direct estimates of free radicals production by cells. Some of the more accurate bioassays for free radicals, e.g. by boronates, dihydroethidium (DHE) [46,137], require specialized expertise in redox biology. Peroxide-activated reporter genes can also be included in genomic responses, discussed below. For assay of airborne activity, a major caveat is that most radicals have extremely short lifespans and are unlikely to reach the cell surface.
Other accessible assays include eicosanoid metabolites that are end-products of enzymatic lipid peroxidation produced by cyclooxygenase or lipoxygenases. Conveniently, the TBARS assay evaluates production of thiobarbituric acid reacting substances from the oxidation of lipids, as well as polysaccharides, nucleic acids, and small sugars. Other lipid oxidation products (prostaglandins, leukotrienes, compounds derived from arachidonic acid) are best measured by mass spectrometry, or more conveniently in some immunoassays [138]. The non-enzymatic oxidation of polyunsaturated fatty acids can produce unique products, including the F2-isoprostanes, also measured by mass spectrometry [139].
4-hydroxy-2-nonenal (HNE) is another useful marker of oxidative damage to lipids by forming protein carbonyl adducts, as studied for exposure to ozone, AAP, and CS [140][134][141]. HNE adducts are readily measured by immunoassay, while protein carbonyls requires reaction with dinitrophenylhydrazine to form adducts that can be measured spectrophotometrically [142].
Recognizing the immense heterogeneity and variability by place and over time, new approaches are needed to understand how air pollution and cigarettes share so much convergent pathology (Table 1B and Appendix 1). We see two major goals in analyzing the human impact or airborne PM. First, better information is needed to formulate policy on air quality control. Second, we must better understand cell-type specific and systemic responses to various PM components. Some suggest that more comprehensive profiles of cell responses using the ever-expanding ‘omics’ to better define short-term and long-term consequences of exposure. This approach will certainly give a massive amount of new data, but seems unlikely to us to yield actionable hypotheses. The half century of efforts to identify ‘the leading carcinogens and toxicants in CS shows that the bioactivities of individual chemicals does not predict body-wide responses. We propose three systematic and mechanistic approaches to assay the epidemiological associations of AAP, CS, HAP and potential synergies:
I, Cell assays for signaling mechanisms should use the cell types that are the first to encounter inhaled PM. Airway and alveolar epithelial cells, and macrophages seem the best choices. Nasal epithelium includes olfactory neurons that can be cultured. Analysis could include an informed ‘omics’ approach that targets pro- and antiinflammatory responses, rather than a broad undefined search. The lung epithelial cell responses to dung smoke in Section 3 document the value of an expanded panel of inflammatory and redox gene expression responses. We suggest that cell inflammatory responses will prove more fruitful for comparing PM activities than some chemical assays. Synergies (Table 1A) urgently warrant study because of the global importance of household air pollution (Table 1A). For comparison with collected ambient PM, we suggest exposure of cells to PM of known surface composition that represents the prime suspects for toxicity, particularly PAH and transition metals. The composition of the defined particles should include assessment of its surface composition. Ambient PM should be chosen from diverse sites to represent a range of surface particle components. We recommend against reporting correlations that have no biologically relevant chemical basis, as discussed for non-reactive metals in oxidative assays.
II, The animal models should be expanded from the rodent base to include small primates, such as the marmoset which are closer to humans in patterns of aging [143]. New in vivo studies are needed to address the key gap of how ambient PM can impact brain and the embryo, despite the small traces of particles or chemical components that leave the lungs, skin or eyes. We ask, could preventing the transmission beyond the lung be a practical approach defense against air pollution for people trapped in highly polluted, poorly regulated environments.
III, Air quality regulations should also consider the demonstrated pro-inflammatory effects of the PM surface components, rather than only PM mass or bulk composition. We advise against reporting assay associations with elements that have no chemical reactivity. The irreducible conclusion remains: we must globally reduce exposure to airborne toxicants.
Highlights.
-
■
Multiple pollution sources show synergies in disease risk, but assays are lacking
-
■
Assays for air pollution toxicants may misinterpret oxidant activity
-
■
Chemical and cell-based assays often show poor correspondence
-
■
New strategies are needed for cell responses to complex toxicants
Acknowledgments
The authors were supported by grant ES023864 from the National Institute of Environmental Health to HJF and AG040753 from the National Institutes of Health. National Institute on Aging to CEF.
We appreciate critical reading by Frank Gilliland, Todd E Morgan, and Costas Sioutas.
Appendix 1
Hazardous associations of Ambient Air Pollution (AAP) & Cigarette Smoke (CS)
| A. Human | |||
|---|---|---|---|
|
| |||
| AAP | CS | AAP-CS interactions; also see Table 1B | |
|
| |||
| Atherosclerosis | |||
| carotid | [144][145] | [148][149] | |
| coronary | [146][147] | [150][151] | [8] |
|
| |||
| Cancer | |||
| lung | [152][153] | [154][155] | [17][9] |
|
| |||
| Metabolism | |||
| insulin sensitivity, adult | [23][106] | weak or no association | |
| BMI, children | [10] | [105] [10] | |
|
| |||
| Neurodegeneration of normal aging | |||
| grey matter atrophy myelin atrophy | [156][157] | [159] [160] |
|
| cognitive decline | [12][158] | [12] | |
|
| |||
| Alzheimer’s disease | [154][161][162] | [163][164][165] | |
|
| |||
| Stroke | [166][167] | [168] | |
Appendix 2
The proposed reaction scheme for the oxidation of DTT through catalysis by quinones [32].
where Q is a quinone, R-SH is a thiol and Me2SO is dimethylsulfoxide. Superoxide then rapidly dismutes to hydrogen peroxide and oxygen:
As the authors showed, the series of reactions actually requires proximal thiols to proceed. Indeed, reaction 2 would rapidly occur within a single DTT molecule, R(SH)2, far more rapidly than between two separate thiols. More importantly, reaction 3 would be very slow compared with the reaction of RSS• with O2 [183]:
Thus, the authors own evidence suggests that a decrease in DTT by a quinone does not occur through this mechanism.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Finch CE. The Role of Global Air Pollution in Aging and Disease: Reading Smoke Signals. Elsevier; 2018. [Google Scholar]
- 2.Sternberg SPK. Air pollution : engineering, science, and policy. College Publishing; 2015. [Google Scholar]
- 3.Stone V, Miller MR, Clift MJD, Elder A, Mills NL, Møller P, Schins RPF, Vogel U, Kreyling WG, Jensen KA, Kuhlbusch TAJ, Schwarze PE, Hoet P, Pietroiusti A, de Vizcaya-Ruiz A, Baeza-Squiban A, Teixeira JP, Tran CL, Cassee FR. Nanomaterials versus ambient ultrafine particles: An opportunity to exchange toxicology knowledge. Environ Health Perspect. 2017;125 doi: 10.1289/EHP424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Staimer N, Gillen DL, Tjoa T, Schauer JJ, Shafer MM, Hasheminassab S, Pakbin P, Vaziri ND, Sioutas C, Delfino RJ. Associations of oxidative stress and inflammatory biomarkers with chemically-characterized air pollutant exposures in an elderly cohort. Environ Res. 2016;150:306–319. doi: 10.1016/j.envres.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, Balakrishnan K, Brunekreef B, Dandona L, Dandona R, Feigin V, Freedman G, Hubbell B, Jobling A, Kan H, Knibbs L, Liu Y, Martin R, Morawska L, Pope CA, Shin H, Straif K, Shaddick G, Thomas M, van Dingenen R, van Donkelaar A, Vos T, Murray CJL, Forouzanfar MH. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389:1907–1918. doi: 10.1016/S0140-6736(17)30505-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO Household air pollution, WHO. 2017 [Google Scholar]
