

Abstract
In recent years, various studies have demonstrated a role for endogenously derived specialized proresolving mediators such as resolvins in the resolution of inflammation. In exploring the signaling mechanisms, in the present study we show that Resolvin D1 (RvD1) reduces LPS-induced endothelial cell (EC)-monocyte interactions via blocking H2O2-mediated PP2A inactivation, NFκB activation and ICAM1 and VCAM1 expression. In addition, we found that H2O2-mediated SHP2 inhibition leads to tyrosine phosphorylation and inactivation of PP2A by LPS, which in turn, accounts for increased NFκB activation and ICAM1 and VCAM1 expression facilitating EC-monocyte interactions and all these LPS-mediated responses were reduced by RvD1. Furthermore, the suppression of NFκB activation, ICAM1 and VCAM1 expression and EC and monocyte interactions by RvD1 involved its receptors ALX/FPR2 and GPR32 as inhibition or neutralization of these receptors negated its effects. Besides, pertussis toxin completely prevented the effects of RvD1 on inhibition of LPS-induced H2O2 production, SHP2 and PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC and monocyte interactions. Together, these observations suggest that RvD1 via activation of Gi-coupled ALX/FPR2 and GPR32 receptors blocks LPS-induced H2O2-mediated SHP2 and PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions, which could be one of the several possible mechanisms underlying the anti-inflammatory actions of this specialized proresolving mediator.
Graphical Abstract
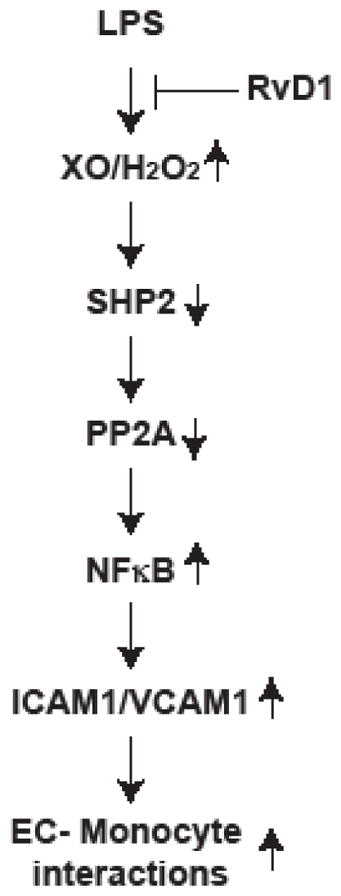
INTRODUCTION
In normal homeostatic conditions the endothelium exerts a negative regulatory effects on the expression of proinflammatory molecules (1, 2). However, cardiovascular risk factors including, hypercholesterolemia, hyperglycemia, hypertension, smoking and aging all alter the endothelial function, with the development of a chronic inflammation (3–5). Dysfunctional endothelium expresses adhesion molecules such as E-selectin, intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1) (6, 7). The expression of adhesion molecules promotes the adherence of lymphocytes, neutrophils and monocytes to the endothelium and their subsequent transmigration into the vessel wall (8, 9). The infiltration of lymphocytes, neutrophils and monocytes into the vessel wall is an early event in the development of vascular inflammation (10, 11). If vascular inflammation progresses unresolved, it can lead to the development of various vascular diseases such as atherosclerosis (12, 13). In recent years, the discovery of specialized proresolving mediators that were found to possess anti-inflammatory properties (14–21), have also been shown to exert atheroprotective effects (22–24). These lipid mediators tend to reduce ROS production, inflammatory cytokines and adhesion molecules expression, leukocyte trafficking and leukocyte-endothelial interactions (25, 26). ROS generation is the most prevailing mechanism of altered endothelial cell function (4, 5). ROS, particularly H2O2 inhibits protein tyrosine phosphatases, which in turn, by promoting the activation of protein tyrosine kinases can lead to activation of cellular signaling events of cell proliferation, migration, apoptosis, and inflammation (27–30). Recently, we have demonstrated a role for RvD1 in the protection of EC barrier function from LPS-induced disruption by preventing AJ disruption (31). Interestingly, the prevention of LPS-induced AJ disruption by RvD1 was dependent on the suppression of XO-mediated ROS production, SHP2 inactivation, Frk activation and AJ protein tyrosine phosphorylation (31).
To understand the anti-inflammatory mechanisms of RvD1 in the present study we have tested the role of phosphatases. Having demonstrated that RvD1 prevents ROS-mediated SHP2 inactivation in the protection of endothelial AJs and its barrier function, in the present study we tested the role of SHP2 in LPS-induced endothelial cell (EC)-monocyte interactions and the efficacy of RvD1 in suppressing these effects. Our findings show that RvD1 abrogates LPS-induced EC-monocyte interactions via inhibiting NFκB activation and ICAM1 and VCAM1 expression and these effects were dependent on the protection of SHP2 and its downstream effector PP2A from H2O2-mediated inactivation. Similarly, the protective effects of RvD1 against the EC-monocyte interactions were dependent on activation of its Gi-coupled ALX/FPR2 and/or GPR32 receptors, as inhibition or neutralization of these receptors or Giα suppressed the efficacy of RvD1 on blockade of LPS-induced H2O2-mediated SHP2 and PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions.
