

Abstract
Inducible nitric oxide synthase (iNOS) plays important roles in cell injury and host defense. Our early study demonstrated that heat shock protein 90 (Hsp90) interacts with iNOS and this interaction enhances iNOS function. Recently, we reported that Hsp90 is also essential for iNOS gene transactivation. In the present study, we investigate the role of Hsp90 in controlling iNOS protein stability. In mouse macrophages, Hsp90 inhibition dissociated Hsp90 from iNOS and the latter subsequently formed aggregates. Aggregation deactivated iNOS. iNOS aggregates were cleared by the ubiquitin-proteasome system (UPS) inside cells. CHIP, an Hsp90-dependent E3 ligase, was previously implicated in iNOS turnover. However, CHIP knockdown had little effect on iNOS degradation in Hsp90-inhibited cells, indicating that other E3 ligases accounted for the clearance of iNOS aggregates. Further studies revealed that the SPRY domain-containing SOCS box protein 2 (SPSB2), an E3 ligase-recruiting protein, was essential for the ubiquitination of iNOS aggregates. SPSB2 knockdown or deleting the SPSB2-interacting domain on iNOS prevented the clearance of iNOS aggregates in Hsp90-inhibited cells. Thus, besides modulating iNOS function and gene transcription, Hsp90 is also essential for the protein stability of iNOS. Hsp90 blockade induces iNOS aggregation and SPSB2 is required for UPS degradation of iNOS aggregates.
Keywords: Nitric oxide, inducible nitric oxide synthase, heat shock protein 90, protein aggregation, ubiquitination, SPRY domain-containing SOCS box protein 2
Graphical abstract
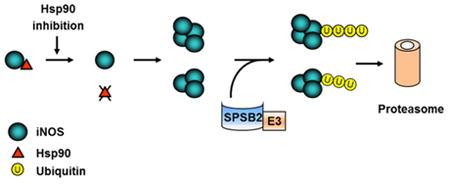
1. Introduction
Nitric oxide (NO) is a gaseous free radical. Although physiological concentrations of NO serve as a signaling molecule in neuronal transmission and cardiovascular regulation, high levels of NO harm cells (1-4). Hence NO is employed by the immune system in the fight against microbe invaders and tumor cells (5-7). In mammals, NO is derived from L-arginine via a reaction catalyzed by a family of NO synthase (NOS) including neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) (7-9). Among them, iNOS participates in cell injury and host defense (10, 11). In contrast to the constitutively existing nNOS and eNOS, little iNOS can be detected in normal cells and tissues. Inflammatory mediators, such as bacterial product lipopolysaccharide (LPS) and cytokines including interferon-γ (IFN-γ), are potent inducers of iNOS gene expression. All NOS isoforms are activated by their binding with the cofactor calmodulin (CaM) (8, 12, 13). For nNOS and eNOS, the binding to CaM is facilitated by the rise of intracellular Ca2+. iNOS, however, contains an intrinsically bound CaM and stays constantly active once expressed (8, 12, 13). The continuous activity along with the high NO-generating efficacy is thought to suit iNOS for its functions in host defense and cell injury.
Because iNOS is constantly active, the regulation of NO production from iNOS was thought to primarily occur at the enzyme transcriptional level. This notion evolves as recent studies show that iNOS function can be affected by protein-protein interactions and posttranslational modifications (14). In an early study, we showed that iNOS associates with heat shock protein 90 (Hsp90) in cells and this association enhances iNOS activity (14). The interaction with Hsp90 is found to be important in the cytotoxic effect of iNOS on cells. Recently, we reported that Hsp90 is necessary for the transcriptional activation of iNOS gene in cells stimulated by both LPS and IFN-γ (15). Hsp90 appeared to be essential for transcriptional factor NF-κB and STAT1 to bind with the iNOS promoter during gene transactivation. The necessary role of Hsp90 in iNOS induction was confirmed in vivo in myocardium infarction (15). Together, these studies demonstrate the importance of Hsp90 in regulating iNOS function and gene expression.