- 7.Reitsma MB, Fullman N, Ng M, Salama JS, Abajobir A, Abate KH, Abbafati C, Abera SF, Abraham B, Abyu GY, Adebiyi AO, Al-Aly Z, Aleman AV, Ali R, Al Alkerwi A, Allebeck P, Al-Raddadi RM, Amare AT, Amberbir A, Ammar W, Amrock SM, Antonio CAT, Asayesh H, Atnafu NT, Azzopardi P, Banerjee A, Barac A, Barrientos-Gutierrez T, Basto-Abreu AC, Bazargan-Hejazi S, Bedi N, Bell B, Bello AK, Bensenor IM, Beyene AS, Bhala N, Biryukov S, Bolt K, Brenner H, Butt Z, Cavalleri F, Cercy K, Chen H, Christopher DJ, Ciobanu LG, Colistro V, Colomar M, Cornaby L, Dai X, Damtew SA, Dandona L, Dandona R, Dansereau E, Davletov K, Dayama A, Degfie TT, Deribew A, Dharmaratne SD, Dimtsu BD, Doyle KE, Endries AY, Ermakov SP, Estep K, Faraon EJA, Farzadfar F, Feigin VL, Feigl AB, Fischer F, Friedman J, G/hiwot TT, Gall SL, Gao W, Gillum RF, Gold AL, Gopalani SV, Gotay CC, Gupta R, Gupta R, Gupta V, Hamadeh RR, Hankey G, Harb HL, Hay SI, Horino M, Horita N, Hosgood HD, Husseini A, Ileanu BV, Islami F, Jiang G, Jiang Y, Jonas JB, Kabir Z, Kamal R, Kasaeian A, Kesavachandran CN, Khader YS, Khalil I, Khang YH, Khera S, Khubchandani J, Kim D, Kim YJ, Kimokoti RW, Kinfu Y, Knibbs LD, Kokubo Y, Kolte D, Kopec J, Kosen S, Kotsakis GA, Koul PA, Koyanagi A, Krohn KJ, Krueger H, Defo BK, Bicer BK, Kulkarni C, Kumar GA, Leasher JL, Lee A, Leinsalu M, Li T, Linn S, Liu P, Liu S, Lo LT, Lopez AD, Ma S, El Razek HMA, Majeed A, Malekzadeh R, Malta DC, Manamo WA, Martinez-Raga J, Mekonnen AB, Mendoza W, Miller TR, Mohammad KA, Morawska L, Musa KI, Nagel G, Neupane SP, Nguyen Q, Nguyen G, Oh IH, Oyekale AS, PA M, Pana A, Park EK, Patil ST, Patton GC, Pedro J, Qorbani M, Rafay A, Rahman M, Rai RK, Ram U, Ranabhat CL, Refaat AH, Reinig N, Roba HS, Rodriguez A, Roman Y, Roth G, Roy A, Sagar R, Salomon J, Sanabria J, de Souza Santos I, Sartorius B, Satpathy M, Sawhney M, Sawyer S, Saylan M, Schaub MP, Schluger N, Schutte AE, Sepanlou SG, Serdar B, Shaikh MA, She J, Shin MJ, Shiri R, Shishani K, Shiue I, Sigfusdottir ID, Silverberg JI, Singh J, Singh V, Slepak EL, Soneji S, Soriano JB, Soshnikov S, Sreeramareddy CT, Stein DJ, Stranges S, Subart ML, Swaminathan S, Szoeke CEI, Tefera WM, Topor-Madry R, Tran B, Tsilimparis N, Tymeson H, Ukwaja KN, Updike R, Uthman OA, Violante FS, Vladimirov SK, Vlassov V, Vollset SE, Vos T, Weiderpass E, Wen CP, Werdecker A, Wilson S, Wubshet M, Xiao L, Yakob B, Yano Y, Ye P, Yonemoto N, Yoon SJ, Younis MZ, Yu C, Zaidi Z, El Sayed Zaki M, Zhang AL, Zipkin B, Murray CJL, Forouzanfar MH, Gakidou E. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990-2015: a systematic analysis from the Global Burden of Disease Study 2015. Lancet. 2017;389:1885–1906. doi: 10.1016/S0140-6736(17)30819-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner MC, Cohen A, Burnett RT, Jerrett M, Diver WR, Gapstur SM, Krewski D, Samet JM, Pope CA., 3rd Interactions between cigarette smoking and ambient PM2.5 for cardiovascular mortality. Env Res. 2017;154:304–310. doi: 10.1016/j.envres.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 9.Turner MC, Cohen A, Jerrett M, Gapstur SM, Diver WR, Pope CA, Krewski D, Beckerman BS, Samet JM. Interactions between cigarette smoking and fine particulate matter in the risk of lung cancer mortality in cancer prevention study II. Am J Epidemiol. 2014;180:1145–1149. doi: 10.1093/aje/kwu275. [DOI] [PubMed] [Google Scholar]
- 10.McConnell R, Shen E, Gilliland FD, Jerrett M, Wolch J, Chih-Chieh C, Lurmann F, Berhane K, Chang CC, Lurmann F, Berhane K, Chih-Chieh C, Lurmann F, Berhane K. Research: Children’s Health. A Longitudinal Cohort Study of Body Mass Index and Childhood Exposure to Secondhand Tobacco Smoke and Air Pollution: The Southern California Children’s Health Study. Environ Health Perspect. 2015;123:360–366. doi: 10.1289/ehp.1307031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riedel C, Schönberger K, Yang S, Koshy G, Chen YC, Gopinath B, Ziebarth S, von Kries R. Parental smoking and childhood obesity: Higher effect estimates for maternal smoking in pregnancy compared with paternal smoking-a meta-analysis. Int J Epidemiol. 2014;43:1593–1606. doi: 10.1093/ije/dyu150. [DOI] [PubMed] [Google Scholar]
- 12.Ailshire JA, Crimmins EM. Fine particulate matter air pollution and cognitive function among older US adults. Am J Epidemiol. 2014;180:359–366. doi: 10.1093/aje/kwu155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Q, Wang Y, Zanobetti A, Wang Y, Koutrakis P, Choirat C, Dominici F, Schwartz JD. Air Pollution and Mortality in the Medicare Population. N Engl J Med. 2017;376:2513–2522. doi: 10.1056/NEJMoa1702747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crouse DL, Peters PA, van Donkelaar A, Goldberg MS, Villeneuve PJ, Brion O, Khan S, Atari DO, Jerrett M, Pope CA, Brauer M, Brook JR, Martin RV, Stieb D, Burnett RT, Van Donkelaar A, Goldberg MS, Villeneuve PJ, Brion O, Khan S, Atari DO, Jerrett M, Pope CA, III, Brauer M, Brook JR, lii CAP, Brauer M, Brook JR, Pope CA, van Donkelaar A, Goldberg MS, Villeneuve PJ, Brion O, Khan S, Atari DO, Jerrett M, Pope CA, Brauer M, Brook JR, Martin RV, Stieb D, Burnett RT. Risk of non accidental and cardiovascular mortality in relation to long-term exposure to low concentrations of fine particulate matter: a Canadian national-level cohort study. Environ Health Perspect. 2012;708:708–714. doi: 10.1289/ehp.1104049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue-Choi M, Liao LM, Reyes-Guzman C, Hartge P, Caporaso N, Freedman ND, Jiang N, Jamal A, Wortley P, Schane R, KM H, P F, CA H, R D, WD F, JH L, JH L, SM A, L G, J B, A R, R L, E P, K B, I K, SH Z, TR H, RE S, A S, W W, RS C, S S, SM C, J B, EA K. Association of Long-term, Low-Intensity Smoking With All-Cause and Cause-Specific Mortality in the National Institutes of Heath-AARP Diet and Health Study. JAMA Intern Med. 2016;10:e0137023. doi: 10.1001/jamainternmed.2016.7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pope CA, Burnett RT, Turner MC, Cohen AJ, Krewski D, Jerrett M, Gapstur SM, Thun MJ. Lung cancer and cardiovascular disease mortality associated with ambient air pollution and cigarette smoke: Shape of the exposure-response relationships. Environ Health Perspect. 2011;119:1616–1621. doi: 10.1289/ehp.1103639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnett RT, Pope CA, 3rd, Ezzati M, Olives C, Lim SS, Mehta S, Shin HH, Singh G, Hubbell B, Brauer M, Anderson HR, Smith KR, Balmes JR, Bruce NG, Kan H, Laden F, Pruss-Ustun A, Turner MC, Gapstur SM, Diver WR, Cohen A. An integrated risk function for estimating the global burden of disease attributable to ambient fine particulate matter exposure. Env Heal Perspect. 2014;122:397–403. doi: 10.1289/ehp.1307049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goudie AS. Desert dust and human health disorders. Environ Int. 2014;63:101–113. doi: 10.1016/j.envint.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Lelieveld J, Evans JS, Fnais M, Giannadaki D, Pozzer A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature. 2015;525:367–71. doi: 10.1038/nature15371. [DOI] [PubMed] [Google Scholar]