MATERIALS AND METHODS
Reagents
Pyrrolidinedithiocarbamic acid (PDTC) (20713), Pertussis toxin (19546), QNZ (10006734) and Resolvin D1 (10012554) were purchased from Cayman Chemical Company (Ann Arbor, MI). Growth factor-reduced Matrigel (354520) and anti-SHP2 antibodies (610622) were obtained from BD Biosciences (Bedford, MA). Allopurinol (PHR1377), LPS (L4391), PEG-catalase (C4963) and PHPS1 (P0039) were bought from Sigma Aldrich Company (St. Louis, MO). Anti-IKKα/β (SC-7607), anti-IκBα (SC-341), anti-NFκB (SC-372 and SC-8008), anti-pPP2A-C(α/β) (SC-12615R), anti-PP2A-C(α/β) (SC-56950), anti-ICAM1 (SC-1511R), anti-VCAM1 (SC-13160) and anti-α-Tubulin (SC-23928) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-pIKKα/β (2697), anti-pIκBα (2859), anti-pNFκB (3033) antibodies were obtained from Cell Signaling Technology (Beverly, MA). Okadaic acid (459620), PP2A inhibitor (539620), anti-FPR2 (ABF118) antibodies and Ser/Thr phosphatase assay kit (17127) were bought from Millipore (Temecula, CA). Anti-GPR32 neutralizing antibody (GTX71225) was obtained from Genetex (Irvine, CA). Anti-cysteine sulfonate antibodies were bought from Enzo Lifesciences (Farmingdale, NY). Neutralizing ICAM1 (ab171123) and VCAM1 (ab47159) antibodies were obtained from Abcam (Cambridge, MA). BOC2 (07201) was purchased from Phoenix Pharmaceuticals (Burlingame, CA). Amplex Red Hydrogen Peroxide Assay kit (A22188), Medium 200 (M200500), low serum growth supplements (S003K), BCECF (B1170) and gentamycin/amphotericin solution (R01510) were bought from ThermoFisher Scientific (Waltham, MA). The enhanced chemiluminescence (ECL) Western blotting detection reagents (RPN2106) were obtained from GE Healthcare (Pittsburg, PA).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Invitrogen (C0035C) and cultured in Medium 200 containing low serum growth supplements (LSGS), 10 μg/ml gentamycin and 0.25 μg/ml amphotericin B. Human THP1 cells (TIB-202) were purchased from American Type Culture Collection (Manassas, VA) grown in RPMI 1640 medium containing 50 μM 2-mercaptoethanol, 10% fetal bovine serum, 100 IU/ml penicillin and 100 μg/ml streptomycin and used in the adhesion and transmigration assays. Cultures were maintained at 37°C in a humidified 95% air and 5% CO2 atmosphere. HUVECs between 6–10 passages were used to perform the experiments unless otherwise indicated.
H2O2 production assay
HUVECs with and without the indicated treatments were collected by scraping and 50 μl of the cell suspension was incubated with 50 μl of 100 μM Amplex Red along with 0.2 U/ml of HRP for 30 min at 37°C in the dark. Following the incubation, the fluorescence intensities were measured in SpectraMax Gemini XS Spectrofluorometer (Molecular Devices) with excitation at 530 nm and emission at 590 nm. The H2O2 production was expressed as RFU.
Immunoprecipitation
Immunoprecipitation was performed as described by us previously (31). Cell extracts containing equal amounts of protein from control and the indicated treatments were incubated with the indicated primary antibodies at 1:100 dilution overnight at 4°C. Protein A/G-conjugated Sepharose CL-4B beads were added and incubation continued for an additional 1 hr at room temperature and the beads were collected by centrifugation at 1000 rpm for 1 min at 4°C. The beads were washed three times with lysis buffer and once with PBS, boiled in SDS sample buffer and analyzed by immunoblotting.
Western blot analysis
After appropriate treatments, cell extracts were prepared and equal amount of protein from control and the indicated treatments were resolved by SDS-PAGE. The proteins were transferred electrophoretically to a nitrocellulose membrane. After blocking in either 5% (w/v) nonfat dry milk or 5% (w/v) BSA, the membrane was probed with appropriate primary antibodies followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The antigen-antibody complexes were detected using enhanced chemiluminescence detection reagents.
Ser/Thr phosphatase assay
Ser/Thr phosphatase activity was measured by dephosphorylation of Ser/Thr phosphatase-specific phosphopeptide and the inorganic phosphate released was detected by malachite green reagent kit. To measure PP2A/B activity, the cell extracts containing equal amounts of protein were immunoprecipitated with anti-PP2A/B antibodies and the immunocomplexes were assayed for phosphatase activity as described by Narayanan et al. (32).
Monocyte adhesion
The adhesion of THP1 cells to HUVEC monolayer was measured with a fluorometric method (33). HUVEC monolayer was grown to confluency, quiesced, treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml). Wherever pharmacological inhibitors or neutralizing antibodies were used, they were added to cells 30 minutes prior to the stimulant. THP1 cells were labeled with 10 μM BCECF in serum-free medium for 30 min and the labeled cells were placed on the HUVEC monolayer at 8 × 104 cells/well, and incubation continued for another 1 hr. After incubation, the nonadherent cells were washed off with PBS and the adherent cells were lysed in 0.2 ml of 0.1 M Tris-HCl buffer, pH 8.0, containing 0.1% Triton X-100. The fluorescence intensity was measured in a Spectra Max Gemini XS Spectroflurometer (Molecular Devices) with excitation at 485 nm and emission at 535 nm. Cell adhesion was expressed as relative fluorescence units.
Monocyte transmigration
THP1 cell transmigration was measured as described previously (34). Wherever pharmacological inhibitors or neutralizing antibodies were used, they were added to cells 30 min prior to the stimulant. HUVEC monolayer was treated with and without LPS in the presence and absence of RvD1 (200 ng/ml) for 1 hr at which time the BCECF-labeled quiescent THP1 cells (1 × 105 cells/well) were added and incubation continued overnight at 37°C. The transmigration of THP1 cells through the HUVEC monolayer was measured by capturing the images by an inverted Zeiss fluorescence microscope (AxioObserver. Z1) via a 10X/NA 0.6 objective and AxioCam MRm camera without any enhancements using the microscope operating and image analysis software AxioVision Version 4.7.2 (Carl Zeiss Imaging Solutions GmbH).
Statistics
All the experiments were performed three times and the data were presented as Mean ± SD. The treatment effects were analyzed by one-way ANOVA followed by Student t test and the p values < 0.05 were considered to be statistically significant.