In addition to gene expression, the levels of active iNOS in cells are also determined by its protein stability and turnover (16-18). Whether or not Hsp90 affects iNOS protein stability, and if it does, how changed iNOS stability is coped with inside cells are the remaining questions in the study of Hsp90 regulation of iNOS. In the present study, we address these issues in mouse macrophages which are stimulated to express iNOS. Our studies find Hsp90 vital for iNOS protein stability. Loss of the interaction with Hsp90 leads to iNOS aggregation and deactivation. Cells employ the ubiquitin-proteasome system (UPS) to eliminate aggregated iNOS proteins. We further reveal that the SPRY domain-containing SOCS box protein 2 (SPSB2), an E3 ligase-recruiting protein, is essential for the proteasomal clearance of iNOS aggregates in cells.
2. Materials and Methods
Materials
Cell culture materials were purchased from Invitrogen (Carlsbad, CA). The antibody against iNOS was from BD Transduction Laboratories. Antibody against Hsp90 was a product of Cell Signaling Technology (Beverly, MA). The antibody against SPSB2 was from Santa Cruz Biotechnology (Santa Cruz, CA). LPS, recombinant mouse IFN-γ, geldanamycin, radicicol, anti-GAPDH and anti-flag antibodies were products of Sigma (St. Louis, MO). Unless otherwise indicated, all other chemicals used in this study were from Sigma.
Cell culture
Mouse macrophage (RAW 264.7, ATCC), human embryonic kidney 293 (HEK293), and African green monkey SV40-transfected kidney fibroblast (COS-7) cells were grown in Dulbecco's modified Eagle's medium with 10% fetal calf serum in a 37°C humidified atmosphere of 95% air and 5% CO2. Expression of iNOS in RAW 264.7 cells was induced by LPS (2 μg/ml, serotype 026:B6) and IFN-γ (100 U/ml).
shRNA
HuSH 29mer shRNA constructs against CHIP gene (Origene Technologies) were transfected into HEK293 cells by using Lipofectamine 2000 reagents (Invitrogen). The CHIP knockdown efficiency was confirmed by Western blotting and the CHIP-depleted cells were subjected to further treatments and analyses.
siRNA
Small interfering RNA (siRNA) oligonucleotides targeting SPSB2 and control nonspecific siRNA were purchased from Santa Cruz Biotechnology. In twelve-well plates, cells were seeded the day before transfection and grown to 30% confluence. siRNA oligonucleotides (100 nM) were transfected into cells by using Lipofectamine 2000 reagents. After 48 h of transfection, cells were subjected to further experiments.
Plasmid construction
To construct the plasmid encoding 50-1144 truncated iNOS, the 50-1144 region of iNOS was PCR-amplified from previously constructed pCMV-iNOS plasmid using primers 5′-CCCAAGCTTGGGATGGGCTCCCCGCAGC and 5′-CCGCTCGAGCGGGCCAGAAGCTGGAAC. After overnight incubation with HindIII and XhoI, 50-1144 iNOS cDNA was cloned into the mammalian expression vector pCMV-Flag-Tag2B using the standard molecular biology procedures. To construct pEGFP-C3/iNOS plasmid encoding GFP-iNOS fusion protein, the HindIII-XhoI fragment of pCMV-iNOS plasmid containing iNOS cDNA was cloned into HindIII-SalI sites of pEGFP-C3 vector.
Cell fractionation
Cells were rinsed with phosphate-buffered saline and lysed on ice for 30 min in a lysis buffer containing moderate detergents (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 50 mM NaF, 1 mM Na3VO4, 5 mM sodium pyrophosphate, 1 mM EDTA and protease inhibitor tablet). After a centrifugation at 14,000×g for 15 min at 4°C, the supernatants and pellets were recovered as soluble and insoluble fractions, respectively. The insoluble pellets were washed by PBS, and boiled in 1.5×SDS/PAGE sample buffer (90 mM Tris-HCl, pH 6.8, 3% SDS, 15% glycerol, 0.01% Bromophenol blue and 62.5 mM dithiothreitol) for 7 min. Total cell samples were obtained by passing the lysates through 30G needles after 30 min incubation in high-detergent lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1% SDS, 1 mM EDTA and protease inhibitor tablet).
Western blotting
Cells were harvested and lysed on ice for 30 min in lysis buffer, followed by 15 min centrifugation at 14,000×g. Protein concentrations were determined by using the detergent-compatible protein assay kit (Bio-Rad). After 5 min boiling in the SDS/PAGE sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 40 mM dithiothreitol, 10% glycerol, and 0.01% Bromophenol blue), the proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the appropriate primary antibodies. Membrane-bound primary antibodies were detected with secondary antibodies conjugated with horseradish peroxidase. Immunoblots were developed on films using the enhanced chemiluminescence technique (SuperSignal West Pico, Pierce).