- 20.Stafoggia M, Zauli-Sajani S, Pey J, Samoli E, Alessandrini E, Basagana X, Cernigliaro A, Chiusolo M, Demaria M, Díaz J, Faustini A, Katsouyanni K, Kelessis AG, Linares C, Marchesi S, Medina S, Pandolfi P, Pérez N, Querol X, Randi G, Ranzi A, Tobias A, Forastiere F, Alessandrini E, Angelini P, Berti G, Bisanti L, Cadum E, Catrambone M, Chiusolo M, Davoli M, de Donato F, Demaria M, Gandini M, Grosa M, Faustini A, Ferrari S, Forastiere F, Pandolfi P, Pelosini R, Perrino C, Pietrodangelo A, Pizzi L, Poluzzi V, Priod G, Randi G, Ranzi A, Rowinski M, Scarinzi C, Stafoggia M, Stivanello E, Zauli-Sajani S, Dimakopoulou K, Elefteriadis K, Katsouyanni K, Kelessis A, Maggos T, Michalopoulos N, Pateraki S, Petrakakis M, Rodopoulou S, Samoli E, Sypsa V, Agis D, Alguacil J, Artiñano B, Barrera-Gómez J, Basagaña X, de la Rosa J, Diaz J, Fernandez R, Jacquemin B, Karanasiou A, Linares C, Ostro B, Perez N, Pey J, Querol X, Salvador P, Sanchez AM, Sunyer J, Bidondo M, Declercq C, Le Tertre A, Lozano P, Medina S, Pascal L, Pascal M. Desert dust outbreaks in Southern Europe: Contribution to daily PM10concentrations and short-term associations with mortality and hospital admissions. Environ Health Perspect. 2016;124:413–419. doi: 10.1289/ehp.1409164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin W, Su S, Wang B, Zhu X, Chen Y, Shen G, Liu J, Cheng H, Wang X, Wu S, Zeng E, Xing B, Tao S. Properties and cellular effects of particulate matter from direct emissions and ambient sources. J Environ Sci Heal - Part A Toxic/Hazardous Subst Environ Eng. 2016:1–9. doi: 10.1080/10934529.2016.1198632. [DOI] [PubMed] [Google Scholar]
- 22.Fariss MW, Gilmour MI, Reilly CA, Liedtke W, Ghio AJ. Emerging mechanistic targets in lung injury induced by combustion-generated particles. Toxicol Sci. 2013;132:253–267. doi: 10.1093/toxsci/kft001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolf K, Popp A, Schneider A, Breitner S, Hampel R, Rathmann W, Herder C, Roden M, Koenig W, Meisinger C, Peters A. KORA-Study Group, Association Between Long-term Exposure to Air Pollution and Biomarkers Related to Insulin Resistance, Subclinical Inflammation, and Adipokines. Diabetes. 2016;65:3314–3326. doi: 10.2337/db15-1567. [DOI] [PubMed] [Google Scholar]
- 24.Johnson TJ, Olfert JS, Cabot R, Treacy C, Yurteri CU, Dickens C, McAughey J, Symonds JPR. Steady-state measurement of the effective particle density of cigarette smoke. J Aerosol Sci. 2014;75:9–16. [Google Scholar]
- 25.Hu S, Polidori A, Arhami M, Shafer MM, Schauer JJ, Cho A, Sioutas C. Redox activity and chemical speciation of size fractioned PM in the communities of the Los Angeles – Long Beach Harbor. Atmos Chem Phys Discuss. 2008;8:11643–11672. [Google Scholar]
- 26.Shirmohammadi F, Hasheminassab S, Wang D, Saffari A, Schauer JJ, Shafer MM, Delfino RJ, Sioutas C. Oxidative potential of coarse particulate matter (PM10-2.5) and its relation to water solubility and sources of trace elements and metals in the Los Angeles Basin. Environ Sci Process Impacts. 2015;17:2110–2121. doi: 10.1039/c5em00364d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirmohammadi F, Hasheminassab S, Wang D, Schauer JJ, Shafer MM, Delfino RJ, Sioutas C. The relative importance of tailpipe and non-tailpipe emissions on the oxidative potential of ambient particles in Los Angeles, CA. Faraday Discuss. 2016;189:361–380. doi: 10.1039/c5fd00166h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawanaka Y, Tsuchiya Y, Yun SJ, Sakamoto K. Size distributions of polycyclic aromatic hydrocarbons in the atmosphere and estimation of the contribution of ultrafine particles to their lung deposition. Environ Sci Technol. 2009;43:6851–6856. doi: 10.1021/es900033u. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Li X, Guo J, Peng B, Cui H, Liu K, Wang S, Qin Y, Sun P, Zhao L, Xie F, Liu H. Distribution of toxic chemicals in particles of various sizes from mainstream cigarette smoke. Inhal Toxicol. 2016;28 doi: 10.3109/08958378.2016.1140851. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Kong H, Zhang X, Peng B, Nie C, Shen G, Liu H. Characterization of particle size distribution of mainstream cigarette smoke generated by smoking machine with an electrical low pressure impactor. J Environ Sci (China) 2014;26:827–833. doi: 10.1016/S1001-0742(13)60472-6. [DOI] [PubMed] [Google Scholar]
- 31.Künzli N, Mudway IS, Götschi T, Shi T, Kelly FJ, Cook S, Burney P, Forsberg B, Gauderman JW, Hazenkamp ME, Heinrich J, Jarvis D, Norbäck D, Payo-Losa F, Poli A, Sunyer J, Borm PJA. Comparison of oxidative properties, light absorbance, and total and elemental mass concentration of ambient PM2.5 collected at 20 European sites. Environ Health Perspect. 2006;114:684–690. doi: 10.1289/ehp.8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumagai Y, Koide S, Taguchi K, Endo A, Nakai Y, Yoshikawa T, Shimojo N. Oxidation of proximal protein sulfhydryls by phenanthraquinone, a component of diesel exhaust particles. Chem Res Toxicol. 2002;15:483–489. doi: 10.1021/tx0100993. [DOI] [PubMed] [Google Scholar]
- 33.Xia T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C, Nel A. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity. Environ Health Perspect. 2004;112:1347–1358. doi: 10.1289/ehp.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med. 1995;19:161–167. doi: 10.1016/0891-5849(94)00236-d. [DOI] [PubMed] [Google Scholar]
- 35.Pryor WA. Biological effects of cigarette smoke, wood smoke, and the smoke from plastics: the use of electron spin resonance. Free Radic Biol Med. 1992;13:659–676. doi: 10.1016/0891-5849(92)90040-n. [DOI] [PubMed] [Google Scholar]
- 36.Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Public Health. 2009;6:445–62. doi: 10.3390/ijerph6020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snell JM, Weissberger A. The Reaction of Thiol Compounds with Quinones. J Am Chem Soc. 1939;61:450–453. [Google Scholar]
- 38.Crobeddu B, Aragao-Santiago L, Bui LC, Boland S, Baeza Squiban A. Oxidative potential of particulate matter 2.5 as predictive indicator of cellular stress. Environ Pollut. 2017;230:125–133. doi: 10.1016/j.envpol.2017.06.051. [DOI] [PubMed] [Google Scholar]
- 39.Saffari A, Daher N, Shafer MM, Schauer JJ, Sioutas C. Seasonal and spatial variation of trace elements and metals in quasi-ultrafine (PM0.25) particles in the Los Angeles metropolitan area and characterization of their sources. Environ Pollut. 2013;181:14–23. doi: 10.1016/j.envpol.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Janssen NAH, Yang A, Strak M, Steenhof M, Hellack B, Gerlofs-Nijland ME, Kuhlbusch T, Kelly F, Harrison R, Brunekreef B, Hoek G, Cassee F. Oxidative potential of particulate matter collected at sites with different source characteristics. Sci Total Environ. 2014;472:572–581. doi: 10.1016/j.scitotenv.2013.11.099. [DOI] [PubMed] [Google Scholar]
- 41.Moreno T, Querol X, Martins V, Minguillón MC, Reche C, Ku LH, Eun HR, Ahn KH, Capdevila M, de Miguel E. Formation and alteration of airborne particles in the subway environment. Environ Sci Process Impacts. 2017;19:59–64. doi: 10.1039/c6em00576d. [DOI] [PubMed] [Google Scholar]
- 42.Wills ED. Lipid peroxide formation in microsomes. General considerations. Biochem J. 1969;113:315–24. doi: 10.1042/bj1130315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keston AS, Brandt R. The fluorometric analysis of ultramicro quantities of hydrogen peroxide. Anal Biochem. 1965;11:1–5. doi: 10.1016/0003-2697(65)90034-5. [DOI] [PubMed] [Google Scholar]
- 44.Rota C, Chignell CF, Mason RP. Evidence for free radical formation during the oxidation of 2′-7′-dichlorofluorescin to the fluorescent dye 2′-7′-dichlorofluorescein by horseradish peroxidase: Possible implications for oxidative stress measurements. Free Radic Biol Med. 1999;27:873–881. doi: 10.1016/s0891-5849(99)00137-9. [DOI] [PubMed] [Google Scholar]
- 45.Balcerczyk A, Kruszewski M, Bartosz G. Does the cellular labile iron pool participate in the oxidation of 2′, 7′-dichlorodihydroflourescein? Free Radic Res. 2007;41:563–570. doi: 10.1080/10715760601175353. [DOI] [PubMed] [Google Scholar]