RESULTS
RvD1 prevents LPS-induced EC-monocyte interactions by suppression of NFκB-mediated cell adhesion molecules expression
To understand the mechanisms by which RvD1 counters inflammatory signals, we tested its effects on LPS-induced expression of ICAM1 and VCAM1. We observed that LPS induces ICAM1 and VCAM1 expression in HUVECs in a time dependent manner (Figure 1A), and RvD1 inhibited this effect (Figure 1B). NFκB mediates the expression of ICAM1 and VCAM1 (6, 9) and its activation is dependent on IKKα/β-mediated IκBα phosphorylation and its degradation (35). To understand the mechanism by which RvD1 prevents ICAM1 and VCAM1 expression, we tested the effect of RvD1 on NFκB activation. We found that LPS induces IKKα/β, IκBα and NFκB phosphorylation in a time dependent manner (Figure 1C) and RvD1 attenuates these effects (Figure 1D). Next, we studied the effect of RvD1 on LPS-induced adhesion and transmigration of THP1 cells through HUVEC monolayer. RvD1 blocked the adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 1E & F). As expected pharmacological inhibitors of NFκB also blocked LPS-induced ICAM1 and VCAM1 expression in HUVECs and the adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 1G–I). Furthermore, neutralizing ICAM1 and VCAM1 antibodies also suppressed both the adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 1H & I).
Figure 1. RvD1 attenuates LPS-induced EC-monocyte interactions via suppression of NFκB-mediated ICAM1 and VCAM1 expression.


A & C. Quiescent HUVECs were treated with and without LPS (500 ng/ml) for the indicated time periods and equal amount of protein from each condition was analyzed by Western blotting for the indicated proteins using their specific antibodies. B & D. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) for the indicated time periods and equal amounts of proteins from each condition were analyzed by Western blotting for the indicated proteins using their specific antibodies. E, F, H & I. Quiescent HUVEC monolayer was treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml), PDTC (50 μM), QNZ (10 μM) or neutralizing ICAM1 or VCAM1 antibodies (2 μg/dish) for 2 hrs. After the treatments, the HUVEC monolayer was washed and BCECF-AM-labeled THP1 cells were seeded onto the monolayer and incubated for 1 hr for adhesion (E & H) and overnight for transmigration (F & I) assays. G. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of PDTC (50 μM) or QNZ (10 μM) for 1 hr and equal amount of protein from each condition was analyzed by Western blotting for the indicated proteins using their specific antibodies. The bar graphs represent quantitative analysis of three experiments. The values are expressed as Means ± SD. *, p < 0.05 vs control; **, p < 0.05 vs LPS.
RvD1 inhibits NFκB-mediated cell adhesion molecules expression by suppression of ROS-mediated PP2A inactivation
Since, we found that RvD1 inhibits LPS-induced Ser/Thr phosphorylation of IKKα/β, IκBα and NFκB, it could be speculated that RvD1 might be influencing the activity of Ser/Thr phosphatases. To address this possibility, we first tested the effect of RvD1 on Ser/Thr phosphatases activity. LPS inhibited the Ser/Thr phosphatases activity and RvD1 prevented this effect (Figure 2A). To identify the Ser/Thr phosphatase that is influenced by RvD1, we studied the time course effect of RvD1 on PP2A and PP2B activities in HUVECs. RvD1 while having no effect on PP2B activity increased PP2A activity in a time dependent manner (Figure 2B). Based on this result, we next tested the effect of LPS on PP2A activity. LPS inhibited PP2A activity in a time dependent manner and RvD1 prevented this effect (Figure 2C). It was reported that Tyr phosphorylation of the catalytic subunit of PP2A leads to its inhibition (36). Therefore, we wanted to explore the mechanism by which RvD1 prevents LPS-induced inhibition of PP2A. We found that LPS induces Tyr phosphorylation of PP2A-C(α/β) in a time dependent manner (Figure 2D) and RvD1 suppresses this effect (Figure 2E). To find whether blockade of PP2A Tyr phosphorylation is a mechanism underlying RvD1-mediated downregulation of NFκB activation and cell adhesion molecules expression, we tested the effect of okadaic acid, a potent inhibitor of Ser/Thr phosphatases (37). In presence of okadaic acid, RvD1 was not able to prevent LPS-induced IKKα/β, IκBα and NFκB phosphorylation as well as ICAM1 and VCAM1 expression (Figure 3A & B). To further confirm the role of PP2A in the reversal of LPS-induced IKKα/β, IκBα and NFκB phosphorylation and ICAM1 and VCAM1 expression by RvD1, we tested the effect of PP2AI, a specific inhibitor of PP2A (38). In line with the above observations, in the presence of PP2AI, RvD1 failed to prevent LPS-induced IKKα/β, IκBα and NFκB phosphorylation and ICAM1 and VCAM1 expression (Figure 3C & D). Consistent with these observations, inhibition of PP2A by PP2AI completely negated the effect of RvD1 in blocking LPS-induced adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 3E & F).
Figure 2. RvD1 prevents PP2A inactivation to block LPS-induced EC-monocyte interactions.

A. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) for 30 min and analyzed for Ser/Thr phosphatase activity using Ser/Thr phosphatase-specific phosphopeptide as a substrate. B. Quiescent HUVECs were treated with and without RvD1 (200 ng/ml) for the indicated time periods and equal amount of protein from each condition was immunoprecipitated with anti-PP2A or anti-PP2B antibodies and the immunocomplexes were assayed for Ser/Thr phosphatase activity as described in panel A. C. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) for the indicated time periods and equal amount of protein from each condition was immunoprecipitated with anti-PP2A antibodies and the immunocomplexes were assayed for Ser/Thr phosphatase activity as described in panel A. D. Quiescent HUVECs were treated with and without LPS (500 ng/ml) for the indicated time periods and equal amount of protein from each condition was analyzed by Western blotting for PP2A-C(α/β) tyrosine phosphorylation using its phospho-specific antibody and the blot was normalized for its total levels. E. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) for the indicated time periods and analyzed by Western blotting for phospho and total levels of PP2A as described in panel D.
Figure 3. Pharmacological inhibition of PP2A prevents the efficacy of RvD1 to block LPS-induced NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions.