Fluorescence imaging
Plasmids encoding GFP-iNOS fusion proteins were transfected into HEK293 or COS-7 cells with Lipofectamine 2000 reagents. After 20 h of plasmid transfection, the cells were treated with MG-132 (20 μM) for 30 min prior to 8 h incubation with geldanamycin (GA, 5 μM) or radicicol (20 μM). Imaging was performed on a fluorescence microscope (Nikon Eclipse 80i) with the Qimaging Retiga-2000R digital camera (Qimaging).
iNOS activity assay
iNOS activity was measured by the L-[14C]arginine to L-[14C]citrulline conversion assay (19). iNOS expression was induced in RAW 264.7 by LPS/IFN-γ for 12h. Cells were then homogenated in homogenate buffer (50 mM Tris-HCl, pH7.4, 50 mM NaF, 1 mM Na3VO4, and protease inhibitor mixture). After centrifugation (14,000×g for 30 min at 4°C), the supernatants were harvested and used for measuring soluble iNOS activity. To obtain aggregated iNOS, LPS/IFN-γ-stimulated cells were incubated with GA (5 μM) or radicicol (20 μM) in the presence of MG-132 (20 μM) for 18 h. The cells were then harvested and homogenated in homogenate buffer. After centrifugation and stringent wash, the pellets were resuspended in the homogenate buffer and used for activity measurements of aggregated iNOS. The cell lysates were added to the reaction mixture containing 50 mM Tris-HCl, pH 7.4, 0.5 mM NADPH, 10 nM CaCl2, 10 μg/ml CaM, 10 μM BH4, 2 μM L-[14C]arginine, and 36 μM L-arginine. After 15 min incubation at 37 °C, the reactions were terminated by ice-cold stop buffer. L-[14C]Citrulline was separated by passing the reaction mixture through Dowex AG 50W-X8 (Na+ form; Sigma) cation exchange columns and quantitated by liquid scintillation counting.
Nitrite assay
Total nitrite released in cell culture medium was measured with a Griess reagent kit (Invitrogen). The reaction consisted of 20 μl of Griess reagent, 150 μl of medium, and 130 μl of deionized water. After incubation of the mixture for 30 min at room temperature, nitrite levels were measured at 548 nm using a M2 spectrophotometric microplate reader (Molecular Devices).
Hsp90-iNOS dissociation assay
Expression of iNOS was induced in RAW 264.7 cells by LPS/IFN-γ for 12 h. Cells were harvested and lysed for 30 min in the following lysis buffer: 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA, 50 mM NaF, 1 mM Na3VO4, and protease inhibitor tablet. The cell lysates were then centrifuged at 14,000×g for 15 min, and the supernatants were incubated with 2′, 5′-ADP-Sepharose affinity resins for 2 hours at 4°C. The beads were washed three times with high salt buffer (50 mM Tris-HCl, 500 mM NaCl, 1 mM EDTA and 0.5% Nonidet P-40), followed by one time wash with regular washing buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA and 0.5% Nonidet P-40). Afterwards, the beads were resuspended in 50 μl washing buffer containing GA (10 μM) or radicicol (20 μM) and then harvested at designated time point. After 1 min centrifugation at 14,000×g, the supernatants were recovered and the proteins bound to beads were eluted by 5 min boiling of the beads in the SDS/PAGE sample buffer. Both supernatant and eluted samples were characterized by Western blotting with appropriate antibodies.
Statistics
Data were expressed as Mean ± SE. Comparisons were made using a two-tailed Student's paired or unpaired t test. Differences were considered statistically significant at P < 0.05.