- 46.Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LeBel CP, Tschiropoulos H, Bondy SC. Evaluation of the Probe 2′,7′-Dlchlorofluorescin as an Indicator of Reactive Oxygen Species Formation and Oxidative Stress Carl. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- 48.Bonini MG, Rota C, Tomasi A, Mason RP. The oxidation of 2′,7′-dichlorofluorescin to reactive oxygen species: a self-fulfilling prophesy? Free Radic Biol Med. 2006;40:968–975. doi: 10.1016/j.freeradbiomed.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 49.Wang HS, Xiao FN, Li ZQ, Ouyang J, Wu ZQ, Xia XH, Zhou GJ. Sensitive determination of reactive oxygen species in cigarette smoke using microchip electrophoresis-localized surface plasmon resonance enhanced fluorescence detection. Lab Chip. 2014;14:1123–8. doi: 10.1039/c3lc51220g. [DOI] [PubMed] [Google Scholar]
- 50.Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc. 2008;3:8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- 51.Babior BM, Kipnes RS, Curnutte JT. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryer-Powder JE, Forman HJ. Adhering lung macrophages produce superoxide demonstrated with desferal-Mn(IV) Free Radic Biol Med. 1989;6:513–518. doi: 10.1016/0891-5849(89)90044-0. [DOI] [PubMed] [Google Scholar]
- 53.Green PS, Perez EJ, Calloway T, Simpkins JW. Estradiol attenuation of beta-amyloid-induced toxicity: a comparison o. J Neurocytol. 2000;29:419–23. doi: 10.1023/a:1007173509470. [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Sun P, Bao Y, Liu J, An L. Cytotoxicity of single-walled carbon nanotubes on PC12 cells. Toxicol Vitr. 2011;25:242–250. doi: 10.1016/j.tiv.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Giordano G, Hong S, Faustman E, Costa L. Measurements of cell death in neuronal and glial cells. 2011 doi: 10.1007/978-1-61779-170-3_11. [DOI] [PubMed] [Google Scholar]
- 56.Visentin M, Pagnoni A, Sarti E, Pietrogrande MC. Urban PM2.5 oxidative potential: Importance of chemical species and comparison of two spectrophotometric cell-free assays. Environ Pollut. 2016;219:72–79. doi: 10.1016/j.envpol.2016.09.047. [DOI] [PubMed] [Google Scholar]
- 57.Morgan TE, Davis DA, Iwata N, Tanner JA, Snyder D, Ning Z, Kam W, Hsu YT, Winkler JW, Chen JC, Petasis NA, Baudry M, Sioutas C, Finch CE. Glutamatergic neurons in rodent models respond to nanoscale particulate urban air pollutants in vivo and in vitro. Environ Health Perspect. 2011;119:1003–1009. doi: 10.1289/ehp.1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Behndig AF, Blomberg A, Helleday R, Duggan ST, Kelly FJ, Mudway IS. Antioxidant responses to acute ozone challenge in the healthy human airway. Inhal Toxicol. 2009;21:933–42. doi: 10.1080/08958370802603789. [DOI] [PubMed] [Google Scholar]
- 59.Pryor WA. Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu Rev Physiol. 1986;48:657–667. doi: 10.1146/annurev.ph.48.030186.003301. [DOI] [PubMed] [Google Scholar]
- 60.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction., Free Radic. Biol Med. 2007;42:926–32. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aust AE, Ball JC, Hu AA, Lighty JS, Smith KR, Straccia AM, Veranth JM, Young WC. Particle characteristics responsible for effects on human lung epithelial cells. Res Rep Heal Eff Inst. 2002:1-65–76. [PubMed] [Google Scholar]
- 62.Premasekharan G, Nguyen K, Contreras J, Ramon V, Leppert VJ, Forman HJ. Iron-mediated lipid peroxidation and lipid raft disruption in low-dose silica-induced macrophage cytokine production. Free Radic Biol Med. 2011;51:1184–94. doi: 10.1016/j.freeradbiomed.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 63.Thomson EM, Breznan D, Karthikeyan S, MacKinnon-Roy C, Vuong NQ, Dabek-Zlotorzynska E, Celo V, Charland J-P, Kumarathasan P, Brook JR, Vincent R, Dockery D, Pope C, Xu X, Spengler J, Ware J, Fay M, Ferris B, Speizer F, Pope C, Burnett R, Thurston G, Thun M, Calle E, Krewski D, Godleski J, Burnett R, Brook J, Dann T, Delocla C, Philips O, Cakmak S, Vincent R, Goldberg M, Krewski D, Crouse D, Peters P, van D, Goldberg M, Villeneuve P, Brion O, Khan S, Atari D, Jerrett M, Pope C, Grahame T, Schlesinger R, Zanobetti A, Franklin M, Koutrakis P, Schwartz J, Peng R, Dominici F, Pastor-Barriuso R, Zeger S, Samet J, Vincent R, Goegan P, Johnson G, Brook J, Kumarathasan P, Bouthillier L, Burnett R, Steenhof M, Gosens I, Strak M, Godri K, Hoek G, Cassee F, Mudway I, Kelly F, Harrison R, Lebret E, Dergham M, Lepers C, Verdin A, Billet S, Cazier F, Courcot D, Shirali P, Garcon G, Mirowsky J, Hickey C, Horton L, Blaustein M, Galdanes K, Peltier R, Chillrud S, Chen L, Ross J, Nadas A, Thomson E, Breznan D, Karthikeyan S, MacKinnon-Roy C, Charland J, Dabek-Zlotorzynska E, Celo V, Kumarathasan P, Brook J, Vincent R, Adamson I, Prieditis H, Hedgecock C, Vincent R, Dreher K, Jaskot R, Lehmann J, Richards J, McGee J, Ghio A, Costa D, Wallenborn J, Schladweiler M, Richards J, Kodavanti U, Schins R, Lightbody J, Borm P, Shi T, Donaldson K, Stone V, Imrich A, Ning Y, Kobzik L, Guastadisegni C, Kelly F, Cassee F, Gerlofs-Nijland M, Janssen N, Pozzi R, Brunekreef B, Sandstrom T, Mudway I, Pope C, Schwartz J, Ransom M, Frampton M, Ghio A, Samet J, Carson J, Carter J, Devlin R, Jerrett M, Buzzelli M, Burnett R, DeLuca P, Mattson M, Guidotti T, Pope C, Rodermund D, Gee M, Grahame T, Lippmann M, Ito K, Hwang J, Maciejczyk P, Chen L, Demokritou P, Kavouras I, Ferguson S, Koutrakis P, Thomson E, Williams A, Yauk C, Vincent R, Fang T, Guo H, Verma V, Peltier R, Weber R, Oakes M, Burke J, Norris G, Kovalcik K, Pancras J, Landis M, Jeong C-H, McGuire M, Herod D, Dann T, Dabek-Zlotorzynska E, Wang D, Ding L, Celo V, Mathieu D, Evans G, Celo V, Dabek-Zlotorzynska E, Becker S, Mundandhara S, Devlin R, Madden M, Schwarze P, Ovrevik J, Hetland R, Becher R, Cassee F, Lag M, Lovik M, Dybing E, Refsnes M, Monn C, Becker S, Oberdorster G, Oberdorster E, Oberdorster J, Chen L, Lippmann M, Wang B, Li K, Jin W, Lu Y, Zhang Y, Shen G, Wang R, Shen H, Li W, Huang Y, Gerlofs-Nijland M, Rummelhard M, Boere A, Leseman D, Duffin R, Schins R, Borm P, Sillanpaa M, Salonen R, Cassee F, Ghio A, Stonehuerner J, Dailey L, Carter J, Gliga A, Skoglund S, Wallinder I, Fadeel B, Karlsson H, Dye J, Lehmann J, McGee J, Winsett D, Ledbetter A, Everitt J, Ghio A, Costa D, O’Neal S, Zheng W, Zhao P, Zhong W, Ying X, Yuan Z, Fu J, Zhou Z, Kim Y, Fazlollahi F, Kennedy I, Yacobi N, Hamm-Alvarez S, Borok Z, Kim K, Crandall E, Riley M, Boesewetter D, Turner R, Kim A, Collier J, Hamilton A, Becker S, Dailey L, Soukup J, Grambow S, Devlin R, Huang Y, Hetland R, Cassee F, Lag M, Refsnes M, Dybing E, Schwarze P. Contrasting biological potency of particulate matter collected at sites impacted by distinct industrial sources. Part Fibre Toxicol. 2016;13:65. doi: 10.1186/s12989-016-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinberg ED. Tobacco smoke iron: an initiator/promoter of multiple diseases. Biometals. 2009;2:207–10. doi: 10.1007/s10534-008-9156-5. [DOI] [PubMed] [Google Scholar]
- 65.Ghio AJ, Stonehuerner J, Quigley DR. Humic-like substances in cigarette smoke condensate and lung tissue of smokers. Am J Physiol. 1994;266:L382–8. doi: 10.1152/ajplung.1994.266.4.L382. [DOI] [PubMed] [Google Scholar]
- 66.Ghio AJ, Stonehuerner J, Pritchard RJ, Piantadosi CA, Quigley DR, Dreher KL, Costa DL. Humic-Like Substances in Air Pollution Particulates Correlate with Concentrations of Transition Metals and Oxidant Generation. Inhal Toxicol. 1996;8:479–494. [Google Scholar]
- 67.Zheng G, He K, Duan F, Cheng Y, Ma Y. Measurement of humic-like substances in aerosols: A review. Environ Pollut. 2013;181:301–314. doi: 10.1016/j.envpol.2013.05.055. [DOI] [PubMed] [Google Scholar]
- 68.Chen Q, Ikemori F, Higo H, Asakawa D, Mochida M. Chemical Structural Characteristics of HULIS and Other Fractionated Organic Matter in Urban Aerosols: Results from Mass Spectral and FT-IR Analysis. Environ Sci Technol. 2016;50:1721–30. doi: 10.1021/acs.est.5b05277. [DOI] [PubMed] [Google Scholar]
- 69.Qiao T, Zhao M, Xiu G, Yu J. Simultaneous monitoring and compositions analysis of PM1 and PM2.5 in Shanghai: Implications for characterization of haze pollution and source apportionment. Sci Total Environ. 2016;557-558:386–394. doi: 10.1016/j.scitotenv.2016.03.095. [DOI] [PubMed] [Google Scholar]