A–D. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) alone or in combination with and without okadaic acid (OA) (10 nM), a Ser/Thr phosphatase inhibitor, or PP2AI (10 μM), a specific inhibitor of PP2A, for 30 min or 1 hr and equal amounts of protein from each condition of 30 min treatment samples were analyzed by Western blotting for pIκBα, pIKKα/β and pNFκB levels, and 1 hr treatments samples were analyzed for ICAM1 and VCAM1 levels and the blots were normalized for their total levels or α-tubulin. E & F. All the conditions were the same as in panel C except that the quiescent HUVEC monolayer after the treatments was tested for THP1 cell adhesion (E) or transmigration (F) as described in Figure 1E and F, respectively. The bar graphs represent Mean ± SD values of three experiments. *, p < 0.05 vs control; **, p < 0.05 vs LPS.
RvD1 protects PP2A from inhibition via suppression of ROS-mediated SHP2 inactivation
Previously, we have shown that RvD1 by blockade of SHP2 inactivation suppresses LPS-induced Tyr phosphorylation of Frk and AJ proteins, VE-cadherin and α-catenin, thereby protecting AJ integrity (31). Hence, we tested whether the suppression of LPS-induced SHP2 inactivation by RvD1 also blocks LPS-induced PP2A-C(α/β) Tyr phosphorylation and its inactivation. In the presence of PHPS1, a specific inhibitor SHP2 (39), RvD1 failed to block LPS-induced PP2A-C(α/β) Tyr phosphorylation and its inactivation (Figure 4A & B). Similarly, in the presence of PHPS1, RvD1 was not able to block LPS-induced IKKα/β, IκBα and NFκB phosphorylation and ICAM1 and VCAM1 expression (Figure 4C & D). In line with these observations, RvD1 also failed to suppress LPS-induced adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 4E & F).
Figure 4. Pharmacological inhibition of SHP2 suppresses the efficacy of RvD1 to block PP2A inhibition, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions.

A & B. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) alone or in combination with and without PHPS1 (10 μM), a potent inhibitor of SHP2, for 30 min and an equal amount of protein from each condition was either assayed for PP2A activity as described in Figure 2, panel C (A) or analyzed by Western blotting for phospho and total PP2A levels (B). C & D. All the conditions were the same as in panel A except that cells were treated for 1 hr or 30 min and cell extracts of 1 hr treatment were analyzed by Western blotting for ICAM1 and VCAM1 levels and 30 min treatment samples were analyzed for pIκBα, pIKKα/β and pNFκB levels and the blots were normalized for their total levels or α-tubulin. E & F. Quiescent HUVEC monolayer was treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) alone or in combination with and without PHPS1 (10 μM) for 2 hrs and assayed for THP1 cell adhesion (E) or transmigration (F) as described in Figure 1E and F, respectively. The bar graphs represent Mean ± SD values of three experiments. *, p < 0.05 vs control; **, p < 0.05 vs LPS.
XO mediates PP2A inactivation, NFκB activation and cell adhesion molecules expression
Previously, we have reported that RvD1 prevents LPS-induced XO activity and ROS production and thus protects SHP2 from oxidation and inactivation (31). In view of these observations, we wanted to find whether RvD1 by inhibition of LPS-induced XO-mediated ROS production prevents PP2A inactivation. Allopurinol, a specific inhibitor of XO (40), suppressed LPS-induced H2O2 production (Figure 5A), PP2A-C(α/β) Tyr phosphorylation and its inactivation, (Figure 5B & C), IKKα/β, IκBα and NFκB phosphorylation (Figure 5D) and ICAM1 and VCAM1 expression (Figure 5E). In accordance with these results, Allopurinol also inhibited LPS-induced adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 5F & G). To find whether H2O2 mediates LPS-induced SHP2 cysteine oxidation and PP2A-C(α/β) Tyr phosphorylation, we tested the effect of PEG-catalase. Preincubation of HUVECs with PEG-catalase (50 U/ml) inhibited LPS-induced H2O2 production, SHP2 cysteine oxidation and PP2A-C(α/β) Tyr phosphorylation (Figure 5H & I). Together, results suggest that XO-mediated H2O2 production is required for LPS-induced inactivation of SHP2 and PP2A.
Figure 5. RvD1 inhibits LPS-induced XO-mediated ROS production to prevent PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions.

A. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of Allopurinol (50 μM), a specific inhibitor of XO, for 10 min and assayed for H2O2 production. B, D & E. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of Allopurinol (50 μM) for 30 min or 1 hr and equal amounts of protein from each condition of 30 min treatment samples were analyzed by Western blotting for pPP2A-C(α/β), pIκBα, pIKKα/β and pNFκB levels and 1 hr treatment samples were analyzed for ICAM1 and VCAM1 levels and the blots were normalized for their total levels or α-tubulin. C. The cell extracts of panel B were assayed for PP2A activity. F & G. Quiescent HUVEC monolayer was treated with and without LPS (500 ng/ml) in the presence and absence of Allopurinol (50 μM) for 2 hrs and assayed for THP1 cell adhesion (F) or transmigration (G) as described in Figure 1E and F, respectively. H & I. Quiescent HUVEC monolayer was treated with and without LPS (500 ng/ml) in the presence and absence of PEG-catalase (50 U/ml) for 10 min for H2O2 production or 30 min for cell extracts preparation. To measure SHP2 cysteine oxidation, cell extracts containing equal amounts of protein from each condition were immunoprecipitated with cysteine sulfonate (CSN) antibodies and the immunocomplexes were immunoblotted for SHP2 using its specific antibodies. The same cell extracts were also analyzed by Western blotting for total SHP2. PP2A-C(α/β) Tyr phosphorylation was measured by Western blotting using its phospho-specific antibodies and the blot was normalized to its total levels. *, p < 0.05 vs control; **, p < 0.05 vs LPS. PEG-Cat, PEG-catalase.