3. Results
3.1 Hsp90 inhibition causes iNOS aggregation and iNOS aggregates are cleared by proteasomes
To determine the effects of Hsp90 on iNOS protein stability and turnover, mouse macrophage RAW 264.7 cells were exposed to LPS/IFN-γ for 12 hours to induce iNOS expression. Then LPS/IFN-γ were removed and iNOS expression was suspended by cycloheximide. After Hsp90 inhibitor or other treatments, cells were fractioned into supernatant (soluble) and pellet (insoluble) fractions in a buffer containing moderate detergent (1% NP-40). As shown in Fig. 1A, normally iNOS stayed as a stable and soluble protein in cytosol. Little iNOS was detected in the pellet fractions of cell lysates. Radicicol, an Hsp90 specific inhibitor, reduced soluble iNOS levels in a time-dependent manner. Interestingly, pretreatment of cells with the proteasome blocker MG-132 failed to prevent the loss of iNOS in the soluble fractions of Hsp90-inhibted cells; instead, a progressive iNOS accumulation was observed in the insoluble fractions following proteasomal blockade. With high levels of strong detergent (1% SDS), the insoluble iNOS proteins could be extracted into soluble fractions (Fig. 1B). Under this condition, Hsp90 inhibition-induced iNOS depletion was seen to be blocked by MG-132 treatment, a characteristic feature expected to occur to the proteins that are degraded by the proteasome. These data suggested that Hsp90 inhibition caused iNOS to form aggregates and these iNOS aggregates were subsequently cleared by proteasomes.
Figure 1.
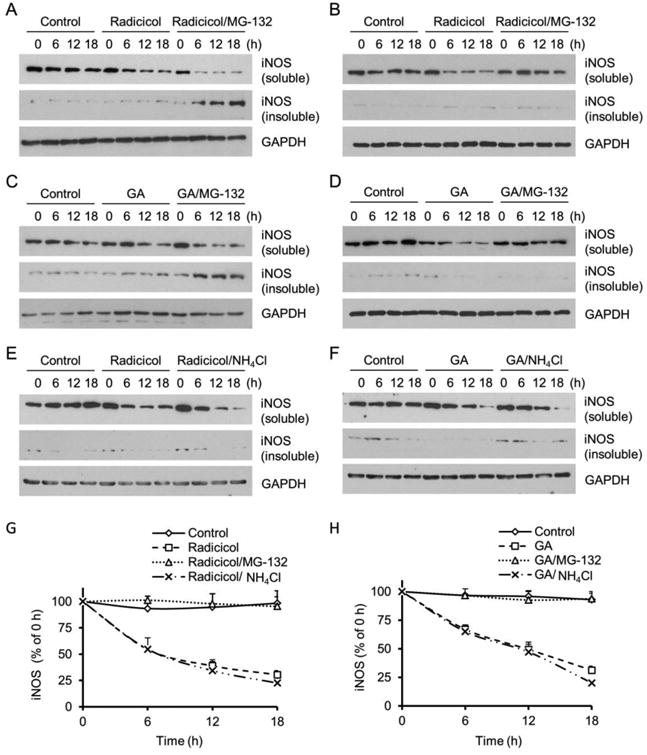
Hsp90 control of iNOS protein stability and turnover in mouse macrophage. (A) iNOS expression was induced in RAW 264.7 cells by LPS (2 μg/ml)/IFN-γ (100 U/ml). Hsp90 inhibition with radicicol (20 μM) caused iNOS aggregation, shown in the insoluble fractions of MG-132-treated cells. In the absence of MG-132 (20 μM), the iNOS aggregates were cleared by the proteasome. (B) Insoluble iNOS aggregates in radicicol-treated cells were dissolved in strong detergent containing buffer (1% SDS) and the characteristic MG-132-sensitive feature of the proteins degraded by the proteasome was shown. (C, D) Geldanamycin (GA, 5 μM), another Hsp90 inhibitor that structurally differed from radicicol, also caused iNOS aggregation and the clearance of iNOS aggregates was blocked by proteasomal inhibition. (E, F) Inhibition of lysosome proteases with NH4Cl (20 mM) had no effect on the degradation of iNOS in Hsp90-inhibited cells. Representative blots shown from 3-5 experiments. (G, H) Quantitative analyses of iNOS degradation in the absence and presence of Hsp90, proteasome, and lysosome inhibitors. Data are means ± SE, n=4.
Because iNOS induction requires Hsp90 (15), siRNA approach to knock down Hsp90 could not be used to corroborate the finding on the effect of radicicol on iNOS protein stability. Radicicol is a specific Hsp90 inhibitor (14, 20). To further eliminate the concern on the potential off-target effects of pharmacological inhibitors, we repeated the above experiments with geldanamycin (GA), another Hsp90 selective inhibitor whose structure is different from that of radicicol. In accordance with the findings obtained with radicicol, Hsp90 inhibition with GA also rendered iNOS aggregation and the clearance of aggregated iNOS was blocked by proteasomal inhibition (Fig. 1C and 1D).