- 70.Ghio AJ, Soukup JM, Dailey LA, Tong H, Kesic MJ, Budinger GRS, Mutlu GM. Wood Smoke Particle Sequesters Cell Iron to Impact a Biological Effect. Chem Res Toxicol. 2015;28:2104–2111. doi: 10.1021/acs.chemrestox.5b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Eijl S, Mortaz E, Ferreira AF, Kuper F, Nijkamp FP, Folkerts G, Bloksma N. Humic acid enhances cigarette smoke-induced lung emphysema in mice and IL-8 release of human monocytes. Pulm Pharmacol Ther. 2011;24:682–689. doi: 10.1016/j.pupt.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 72.McCarthy CE, Duffney PF, Gelein R, Thatcher TH, Elder A, Phipps RP, Sime PJ. Dung biomass smoke activates inflammatory signaling pathways in human small airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2016;311:L1222–L1233. doi: 10.1152/ajplung.00183.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCarthy CE, Duffney PF, Wyatt JD, Thatcher TH, Phipps RP, Sime PJ. Comparison of in vitro toxicological effects of biomass smoke from different sources of animal dung. Toxicol Vitr. 2017;43:76–86. doi: 10.1016/j.tiv.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koizumi Y, Nagase H, Nakajima T, Kawamura M, Ohta K. Toll-like receptor 3 ligand specifically induced bronchial epithelial cell death in caspase dependent manner and functionally upregulated Fas expression. Allergol Int. 2016;65:S30–S37. doi: 10.1016/j.alit.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 75.Kleinman MT, Araujo JA, Nel A, Sioutas C, Campbell A, Cong PQ, Li H, Bondy SC. Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol Lett. 2008;178:127–130. doi: 10.1016/j.toxlet.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol. 2005;207:269–275. doi: 10.1016/j.taap.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 77.Baeza-Squiban A, Bonvallot V, Boland S, Marano F. Airborne particles evoke an inflammatory response in human airway epithelium. Activation of transcription factors. Cell Biol Toxicol. 1999;15:375–380. doi: 10.1023/a:1007653900063. [DOI] [PubMed] [Google Scholar]
- 78.Carter JD, Ghio AJ, Samet JM, Devlin RB. Cytokine production by human airway epithelial cells after exposure to an air pollution particle is metal-dependent. Toxicol Appl Pharmacol. 1997;146:180–188. doi: 10.1006/taap.1997.8254. [DOI] [PubMed] [Google Scholar]
- 79.Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB. Effects of TNF- α on Expression of ICAM-1 in Human Airway Epithelial Cells In Vitro. Am J Respir Cell Mol Biol. 2000;22:685–692. doi: 10.1165/ajrcmb.22.6.3925. [DOI] [PubMed] [Google Scholar]
- 80.Chen C, Chou C, Sun Y, Huang W. Tumor necrosis factor alpha-induced activation of downstream NF-kappa B site of the promoter mediates epithelial ICAM-1 expression and monocyte adhesion. Involvement of PKC alpha, tyrosine kinase, and IKK2, but not MAPKs, pathway. Cell Signal. 2001;13:543–53. doi: 10.1016/s0898-6568(01)00171-1. [DOI] [PubMed] [Google Scholar]
- 81.Garg R, Blando J, Perez CJ, Wang H, Benavides FJ, Kazanietz MG. Activation of nuclear factor κB (NF-κB) in prostate cancer is mediated by protein kinase C epsilon (PKCepsilon) J Biol Chem. 2012;287:37570–82. doi: 10.1074/jbc.M112.398925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin CH, Kuan IH, Lee HM, Lee WS, Sheu JR, Ho YS, Wang CH, Kuo HP. Induction of cyclooxygenase-2 protein by lipoteichoic acid from Staphylococcus aureus in human pulmonary epithelial cells: involvement of a nuclear factor-kappa B-dependent pathway. Br J Pharmacol. 2001;134:543–552. doi: 10.1038/sj.bjp.0704290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen CC, Chen JJ, Chou CY. Protein kinase c alpha but not p44/42 mitogen-activated protein kinase, p38, or c-Jun NH(2)-terminal kinase is required for intercellular adhesion molecule-1 expression mediated by interleukin-1 beta: involvement of sequential activation of tyrosine kinas. Mol Pharmacol. 2000;58:1479–1489. doi: 10.1124/mol.58.6.1479. [DOI] [PubMed] [Google Scholar]
- 84.Woodward N, Levine M, Haghani A, Shirmohammadi F, Saffari A, Sioutas C, Morgan TE, Finch CE. Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J Neuroinflamm. 2017:84. doi: 10.1186/s12974-017-0858-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mann GE, Forman HJ. Introduction to Special Issue on ’Nrf2 Regulated Redox Signaling and Metabolism in Physiology and Medicine. Free Radic Biol Med. 2015;88:91–92. doi: 10.1016/j.freeradbiomed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 86.Wang H, Liu X, Umino T, Skold CM, Zhu Y, Kohyama T, Spurzem JR, Romberger DJ, Rennard SI. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am J Respir Cell Mol Biol. 2001;25:772–779. doi: 10.1165/ajrcmb.25.6.4458. [DOI] [PubMed] [Google Scholar]
- 87.Müller T, Hengstermann A. Nrf2: Friend and Foe in Preventing Cigarette Smoking-Dependent Lung Disease. Chem Res Toxicol. 2012;25:1805–1824. doi: 10.1021/tx300145n. [DOI] [PubMed] [Google Scholar]
- 88.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma. Antioxid Redox Signal. 2006;8:88–98. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 90.Zhang H, Zhou L, Yuen J, Birkner N, Leppert V, O’Day PA, Forman HJ. Delayed Nrf2-regulated antioxidant gene induction in response to silica nanoparticles. Free Radic Biol Med. 2017;108:311–319. doi: 10.1016/j.freeradbiomed.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sekine T, Hirata T, Mine T, Fukano Y. Activation of transcription factors in human bronchial epithelial cells exposed to aqueous extracts of mainstream cigarette smoke in vitro. Toxicol Mech Methods. 2016;26:22–31. doi: 10.3109/15376516.2015.1123788. [DOI] [PubMed] [Google Scholar]
- 92.Schumacher FR, Schubert S, Hannus M, Sönnichsen B, Ittrich C, Kreideweiss S, Kurz T, Rippmann JF. RNAi Screen for NRF2 Inducers Identifies Targets That Rescue Primary Lung Epithelial Cells from Cigarette Smoke Induced Radical Stress. PLoS One. 2016;11:e0166352. doi: 10.1371/journal.pone.0166352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaul N, Forman HJ. Activation of NFκB by the respiratory burst of macrophages. Free Radic Biol Med. 1996;21:401–405. doi: 10.1016/0891-5849(96)00178-5. [DOI] [PubMed] [Google Scholar]
- 94.Ursini F, Maiorino M, Forman HJ. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016;8:205–215. doi: 10.1016/j.redox.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who’s listening? Antioxid Redox Signal. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iu M, Zago M, Rico de Souza A, Bouttier M, Pareek S, White JH, Hamid Q, Eidelman DH, Baglole CJ. RelB attenuates cigarette smoke extract-induced apoptosis in association with transcriptional regulation of the aryl hydrocarbon receptor. Free Radic Biol Med. 2017;108:19–31. doi: 10.1016/j.freeradbiomed.2017.02.045. [DOI] [PubMed] [Google Scholar]
- 97.Vogel CFA, Khan EM, Leung PSC, Gershwin ME, Chang WLW, Wu D, Haarmann-Stemmann T, Hoffmann A, Denison MS. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-kB. J Biol Chem. 2014;289:1866–1875. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sundar IK, Chung S, Hwang JW, Lapek JD, Bulger M, Friedman AE, Yao H, Davie JR, Rahman I. Mitogen- and stress-activated kinase 1 (MSK1) regulates cigarette smoke-induced histone modifications on NF-κB-dependent genes. PLoS One. 2012;7:e31378. doi: 10.1371/journal.pone.0031378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, Moreno-Macias H, Smith JA, Brody JA, Dhingra R, Yousefi P, Pankow JS, Kunze S, Shah SH, McRae AF, Lohman K, Sha J, Absher DM, Ferrucci L, Zhao W, Demerath EW, Bressler J, Grove ML, Huan T, Liu C, Mendelson MM, Yao C, Kiel DP, Peters A, Wang-Sattler R, Visscher PM, Wray NR, Starr JM, Ding J, Rodriguez CJ, Wareham NJ, Irvin MR, Zhi D, Barrdahl M, Vineis P, Ambatipudi S, Uitterlinden AG, Hofman A, Schwartz J, Colicino E, Hou L, Vokonas PS, Hernandez DG, Singleton AB, Bandinelli S, Turner ST, Ware EB, Smith AK, Klengel T, Binder EB, Psaty BM, Taylor KD, Gharib SA, Swenson BR, Liang L, Demeo DL, O’Connor GT, Herceg Z, Ressler KJ, Conneely KN, Sotoodehnia N, Kardia SLR, Melzer D, Baccarelli AA, Van Meurs JBJ, Romieu I, Arnett DK, Ong KK, Liu Y, Waldenberger M, Deary IJ, Fornage M, Levy D, London SJ. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pope CA, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA. 2002;287:1132–1141. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pope CA, Burnett RT, Krewski D, Jerrett M, Shi Y, Calle EE, Thun MJ. Cardiovascular mortality and exposure to airborne fine particulate matter and cigarette smoke: shape of the exposure-response relationship. Circulation. 2009;120:941–8. doi: 10.1161/CIRCULATIONAHA.109.857888. [DOI] [PubMed] [Google Scholar]