Both ALX/FPR2 and GPR32 mediate the protective effects of RvD1 against LPS-induced EC-monocyte interactions
Previously we have demonstrated that RvD1 protects AJs from LPS-induced disruption via its receptors ALX/FPR2 and GPR32 (24). Blockade of either ALX/FPR2 using its specific inhibitor, BOC2 (41) or suppression of GPR32 function by its neutralizing antibodies (10 μg/ml) (42) alone or in combination attenuated the efficacy of RvD1 in preventing LPS-induced PP2A-C(α/β) tyrosine phosphorylation and its inactivation (Figure 6A & B), IKKα/β, IκBα and NFκB phosphorylation (Figure 6C), ICAM1 and VCAM1 expression (Figure 6D) and adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 6E & F). In order to identify the G proteins through which RvD1 exerts its effects, we have tested the role of Gi protein. Pertussis toxin (50 ng/ml), a specific blocker of Gαi (43), completely blocked the effects of RvD1 on LPS-induced H2O2 production, SHP2 cysteine oxidation, PP2A-C(α/β) tyrosine phosphorylation, NFκB activation, ICAM1 and VCAM1 expression and adhesion and transmigration of THP1 cells through the HUVEC monolayer (Figure 7A–E).
Figure 6. The efficacy of RvD1 in the suppression of LPS-induced XO-mediated ROS production, SHP2 and PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions is dependent on activation of its receptors ALX/FPR2 and GPR32.
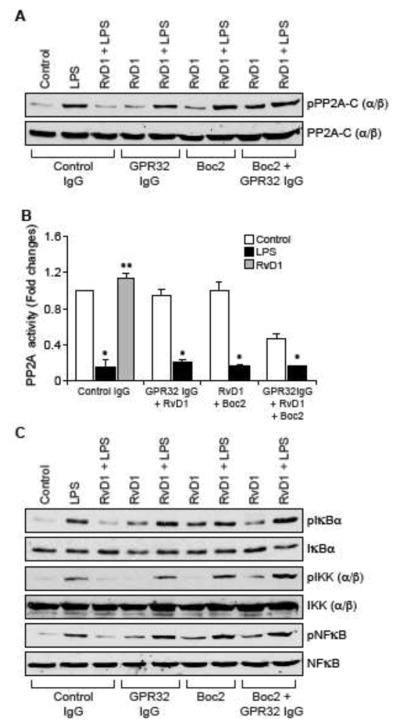
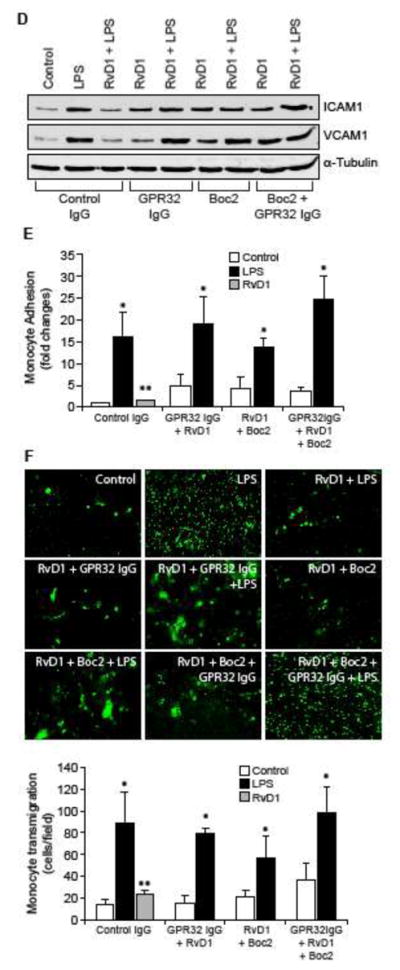
A, C & D. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) along with the indicated combinations of control IgG, GPR32 IgG (10 μg/ml), ALX/FPR2 inhibitor Boc2 (3 μM) for 30 min or 1 hr and cell extracts were prepared. Cell extracts of 30 min treatment were analyzed by Western blotting for pPP2A-C(α/β), pIκBα, pIKKα/β and pNFκB levels and cell extracts of 1 hr treatment were analyzed for ICAM1 and VCAM1 levels and the blots were normalized for their total levels or α-tubulin. B. The cell extracts of 30 min treatment in panel A were analyzed for PP2A activity. E & F. All the conditions were the same as in panel A except that after the treatments, the HUVEC monolayer was subjected to THP1 cell adhesion and transmigration assays as described in Figure 1E and F, respectively. The bar graphs represent Mean ± SD values of three experiments. *, p < 0.05 vs control or control IgG; **, p < 0.05 vs LPS or control IgG + LPS.
Figure 7. Pertussis toxin blocks the efficacy of RvD1 in the suppression of LPS-induced ROS production, SHP2 cysteine oxidation, and PP2A-C(α/β) tyrosine phosphorylation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions.

A. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) in combination with and without pertussis toxin (50 ng/ml) for 10 min and assayed for H2O2 production. B & C. Quiescent HUVECs were treated with and without LPS (500 ng/ml) in the presence and absence of RvD1 (200 ng/ml) in combination with and without pertussis toxin (50 ng/ml) for 30 min or 1 hr and cell extracts were prepared. Cell extracts of 30 min treatment were analyzed for SHP2 cysteine oxidation, PP2A-C(α/β) tyrosine phosphorylation and NFκB activation as described in Figure 5, panels I and D, respectively and the cell extracts of 1 hr treatment were analyzed for ICAM1 and VCAM1 expression as described in Figure 5, panel E and blots were normalized for their total levels or α-tubulin. D & E. All the conditions were the same as in panel A except that after the treatments, the HUVEC monolayer was subjected to THP1 cell adhesion (D) or transmigration (E) assays as described in Figure 1E and F, respectively. The bar graphs represent Mean ± SD values of three experiments. *, p < 0.05 vs control; **, p < 0.05 vs LPS. PTX, pertussis toxin.