To determine the possible contribution of the autophagy-lysosome pathway to the degradation of iNOS aggregates, we treated the cells with the pan autophagy-lysosome inhibitor ammonium chloride (NH4Cl), which changes the acidic environment of lysosomes and inhibits the proteases inside. In contrast to the effect of MG-132, NH4Cl (20 mM) had no significant effect on the clearance of iNOS aggregates in cells treated by either radicicol or GA (Fig. 1E and 1F). The different effects of proteasome and lysosome inhibition on the clearance of iNOS aggregates were compared quantitatively in Fig. 1 G and 1H. These results demonstrated that it was the proteasome, rather than the autophagy-lysosome pathway, responsible for the clearance of iNOS aggregates in Hsp90-inhibited cells.
3.2 GFP-iNOS aggregation and degradation in transfected cells
To visualize iNOS aggregation and degradation under Hsp90 inhibition, a GFP-tagged iNOS plasmid was constructed and transfected into HEK293 cells. Expressed iNOS was evenly distributed in the cytosol (Fig. 2, top panel). Hsp90 inhibition with radicicol or GA depleted intracellular iNOS as evidenced by the reduction of GFP fluorescence in cells. Blocking proteasomes resulted in intense fluorescence cluster formation in cells. With COS-7 cells whose size is bigger than that of HEK293 cells, Hsp90 inhibition-induced iNOS aggregation was clearly demonstrated after blocking proteasomes (Fig 2, lower panel). These aggregates displayed as fluorescent clusters with different sizes and shapes. These imaging data were consistent with the findings from the cell fractionation experiments. These two lines of evidence together demonstrated that Hsp90 was essential for the stability of iNOS as a soluble protein in the cytosol.
Figure 2.
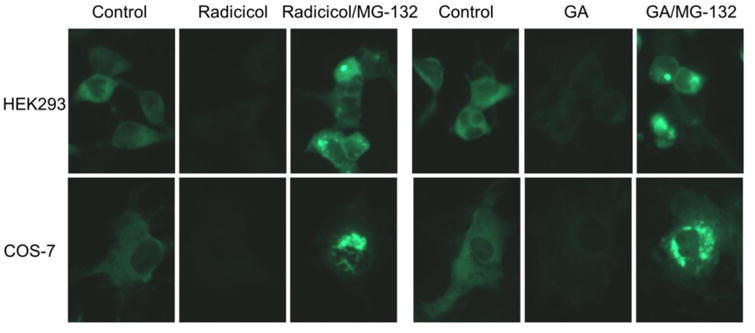
Fluorescence imaging of Hsp90 inhibition-induced GFP-iNOS aggregation and clearance in transfected cells. GFP-iNOS fusion proteins were expressed in HEK293 and COS-7 cells. As shown, Hsp90 inhibition (Radicicol, 20 μM; GA, 5 μM) depleted GFP-iNOS. In the presence of proteasome inhibitor, iNOS aggregates were seen in cells. Representative images are shown from triplicate experiments.
3.3 Aggregation deactivates iNOS
Aggregation usually leads to the loss of protein function (21-23). To determine the effect of protein aggregation on iNOS function inside cells, we measured NO productions from iNOS-induced cells in the absence and presence of Hsp90 inhibitors. Compared to that in the control groups, markedly decreased NO production was seen in Hsp90-inhibited cells in which aggregated iNOS was preserved by MG132 (Fig. 3A). These results suggested that aggregation deactivated iNOS. To further demonstrate the functional consequence of iNOS aggregation, we assayed the catalytic activity of soluble and aggregated iNOS in vitro. As shown in Fig. 3B and 3C, soluble iNOS exhibited robust NO-generating activity. In contrast, aggregated iNOS from either radicicol or GA-treated cells largely lost its catalytic function. Thus, these in vitro and cell culture studies collectively showed that aggregation resulted in iNOS inactivation.
Figure 3.