- 102.Krewski D, Jerrett M, Burnett RT, Ma R, Hughes E, Shi Y, Turner MC, Pope CA, 3rd, Thurston G, Calle EE, Thun MJ, Beckerman B, DeLuca P, Finkelstein N, Ito K, Moore DK, Newbold KB, Ramsay T, Ross Z, Shin H, Tempalski B. Extended follow-up and spatial analysis of the American Cancer Society study linking particulate air pollution and mortality. Res Rep Health Eff Inst. 2009:5-114–36. [PubMed] [Google Scholar]
- 103.Benowitz NL, Jacob P. Daily intake of nicotine during cigarette smoking. Clin Pharmacol Ther. 1984;35:499–504. doi: 10.1038/clpt.1984.67. [DOI] [PubMed] [Google Scholar]
- 104.Baker RR, Dixon M. The retention of tobacco smoke constituents in the human respiratory tract. Inhal Toxicol. 2006;18:255–94. doi: 10.1080/08958370500444163. [DOI] [PubMed] [Google Scholar]
- 105.Kim HW, Kam S, Lee DH. Synergistic interaction between polycyclic aromatic hydrocarbons and environmental tobacco smoke on the risk of obesity in children and adolescents: The U.S. National Health and Nutrition Examination Survey 2003-2008. Environ Res. 2014;135:354–360. doi: 10.1016/j.envres.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 106.Thiering E, Markevych I, Brüske I, Fuertes E, Kratzsch J, Sugiri D, Hoffmann B, Von Berg A, Bauer CP, Koletzko S, Berdel D, Heinrich J. Associations of residential long-term air pollution exposures and satellite-derived greenness with insulin resistance in German adolescents. Environ Health Perspect. 2016;124:1291–1298. doi: 10.1289/ehp.1509967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Holmes D. Endocrine disruptors: Air pollution linked to insulin resistance. Nat Rev Endocrinol. 2016;12:688–688. doi: 10.1038/nrendo.2016.168. [DOI] [PubMed] [Google Scholar]
- 108.Vuong NQ, Breznan D, Goegan P, O’Brien JS, Williams A, Karthikeyan S, Kumarathasan P, Vincent R. In vitro toxicoproteomic analysis of A549 human lung epithelial cells exposed to urban air particulate matter and its water-soluble and insoluble fractions. Part Fibre Toxicol. 2017;14:39. doi: 10.1186/s12989-017-0220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine. 2014;9:51–63. doi: 10.2147/IJN.S26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.De Jong WH, Borm PJA. Drug delivery and nanoparticles:applications and hazards. Int J Nanomedicine. 2008;3:133–149. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xia T, Kovochich M, Liong M, Mädler L, Gilbert B, Shi H, Yeh JI, Zink JI, Nel AE. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano. 2008;2:2121–2134. doi: 10.1021/nn800511k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fazlollahi F, Kim YH, Sipos A, Hamm-Alvarez SF, Borok Z, Kim KJ, Crandall ED. Nanoparticle translocation across mouse alveolar epithelial cell monolayers: Species-specific mechanisms. Nanomedicine Nanotechnology Biol Med. 2013;9:786–794. doi: 10.1016/j.nano.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hamilton RF, Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 2008;44:1246–1258. doi: 10.1016/j.freeradbiomed.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ajmani GS, Suh HS, Pinto JM. Effects of Ambient Air Pollution Exposure on Olfaction: A Review. Environ Health Perspect. 2016;124:1683–1693. doi: 10.1289/EHP136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miller F. Dosimetry of particles: Critical factors having risk assessment implications. Inhal Toxicol. 2000;12:389–395. doi: 10.1080/08958378.2000.11463250. [DOI] [PubMed] [Google Scholar]
- 117.Cheng H, Davis DA, Hasheminassab S, Sioutas C, Morgan TE, Finch CE. Urban traffic-derived nanoparticulate matter reduces neurite outgrowth via TNFα in vitro. J Neuroinflammation. 2016;13:19. doi: 10.1186/s12974-016-0480-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Kreyling W, Cox C, Oberdörster G, Sharp Z, Atudorei V, Elder A, Gelein R, Kreyling W, Cox C. Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol. 2004;16:437–445. doi: 10.1080/08958370490439597. [DOI] [PubMed] [Google Scholar]
- 119.Elder A, Gelein R, Silva V, Feikert T, Opanashuk L, Carter J, Potter R, Maynard A, Ito Y, Finkelstein J, Oberdörster G. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ Health Perspect. 2006;114:1172–1178. doi: 10.1289/ehp.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dorman DC, McManus BE, Parkinson CU, Manuel CA, McElveen AM, Everitt JI. Nasal Toxicity of Manganese Sulfate and Manganese Phosphate in Young Male Rats Following Subchronic (13-Week) Inhalation Exposure. Inhal Toxicol. 2004;16:481–488. doi: 10.1080/08958370490439687. [DOI] [PubMed] [Google Scholar]
- 121.Perge JA, Niven JE, Mugnaini E, Balasubramanian V, Sterling P. Why Do Axons Differ in Caliber? J Neurosci. 2012;32:626–638. doi: 10.1523/JNEUROSCI.4254-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Calderón-Garcidueñas L, Solt AC, Henríquez-Roldán C, Torres-Jardón R, Nuse B, Herritt L, Villarreal-Calderón R, Osnaya N, Stone I, Garcia R, Brooks DM, González-Maciel A, Reynoso-Robles R, Delgado-Chávez R, Reed W. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol. 2008;36:289–310. doi: 10.1177/0192623307313011. [DOI] [PubMed] [Google Scholar]
- 123.Ejaz S, Anwar K, Ashraf M. MRI and neuropathological validations of the involvement of air pollutants in cortical selective neuronal loss. Environ Sci Pollut Res. 2014;21:3351–3362. doi: 10.1007/s11356-013-2294-5. [DOI] [PubMed] [Google Scholar]
- 124.McCallister MM, Li Z, Zhang T, Ramesh A, Clark RS, Maguire M, Hutsell B, Newland MC, Hood DB. Revealing Behavioral Learning Deficit Phenotypes Subsequent to In Utero Exposure to Benzo(a)pyrene. Toxicol Sci. 2016;149:42–54. doi: 10.1093/toxsci/kfv212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Geiser M, Kreyling WG. Deposition and biokinetics of inhaled nanoparticles. Part Fibre Toxicol. 2010;7:2. doi: 10.1186/1743-8977-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ménache MG, Miller FJ, Raabe OG. Particle inhalability curves for humans and small laboratory animals. Ann Occup Hyg. 1995;39:317–28. [PubMed] [Google Scholar]
- 127.Bennett WD, Zeman KL, Jarabek AM. Nasal contribution to breathing and fine particle deposition in children versus adults. J Toxicol Environ Health A. 2008;71:227–37. doi: 10.1080/15287390701598200. [DOI] [PubMed] [Google Scholar]
- 128.Davis DA, Akopian G, Walsh JP, Sioutas C, Morgan TE, Finch CE. Urban air pollutants reduce synaptic function of CA1 neurons via an NMDA/ pathway in vitro. J Neurochem. 2013;127:509–19. doi: 10.1111/jnc.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bolton JL, Auten RL, Bilbo SD. Prenatal air pollution exposure induces sexually dimorphic fetal programming of metabolic and neuroinflammatory outcomes in adult offspring. Brain Behav Immun. 2014;37:30–44. doi: 10.1016/j.bbi.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 130.Muoth C, Aengenheister L, Kucki M, Wick P, Buerki-Thurnherr T. Nanoparticle transport across the placental barrier: pushing the field forward! Nanomedicine. 2016;11:941–957. doi: 10.2217/nnm-2015-0012. [DOI] [PubMed] [Google Scholar]