DISCUSSION
The dysfunctional endothelium expresses several proinflammatory molecules including cell adhesion molecules (1, 2, 5). The adhesion molecules play an important role in lymphocyte, neutrophil and monocyte adhesion to the endothelium and their transmigration into arteries setting a phase in the development of vascular inflammation (7–11). Vascular inflammation is one of the several major factors in the development and progression of cardiovascular diseases such as atherosclerosis (12, 13). It was reported that inhibition of cell adhesion molecules protects from developing atherosclerosis in experimental animal models (44, 45). In homeostatic conditions the inflammatory process itself facilitates its resolution to minimize tissue and organ damage (46). A growing body of evidence points out a role for a defective resolution of inflammation in atherosclerosis (47). However, the mechanisms responsible for the resolution of inflammation are not well understood till recently when specialized lipid mediators namely maresins, protectins and resolvins with anti-inflammatory properties have been discovered (14–21). The levels of these lipid mediators have been found diminished in vulnerable atherosclerotic plaques, thus emphasizing their anti-atherogenic functions (22–24, 48). In fact, maresins and resolvins have been reported to be atheroprotective in experimental animal models albeit the underlying mechanisms were not well understood (22–24, 48). In the present study we show that RvD1 besides its role in resolution of inflammation also inhibits the early events of inflammation. Specifically, our findings show that RvD1 inhibits cell adhesion molecules ICAM1 and VCAM1 expression by LPS. A large body of data shows that NFκB plays a major role in the expression of cell adhesion molecules in response to inflammatory stimulants and oxidants (6, 35, 49). Our findings show that LPS induces both ICAM1 and VCAM1 expression via activation of NFκB. In addition, LPS-induced NFκB-mediated ICAM1 and VCAM1 expression require XO-mediated ROS production. Interestingly, RvD1 via suppressing ROS production blocks the effect of LPS on NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions. The recruitment of circulating monocytes to the dysfunctional endothelium occurs via a tightly regulated multi-step process mediated by a combination of cell surface adhesion molecules (50). The initial adhesive interactions between monocytes and endothelium are tethering and rolling (49). The monocytes migrate into the interstitium through spaces between adjacent ECs (51, 52). Previously, it has been reported that the disruption of cell-cell junctions by pro-inflammatory stimuli facilitates the transmigration of monocytes through the endothelium (51, 53). It was also demonstrated that while VCAM1 plays a role in the initial stages of monocyte recruitment to the endothelium, ICAM1 is believed to participate in adhesion strengthening and spreading of monocytes and their transendothelial migration (54, 55). Interestingly RvD1 suppresses both ICAM1 and VCAM1 expression in preventing the EC-monocyte interactions. This conclusion can be further supported by the findings that inhibition of the function of either ICAM1 or VCAM1 by their neutralizing antibodies completely suppressed EC-monocyte interactions.
Although the role of ROS and NFκB in the regulation of cell adhesion molecules expression has been studied extensively (6, 35, 49), the direct involvement of phosphatases in the modulation of these effects is not clear. In this aspect, the present findings show that LPS inhibits PP2A activity, which in turn, appears to be involved in the phosphorylation of IKKαβ, IκB and NFκB. The kinases IKKα/β upon stimulation by inflammatory molecules, phosphorylate its substrate IκB leading to its ubiquitination and proteasomal degradation and as a result NFκB is relieved from its inhibitory constraint and becomes activated (35, 49). What is exciting is that RvD1 prevents PP2A inactivation by LPS, thus promoting the dephosphorylation and inactivation of NFκB. Previous studies have shown that tyrosine phosphorylation of the catalytic subunit of PP2A leads to its inactivation (36). We found that LPS induces the tyrosine phosphorylation of PP2A catalytic subunit C and this correlates with inhibition of its activity. Surprisingly, RvD1 blocks the tyrosine phosphorylation of PP2A-Cα/β and this correlates with its reversal from inactivation. Since RvD1 fails to prevent NFκB activation and ICAM1 and VCAM1 expression in the presence of PP2A inhibitors, it is likely that RvD1 interferes with EC-monocyte interactions via protecting PP2A activity. Besides inflammation, ROS are linked to the development of atherosclerosis (56, 57). Many cell types including inflammatory cells and dysfunctional endothelial cells produce ROS (56, 57). In addition, H2O2 has been reported to act as a cellular signaling molecule (58). One of the mechanisms by which H2O2 mediates cellular signaling is its capacity to oxidize the cysteine residues in the catalytic sites of protein tyrosine phosphatases (PTPs) rendering them inactive, which in turn, promotes the phosphorylation and activation of protein tyrosine kinases (PTKs) (27–29, 59). Previously, we have shown that LPS via XO-mediated ROS production triggers the oxidation of the catalytic cysteine residue of SHP2 leading to its inactivation (31). It was reported that H2O2 inhibits SHP2 activity via oxidation of its catalytic cysteine residue (27, 59, 60). Suppression of SHP2 phosphatase activity by LPS enhanced AJ protein tyrosine phosphorylation and their disruption. In addition, we have reported that RvD1 plays a crucial role in blocking XO-mediated ROS production and SHP2 inactivation to prevent AJ protein tyrosine phosphorylation and thereby protecting EC barrier function (31). In the present study we found that RvD1 by suppression of protein tyrosine phosphatase SHP2 inactivation further blocks protein phosphatase, PP2A inactivation by LPS. To our knowledge, these findings demonstrate for the first time that RvD1 by prevention of LPS-induced SHP2 and PP2A inactivation suppresses NFκB activation and ICAM1 and VCAM1 expression, which in turn reduce the EC-monocyte interactions. Furthermore, the observations that RvD1 was not able to prevent PP2A-C(α/β) Tyr phosphorylation and its inactivation in the presence of SHP2 inhibitor suggests a crosstalk between protein tyrosine phosphatase SHP2 and protein (Ser/Thr) phosphatase PP2A in LPS-induced NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions.