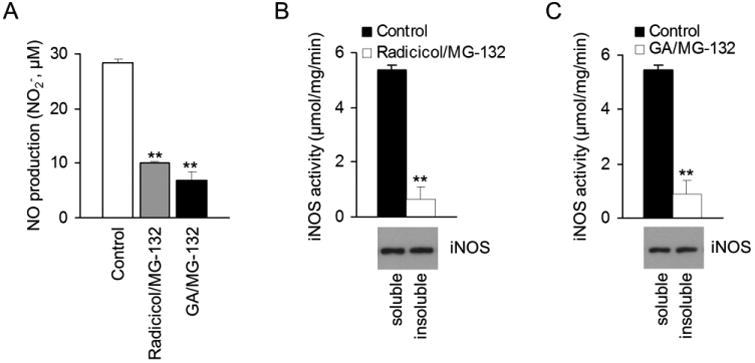
Aggregation causes iNOS to lose catalytic function. (A) NO production from iNOS expressed in RAW 264.7 cells in the absence and presence of Hsp90 and proteasome inhibitors. Data are mean ± SE. ** P < 0.01 vs. control, n=3. (B, C) Under Hsp90 inhibition, the catalytic activity of iNOS was largely lost after aggregation. ** P < 0.01 vs. control, n = 4.
3.4 Hsp90 inhibition induces Hsp90-iNOS dissociation
We then sought to gain further understandings on the process of iNOS aggregation after Hsp90 inhibition. Both radicicol and GA target the ATPase pocket of Hsp90 (20, 24-26). It is unclear whether or not radicicol or GA disrupts Hsp90-iNOS association in the process of iNOS aggregation. To address this question, we isolated iNOS-Hsp90 complexes from cells, exposed them to Hsp90 inhibitors, and monitored the alterations of the Hsp90 bound with iNOS as well as those released into the supernatants. As shown in Fig. 4A and 4B, both radicicol and GA treatment caused a time-dependent decreases in the levels of Hsp90 bound with iNOS. Inversely proportional to the decreases in iNOS-bound Hsp90, the levels of free Hsp90 released into the supernatants were increased. These data demonstrated that Hsp90 inhibition caused iNOS-Hsp90 dissociation. It was noted that the Hsp90-iNOS dissociation occurred before iNOS aggregation, suggesting that Hsp90 dissociation might be required for iNOS aggregation.
Figure 4.
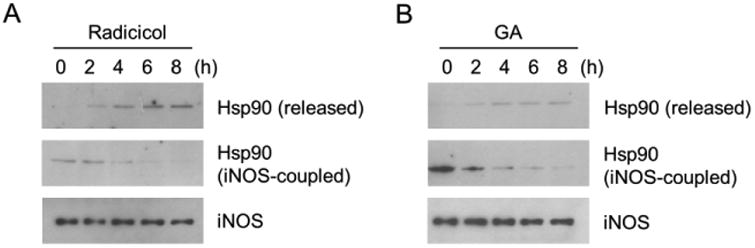
Hsp90 inhibition causes Hsp90 dissociation from iNOS. (A) Radicicol decreased the levels of Hsp90 that bound with iNOS. Meanwhile, the amounts of free Hsp90 released from Hsp90-iNOS complexes were increased following radicicol treatment. (B) Hsp90 inhibition with GA also rendered Hsp90 dissociation from iNOS. Representative data were shown from three independent experiments.
3.5 CHIP is dispensable for the clearance of iNOS aggregates
Previous studies reported that iNOS can be degraded by the UPS and the carboxyl terminus of Hsc70 interacting protein (CHIP) has been implicated in iNOS protein turnover (16, 27-29). CHIP is an Hsp70/Hsp90-dependent E3 ligase. Hsp90 is critical for CHIP to ubiquitinate its client proteins (30, 31). The fact that Hsp90 inhibition promoted iNOS degradation via UPS suggested that other E3 ligases likely accounted for the proteasomal clearance of iNOS aggregates under Hsp90 inhibition condition. To further rule out the involvement of CHIP in the clearance of iNOS aggregates in Hsp90-inhibited cells, we expressed iNOS into control and CHIP knockdown HEK293 cells and exposed them to Hsp90 inhibitor. To focus on the degradation process of iNOS aggregates, we extracted intracellular iNOS with strong detergent-containing buffer (1% SDS) and monitored the changes of total iNOS levels in this experiment and those hereafter. As shown in Fig. 5A, Hsp90 inhibition caused iNOS degradation in both control and CHIP knockdown cells. The time courses of iNOS depletion in control and CHIP knockdown cells were indistinguishable (Fig. 5B). These data unambiguously demonstrated that CHIP was not required in the degradation of iNOS aggregates in Hsp90-inhibited cells.