- 131.Myllynen P, Vähäkangas K. Placental transfer and metabolism: An overview of the experimental models utilizing human placental tissue. Toxicol Vitr. 2012;27:507–512. doi: 10.1016/j.tiv.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 132.Grafmueller S, Manser P, Diener L, Diener PA, Maeder-Althaus X, Maurizi L, Jochum W, Krug HF, Buerki-Thurnherr T, von Mandach U, Wick P. Bidirectional transfer study of polystyrene nanoparticles across the placental barrier in an ex vivo human placental perfusion model. Environ Health Perspect. 2015;123:1280–1286. doi: 10.1289/ehp.1409271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mumaw CL, Levesque S, McGraw C, Robertson S, Lucas S, Stafflinger JE, Campen MJ, Hall P, Norenberg JP, Anderson T, Lund AK, McDonald JD, Ottens AK, Block ML. Microglial priming through the lung-brain axis: The role of air pollution-induced circulating factors. FASEB J. 2016;30:1880–1891. doi: 10.1096/fj.201500047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheng H, Saffari A, Sioutas C, Forman HJHJ, Morgan TETE, Finch CECE. Nanoscale particulate matter from urban traffic rapidly induces oxidative stress and inflammation in olfactory epithelium with concomitant effects on brain. Environ Health Perspect. 2016;124:1537–1546. doi: 10.1289/EHP134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang H, Liu H, Davies KJAKJA, Sioutas C, Finch CECE, Morgan TETE, Forman HJHJ. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic Biol Med. 2012;52:2038–46. doi: 10.1016/j.freeradbiomed.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kadiiska MB, Mason RP, Dreher KL, Costa DL, Ghio AJ. In vivo evidence of free radical formation in the rat lung after exposure to an emission source air pollution particle. Chem Res Toxicol. 1997;10:1104–1108. doi: 10.1021/tx970049r. [DOI] [PubMed] [Google Scholar]
- 137.Forman HJ, Augusto O, Brigelius-Flohe R, Dennery PA, Kalyanaraman B, Ischiropoulos H, Mann GE, Radi R, Roberts LJ, Vina J, Davies KJA. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic Biol Med. 2015;78:233–5. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 138.Tsikas D, Zoerner AA. Analysis of eicosanoids by LC-MS/MS and GC-MS/MS: A historical retrospect and a discussion. J Chromatogr B Anal Technol Biomed Life Sci. 2014;964:79–88. doi: 10.1016/j.jchromb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 139.Milne GL, Musiek ES, Morrow JD. F2-isoprostanes as markers of oxidative stress in vivo: an overview. Biomarkers. 2005;10(Suppl 1):S10–23. doi: 10.1080/13547500500216546. [DOI] [PubMed] [Google Scholar]
- 140.Hamilton RF, Jr, Li L, Eschenbacher WL, Szweda L, Holian A. Potential involvement of 4-hydroxynonenal in the response of human lung cells to ozone. Am J Physiol. 1998;274:L8–16. doi: 10.1152/ajplung.1998.274.1.L8. [DOI] [PubMed] [Google Scholar]
- 141.Sticozzi C, Belmonte G, Pecorelli A, Arezzini B, Gardi C, Maioli E, Miracco C, Toscano M, Forman HJ, Valacchi G. Cigarette smoke affects keratinocytes SRB1 expression and localization via H 2O 2 production and HNE protein adducts formation. PLoS One. 2012;7:e33592. doi: 10.1371/journal.pone.0033592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- 143.Mattison JA, Vaughan KL. An overview of nonhuman primates in aging research. Exp Gerontol. 2017;94:41–45. doi: 10.1016/j.exger.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aguilera I, Dratva J, Caviezel S, Burdet L, de Groot E, Ducret-Stich RE, Eeftens M, Keidel D, Meier R, Perez L, Rothe T, Schaffner E, Schmit-Trucksäss A, Tsai M-YM-Y, Schindler C, Künzli N, Probst-Hensch N, Ducret-Stich RE, Eeftens M, Keidel D, Meier R, Perez L, Rothe T, Schaffner E, Schmit-Trucksäss A, Tsai M-YM-Y, Schindler C, Künzli N, Probst-Hensch N. Particulate matter and subclinical atherosclerosis: Associations between different particle sizes and sources with carotid intima-media thickness in the SAPALDIA study. Environ Health Perspect. 2016;124:1700–1706. doi: 10.1289/EHP161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu X, Lian H, Ruan Y, Liang R, Zhao X, Routledge M, Fan Z. Association of exposure to particular matter and carotid intima-media thickness: A systematic review and meta-analysis. Int J Environ Res Public Health. 2015;12 doi: 10.3390/ijerph121012924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hartiala J, Breton CV, Tang WHW, Lurmann F, Hazen SL, Gilliland FD, Allayee H. Ambient Air Pollution Is Associated With the Severity of Coronary Atherosclerosis and Incident Myocardial Infarction in Patients Undergoing Elective Cardiac Evaluation. J Am Heart Assoc. 2016;5:e003947. doi: 10.1161/JAHA.116.003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kaufman JD, Adar SD, Barr RG, Budoff M, Burke GL, Curl CL, Daviglus ML, Roux AVD, Gassett AJ, Jacobs DR, Kronmal R, Larson TV, Navas-Acien A, Olives C, Sampson PD, Sheppard L, Siscovick DS, Stein JH, Szpiro AA, Watson KE. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): a longitudinal cohort study. Lancet. 2016;388:696–704. doi: 10.1016/S0140-6736(16)00378-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hansen K, Östling G, Persson M, Nilsson PM, Melander O, Engström G, Hedblad B, Rosvall M. The effect of smoking on carotid intima-media thickness progression rate and rate of lumen diameter reduction. Eur J Intern Med. 2016;28:74–9. doi: 10.1016/j.ejim.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 149.Huang LC, Lin RT, Chen CF, Chen CH, Juo SHH, Lin HF. Predictors of Carotid Intima-Media Thickness and Plaque Progression in a Chinese Population. J Atheroscler Thromb. 2016;23:940–9. doi: 10.5551/jat.32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Benziger CP, Roth GA, Moran AE. The Global Burden of Disease Study and the Preventable Burden of NCD. Glob Heart. 2016;11:393–397. doi: 10.1016/j.gheart.2016.10.024. [DOI] [PubMed] [Google Scholar]
- 151.Nicoll R, Wiklund U, Zhao Y, Diederichsen A, Mickley H, Ovrehus K, Zamorano J, Gueret P, Schmermund A, Maffei E, Cademartiri F, Budoff M, Henein M. Gender and age effects on risk factor-based prediction of coronary artery calcium in symptomatic patients: A Euro-CCAD study. Atherosclerosis. 2016;252:32–9. doi: 10.1016/j.atherosclerosis.2016.07.906. [DOI] [PubMed] [Google Scholar]
- 152.Hamra GB, Guha N, Cohen A, Laden F, Raaschou-Nielsen O, Samet JM, Vineis P, Forastiere F, Saldiva P, Yorifuji T, Loomis D. Outdoor particulate matter exposure and lung cancer: A systematic review and meta-analysis. Environ Health Perspect. 2014;122:906–911. doi: 10.1289/ehp/1408092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cui P, Huang Y, Han J, Song F, Chen K. Ambient particulate matter and lung cancer incidence and mortality: A meta-analysis of prospective studies. Eur J Public Health. 2015;25:324–329. doi: 10.1093/eurpub/cku145. [DOI] [PubMed] [Google Scholar]