It was reported that RvD1 mediates its cellular effects via its receptors ALX/FPR2 and GPR32 (19, 61, 62). Previously, we have demonstrated that RvD1 protects EC adherens junction integrity and its barrier function via involving its receptors ALX/FPR2 and GPR32 (31). Here, we found that while inhibition of either ALX/FPR2 or GPR32 alone substantially blocked the effects of RvD1 on the suppression of PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions, interference with both the receptors simultaneously completely prevented its effects. Indeed, RvD1 was not able to prevent LPS-induced H2O2 production, PP2A inactivation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions when both the receptors were blocked. Since pertussis toxin suppressed the efficacy of RvD1 on the attenuation of LPS-induced H2O2 production, SHP2 cysteine oxidation, PP2A-C(α/β) tyrosine phosphorylation, NFκB activation, ICAM1 and VCAM1 expression and EC-monocyte interactions, it is likely that RvD1 mediates its effects via activation of Gαi signaling. These observations were also consistent with a previous report where it was shown that pertussis toxin blocks the effects of RvD1 on polymorphonuclear leukocyte responses (61). It is also noteworthy that inhibition of RvD1 receptors alone led to increased basal expression of ICAM1 and VCAM1, which might suggest an endogenous role for RvD1 and its receptors in the counter regulation of these adhesion molecules. In conclusion, the present study provides the first evidence that RvD1 by preventing H2O2 production and phosphatases SHP2 and PP2A inactivation suppresses NFκB-mediated expression of cell adhesion molecules, ICAM1 and VCAM1, and thereby attenuates the adhesion and transmigration of monocytes through the endothelial monolayer. Since all the afore-mentioned protective effects of RvD1 were linked primarily to its efficacy to suppress LPS-induced H2O2 production and SHP2 cysteine oxidation, it is possible that RvD1 might act as an oxidant scavenger, perhaps indirectly if not directly.
HIGHLIGHTS.
RvD1 blocks NFκB-ICAM1/VCAM1 signaling by inhibition of SHP2 and PP2A inactivation.
ALX and GPR32 mediate the effects of RvD1 on preventing EC-monocyte interactions.
The protective effects of RvD1 on EC function require Gαi signaling activation.
Acknowledgments
This work was supported by a grant HL074860 from NHLBI to GNR.
Footnotes
AUTHORS CONTRIBUTIONS
RC, performed Western blotting, Ser/Thr phosphatase and PP2A activity, monocyte adhesion and transmigration assays and written the initial manuscript draft; AMM, performed Western blotting, ROS assay and monocyte adhesion and transmigration assays; NKS, performed monocyte transmigration assay; GNR conceived the overall goal of the project, designed the experiments, interpreted the data and wrote the manuscript.
CONFLICT OF INTEREST
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- 2.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 3.Lamon BD, Hajjar DP. Inflammation at the molecular interface of atherogenesis:an anthropological journey. Am J Pathol. 2008;173:1253–1264. doi: 10.2353/ajpath.2008.080442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation. 2013;20:239–47. doi: 10.1111/micc.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, García-Cardeña G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and –resistant regions of human vasculature. Proc Natl Acad Sci U S A. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins T. Endothelial nuclear factor-kappa B and the initiation of the atherosclerotic lesion. Lab Invest. 1993;68:499–508. [PubMed] [Google Scholar]
- 7.Langer HF, Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med. 2009;13:1211–1220. doi: 10.1111/j.1582-4934.2009.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107:321–330. doi: 10.1093/cvr/cvv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu HB, Agranoff AB, Nabel EG, Leung K, Duckett CS, Neish AS, Collins T, Nabel GJ. Differential regulation of vascular cell adhesion molecule 1 gene expression by specific NF-kappa B subunits in endothelial and epithelial cells. Mol Cell Biol. 1993;13:6283–6289. doi: 10.1128/mcb.13.10.6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–159. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 11.Cerletti C, Evangelista V, de Gaetano G. P-selectin-beta 2-integrin cross-talk: a molecular mechanism for polymorphonuclear leukocyte recruitment at the site of vascular damage. Thromb Haemost. 1999;82:787–793. [PubMed] [Google Scholar]
- 12.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 13.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol. 2004;122:305–321. doi: 10.1007/s00418-004-0695-8. [DOI] [PubMed] [Google Scholar]
- 15.Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta. 2015;1851:397–413. doi: 10.1016/j.bbalip.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian H, Lu Y, Sherwood AM, Hongqian D, Hong S. Resolvins E1 and D1 in choroid-retinal endothelial cells and leukocytes: biosynthesis and mechanisms of anti-inflammatory actions. Invest Ophthalmol Vis Sci. 2009;50:3613–3620. doi: 10.1167/iovs.08-3146. [DOI] [PubMed] [Google Scholar]
- 18.Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, Arroyo V, Claria J. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol. 2011;187:5408–5418. doi: 10.4049/jimmunol.1100225. [DOI] [PubMed] [Google Scholar]
- 19.Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler Thromb Vasc Biol. 2012;32:1970–1988. doi: 10.1161/ATVBAHA.112.249508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maekawa T, Hosur K, Abe T, Kantarci A, Ziogas A, Wang B, Van Dyke TE, Chavakis T, Hajishengallis G. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3β-C/EBPβ pathway. Nat Commun. 2015;6:8272. doi: 10.1038/ncomms9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang MJ, Sansbury BE, Hellmann J, Baker JF, Guo L, Parmer CM, Prenner JC, Conklin DJ, Bhatnagar A, Creager MA, Spite M. Resolvin D2 Enhances Postischemic Revascularization While Resolving Inflammation. Circulation. 2016;134:666–680. doi: 10.1161/CIRCULATIONAHA.116.021894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008;22:3595–3606. doi: 10.1096/fj.08-112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, Van Dyke TE. Resolvin E1 (RvE1) Attenuates Atherosclerotic Plaque Formation in Diet and Inflammation-Induced Atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35:1123–1133. doi: 10.1161/ATVBAHA.115.305324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Döring Y, Drechsler M, Weber C, Zimmer R, Cenac N, Soehnlein O. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ Res. 2016;119:1030–1038. doi: 10.1161/CIRCRESAHA.116.309492. [DOI] [PubMed] [Google Scholar]
- 25.Damgaard C, Kantarci A, Holmstrup P, Hasturk H, Nielsen CH, Van Dyke TE. Porphyromonas gingivalis-induced production of reactive oxygen species, tumor necrosis factor-α, interleukin-6, CXCL8 and CCL2 by neutrophils from localized aggressive periodontitis and healthy donors: modulating actions of red blood cells and resolvin E1. J Periodontal Res. 2017;52:246–254. doi: 10.1111/jre.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med. 2008;45:1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Oh J, Hur MW, Lee CE. SOCS1 protects protein tyrosine phosphatases by thioredoxin upregulation and attenuates Jaks to suppress ROS-mediated apoptosis. Oncogene. 2009;28:3145–3156. doi: 10.1038/onc.2009.169. [DOI] [PubMed] [Google Scholar]
- 30.Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- 31.Chattopadhyay R, Raghavan S, Rao GN. Resolvin D1 via prevention of ROS-mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol. 2017;12:438–455. doi: 10.1016/j.redox.2017.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, Warren ST. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chattopadhyay R, Tinnikov A, Dyukova E, Singh NK, Kotla S, Mobley JA, Rao GN. 12/15-Lipoxygenase-dependent ROS production is required for diet-induced endothelial barrier dysfunction. J Lipid Res. 2015;56:562–577. doi: 10.1194/jlr.M055566. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Kundumani-Sridharan V, Dyukova E, Hansen DE, 3rd, Rao GN. 12/15-Lipoxygenase mediates high-fat diet-induced endothelial tight junction disruption and monocyte transmigration: a new role for 15(S)-hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J Biol Chem. 2013;288:15830–15842. doi: 10.1074/jbc.M113.453290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Israel A. Signal transduction. IkappaB kinase all zipped up. Nature. 1997;388:519–521. doi: 10.1038/41433. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- 37.Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M, Makkinje A, Damuni Z. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry. 1996;35:6998–7002. doi: 10.1021/bi960581y. [DOI] [PubMed] [Google Scholar]
- 39.Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, Birchmeier W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A. 2008;105:7275–7280. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003;107:416–421. doi: 10.1161/01.cir.0000046448.26751.58. [DOI] [PubMed] [Google Scholar]
- 41.Stenfeldt AL, Karlsson J, Wenneras C, Bylund J, Fu H, Dahlgren C. Cyclosporin H, Boc-MLF and Boc-FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation. 2007;30:224–229. doi: 10.1007/s10753-007-9040-4. [DOI] [PubMed] [Google Scholar]
- 42.Hsiao HM, Thatcher TH, Levy EP, Fulton RA, Owens KM, Phipps RP, Sime PJ. Resolvin D1 attenuates polyinosinic-polycytidylic acid-induced inflammatory signaling in human airway epithelial cells via TAK1. J Immunol. 2014;193:4980–4987. doi: 10.4049/jimmunol.1400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ, Lakatta EG. Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999;84:43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- 44.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2000;191:189–194. doi: 10.1084/jem.191.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. The combined role of P- and E-selectins in atherosclerosis. J Clin Invest. 1998;102:145–152. doi: 10.1172/JCI3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nature immunology. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 47.Viola J, Soehnlein O. Atherosclerosis - A matter of unresolved inflammation. Semin Immunol. 2015;27:184–193. doi: 10.1016/j.smim.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 48.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stamatovic SM, Sladojevic N, Keep RF, Andjelkovic AV. Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells. Mol Cell Biol. 2012;32:3414–3427. doi: 10.1128/MCB.06678-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reijerkerk A, Kooij G, van der Pol SM, Khazen S, Dijkstra CD, de Vries HE. Diapedesis of monocytes is associated with MMP-mediated occludin disappearance in brain endothelial cells. FASEB J. 2006;20:2550–2552. doi: 10.1096/fj.06-6099fje. [DOI] [PubMed] [Google Scholar]
- 53.Reglero-Real N, Colom B, Bodkin JV, Nourshargh S. Endothelial Cell Junctional Adhesion Molecules: Role and Regulation of Expression in Inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:2048–2057. doi: 10.1161/ATVBAHA.116.307610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakai A, Kume N, Nishi E, Tanoue K, Miyasaka M, Kita T. P-selectin and vascular cell adhesion molecule-1 are focally expressed in aortas of hypercholesterolemic rabbits before intimal accumulation of macrophages and T lymphocytes. Arterioscler Thromb Vasc Biol. 1997;17:310–316. doi: 10.1161/01.atv.17.2.310. [DOI] [PubMed] [Google Scholar]
- 55.Kevil CG, Chidlow JH, Bullard DC, Kucik DF. High-temporal-resolution analysis demonstrates that ICAM-1 stabilizes WEHI 274.1 monocytic cell rolling on endothelium. Am J Physiol Cell Physiol. 2003;285:C112–C118. doi: 10.1152/ajpcell.00334.2002. [DOI] [PubMed] [Google Scholar]
- 56.Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A American Heart Association Council on Basic Cardiovascular Sciences. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association. Circ Res. 2016;119:e39–75. doi: 10.1161/RES.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lönn ME, Dennis JM, Stocker R. Actions of “antioxidants” in the protection against atherosclerosis. Free Radic Biol Med. 2012;53:863–884. doi: 10.1016/j.freeradbiomed.2012.05.027. [DOI] [PubMed] [Google Scholar]
- 58.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karisch R, Neel BG. Methods to monitor classical protein-tyrosine phosphatase oxidation. FEBS J. 2013;280:459–475. doi: 10.1111/j.1742-4658.2012.08626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boedtkjer E, Aalkjaer C. Insulin inhibits Na+/H+ exchange in vascular smooth muscle and endothelial cells in situ: involvement of H2O2 and tyrosine phosphatase SHP-2. Am J Physiol Heart Circ Physiol. 2009;296:H247–H255. doi: 10.1152/ajpheart.00725.2008. [DOI] [PubMed] [Google Scholar]
- 61.Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH, Yang R, Petasis NA, Serhan CN. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci U S A. 2010;107:1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiurchiù V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, Serhan CN. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med. 2016;8:353ra111. doi: 10.1126/scitranslmed.aaf7483. [DOI] [PMC free article] [PubMed] [Google Scholar]