- 154.Doll R, Peto R, Boreham J, Sutherland I. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ. 2004;328:1519. doi: 10.1136/bmj.38142.554479.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen Z, Peto R, Zhou M, Iona A, Smith M, Yang L, Guo Y, Chen Y, Bian Z, Lancaster G, Sherliker P, Pang S, Wang H, Su H, Wu M, Wu X, Chen J, Collins R, Li L. China Kadoorie Biobank (CKB) collaborative group, Contrasting male and female trends in tobacco-attributed mortality in China: evidence from successive nationwide prospective cohort studies. Lancet (London, England) 2015;386:1447–56. doi: 10.1016/S0140-6736(15)00340-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen T, Jia G, Wei Y, Li J. Beijing ambient particle exposure accelerates atherosclerosis in ApoE knockout mice. Toxicol Lett. 2013;223:146–153. doi: 10.1016/j.toxlet.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 157.Casanova R, Wang X, Reyes J, Akita Y, Serre ML, Vizuete W, Chui HC, Driscoll I, Resnick SM, Espeland MA, Chen JC. A Voxel-Based Morphometry Study Reveals Local Brain Structural Alterations Associated with Ambient Fine Particles in Older Women. Front Hum Neurosci. 2016;10:495. doi: 10.3389/fnhum.2016.00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cacciottolo M, Wang X, Driscoll I, Woodward N, Saffari A, Reyes J, Serre ML, Vizuete W, Sioutas C, Morgan TE, Gatz M, Chui HC, Shumaker SA, Resnick SM, Espeland MA, Finch CE, Chen JC. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl Psychiatry. 2017;7:e1022. doi: 10.1038/tp.2016.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Karama S, Ducharme S, Corley J, Chouinard-Decorte F, Starr JM, Wardlaw JM, Bastin ME, Deary IJ. Cigarette smoking and thinning of the brain’s cortex. Mol Psychiatry. 2015;20:778–85. doi: 10.1038/mp.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Prom-Wormley E, Maes HHM, Schmitt JE, Panizzon MS, Xian H, Eyler LT, Franz CE, Lyons MJ, Tsuang MT, Dale AM, Fennema-Notestine C, Kremen WS, Neale MC. Genetic and Environmental Contributions to the Relationships Between Brain Structure and Average Lifetime Cigarette Use. Behav Genet. 2015;45:157–170. doi: 10.1007/s10519-014-9704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Oudin A, Forsberg B, Adolfsson AN, Lind N, Modig L, Nordin M, Nordin S, Adolfsson R, Nilsson LG. Traffic-related air pollution and dementia incidence in Northern Sweden: A longitudinal study. Environ Health Perspect. 2016;124:306–312. doi: 10.1289/ehp.1408322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jung CR, Lin YT, Hwang BF. Ozone, particulate matter, and newly diagnosed Alzheimer’s disease: a population-based cohort study in Taiwan. J Alzheimers Dis. 2015;44:573–84. doi: 10.3233/JAD-140855. [DOI] [PubMed] [Google Scholar]
- 163.Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10:819–828. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Durazzo TC, Mattsson N, Weiner MW. Alzheimer’s Disease Neuroimaging Initiative, Smoking and increased Alzheimer’s disease risk: a review of potential mechanisms. Alzheimers Dement. 2014;10:S122–45. doi: 10.1016/j.jalz.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Deochand C, Tong M, Agarwal AR, Cadenas E, de la Monte SM. Tobacco Smoke Exposure Impairs Brain Insulin/IGF Signaling: Potential Co-Factor Role in Neurodegeneration. J Alzheimers Dis. 2016;50:373–86. doi: 10.3233/JAD-150664. [DOI] [PubMed] [Google Scholar]
- 166.Scheers H, Jacobs L, Casas L, Nemery B, Nawrot TS. Long-Term Exposure to Particulate Matter Air Pollution Is a Risk Factor for Stroke: Meta-Analytical Evidence. Stroke. 2015;46:3058–3066. doi: 10.1161/STROKEAHA.115.009913. [DOI] [PubMed] [Google Scholar]
- 167.Wang Y, Eliot MN, Wellenius GA. Short-term changes in ambient particulate matter and risk of stroke: A systematic review and meta-analysis. J Am Heart Assoc. 2014;3 doi: 10.1161/JAHA.114.000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.O’Donnell MJ, Fang J, Mittleman MA, Kapral MK, Wellenius GA, Investigators of the Registry of Canadian Stroke Network Fine particulate air pollution (PM2.5) and the risk of acute ischemic stroke. Epidemiology. 2011;22:422–31. doi: 10.1097/EDE.0b013e3182126580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li R, Navab M, Pakbin P, Ning Z, Navab K, Hough G, Morgan TE, Finch CE, Araujo JA, Fogelman AM, Sioutas C, Hsiai T. Ambient ultrafine particles alter lipid metabolism and HDL anti-oxidant capacity in LDLR-null mice. J Lipid Res. 2013;54:1608–15. doi: 10.1194/jlr.M035014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li R, Mittelstein D, Kam W, Pakbin P, Du Y, Tintut Y, Navab M, Sioutas C, Hsiai T. Atmospheric ultrafine particles promote vascular calcification via the NF-κB signaling pathway. Am J Physiol Cell Physiol. 2013;304:C362–9. doi: 10.1152/ajpcell.00322.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li R, Navab K, Hough G, Daher N, Zhang M, Mittelstein D, Lee K, Pakbin P, Saffari A, Bhetraratana M, Sulaiman D, Wu L, Wu L, Sioutas N, Wine E, Tseng CH, Araujo JA, Fogelman A, Sioutas C, Navab M, Hsiai TK. Effect of exposure to atmospheric ultrafine particles on production of free fatty acids and lipid metabolites in the mouse small intestine. Environ Health Perspect. 2015;123:34–40. doi: 10.1289/ehp.1307036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rao X, Zhong J, Maiseyeu A, Gopalakrishnan B, Villamena FA, Chen LC, Harkema JR, Sun Q, Rajagopalan S. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ Res. 2014;115:770–780. doi: 10.1161/CIRCRESAHA.115.304666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lietz M, Berges A, Lebrun S, Meurrens K, Steffen Y, Stolle K, Schueller J, Boue S, Vuillaume G, Vanscheeuwijck P, Moehring M, Schlage W, De Leon H, Hoeng J, Peitsch M. Cigarette-smoke-induced atherogenic lipid profiles in plasma and vascular tissue of apolipoprotein E-deficient mice are attenuated by smoking cessation. Atherosclerosis. 2013;229:86–93. doi: 10.1016/j.atherosclerosis.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 174.Lo Sasso G, Titz B, Nury C, Boué S, Phillips B, Belcastro V, Schneider T, Dijon S, Baumer K, Peric D, Dulize R, Elamin A, Guedj E, Buettner A, Leroy P, Kleinhans S, Vuillaume G, Veljkovic E, Ivanov NV, Martin F, Vanscheeuwijck P, Peitsch MC, Hoeng J. Effects of cigarette smoke, cessation and switching to a candidate modified risk tobacco product on the liver in Apoe −/− mice–a systems toxicology analysis. Inhal Toxicol. 2016;28:226–40. doi: 10.3109/08958378.2016.1150368. [DOI] [PubMed] [Google Scholar]
- 175.Liu C, Xu X, Bai Y, Wang TY, Rao X, Wang A, Sun L, Ying Z, Gushchina L, Maiseyeu A, Morishita M, Sun Q, Harkema JR, Rajagopalan S. Air pollution-mediated susceptibility to inflammation and insulin resistance: Influence of CCR2 pathways in mice. Environ Health Perspect. 2014;122:17–26. doi: 10.1289/ehp.1306841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Thatcher MO, Tippetts TS, Nelson MB, Swensen AC, Winden DR, Hansen ME, Anderson MC, Johnson IE, Porter JP, Reynolds PR, Bikman BT. Ceramides mediate cigarette smoke-induced metabolic disruption in mice. Am J Physiol Endocrinol Metab. 2014;307:E919–E927. doi: 10.1152/ajpendo.00258.2014. [DOI] [PubMed] [Google Scholar]
- 177.Woodward NC, Pakbin P, Saffari A, Shirmohammadi F, Haghani A, Sioutas C, Cacciottolo M, Morgan TE, Finch CE. Traffic-related air pollution impact on mouse brain accelerates myelin and neuritic aging changes with specificity for CA1 neurons. Neurobiol Aging. 2017;53:48–58. doi: 10.1016/j.neurobiolaging.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Torres LH, Garcia RCT, Blois AMM, Dati LMM, Dur??o AC, Alves AS, Pacheco-Neto M, Mauad T, Britto LRG, Xavier GF, Camarini R, Marcourakis T. Exposure of neonatal mice to tobacco smoke disturbs synaptic proteins and spatial learning and memory from late infancy to early adulthood. PLoS One. 2015;10 doi: 10.1371/journal.pone.0136399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun. 2013;4:1495. doi: 10.1038/ncomms2494. [DOI] [PubMed] [Google Scholar]
- 180.Liu Q, Babadjouni R, Radwanski R, Cheng H, Patel A, Hodis DM, He S, Baumbacher P, Russin JJ, Morgan TE, Sioutas C, Finch CE, Mack WJ. Stroke damage is exacerbated by nano-size particulate matter in a mouse model. PLoS One. 2016;11 doi: 10.1371/journal.pone.0153376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cao L, Xu CB, Zhang Y, Cao YX, Edvinsson L. Secondhand cigarette smoke exposure causes upregulation of cerebrovascular 5-HT1B receptors via the Raf/ERK/MAPK pathway in rats. Acta Physiol. 2013;207:183–193. doi: 10.1111/j.1748-1716.2012.02478.x. [DOI] [PubMed] [Google Scholar]
- 182.Yang JT, Chang CN, Wu JH, Chung CY, Weng HH, Cheng WC, Lee TH. Cigarette smoking decreases neurotrophin-3 expression in rat hippocampus after transient forebrain ischemia. Neurosci Res. 2008;60:431–438. doi: 10.1016/j.neures.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 183.Winterbourn CC. Superoxide as an intracellular radical sink. Free Radic Biol Med. 1993;14:85–90. doi: 10.1016/0891-5849(93)90512-s. [DOI] [PubMed] [Google Scholar]