Figure 5.
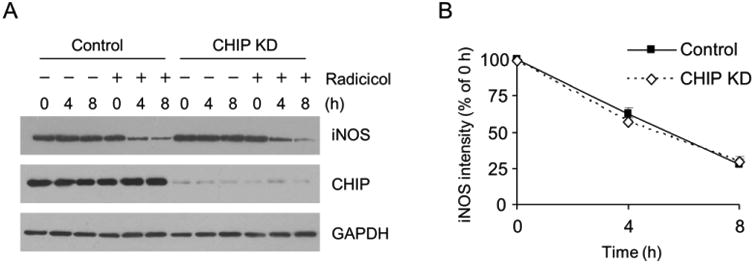
CHIP depletion has no effect on iNOS degradation in Hsp90-inhibited cells. (A) Knockdown of CHIP in HEK293 cells failed to prevent the iNOS depletion in radicicol-treated cells. (B) Quantitative analyses of the effects of CHIP knockdown on iNOS degradation in Hsp90-inhibited cells. Data are mean ± SE, n = 4.
3.6 SPSB2 is essential for UPS clearance of iNOS aggregates
Recent studies reported that the SPRY domain-containing SOCS box protein 2 (SPSB2), an E3 ligase-recruiting protein, participates in iNOS ubiquitination and turnover in normal conditions (32, 33). We thus explored if SPSB2 played roles in the proteasomal clearance of iNOS aggregates in Hsp90-inhibited cells. As shown in Fig 6A and 6B, SPSB2 knockdown largely prevented the degradation of iNOS aggregates, indicating that SPSB2 mediated the ubiquitination and degradation of aggregated iNOS. Indeed, we isolated iNOS aggregates from Hsp90-inhibited cells and found their ubiquitination levels were markedly reduced in SPSB2 knockdown cells (Fig. 6C). These results revealed that the SPSB2 was required for ubiquitination and degradation of iNOS aggregates.
Figure 6.
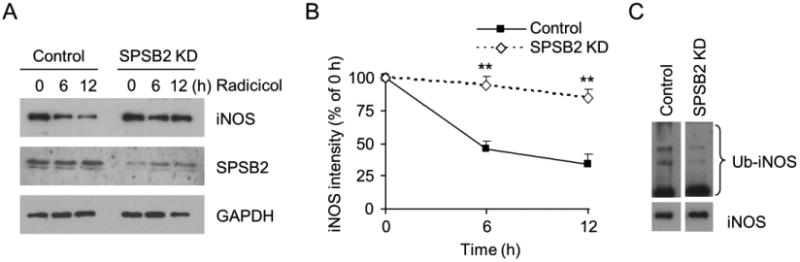
SPSB2 is essential for the ubiquitination and degradation of iNOS aggregates following Hsp90 inhibition. (A) SPSB2 knockdown prevented the loss of iNOS in radicicol-treated cells. (B) Quantitative analyses of the effects of SPSB2 knockdown on iNOS degradation after Hsp90 inhibition. Data are mean ± SE. ** P < 0.01 vs. control, n = 4. (C) Ubiquitination levels of iNOS aggregates were markedly reduced by SPSB2 knockdown in RAW 264.7 cells. Representative images are shown from triplicate experiments.
3.7 Deleting the SPSB2-binding domain on iNOS prevents the clearance of iNOS aggregates
iNOS is known to interact with SPSB2 through a DINNN containing domain (amino acids 23-27) in its N-terminal region (32, 33). To demonstrate the necessity of the interaction with SPSB2 in the clearance of iNOS aggregates, we constructed a SPSB2-binding domain deletion iNOS (iNOS50-1144) and expressed it into HEK293 cells. As shown in Fig. 7A, the removal of the SPSB2-binding domain had no effect on the catalytic function of iNOS as evidenced by the similar NO productions in the cells expressing iNOS or iNOS50-1144. These data suggested that there was no major change in iNOS folding or conformation after removing the SPSB2 binding domain. However, deleting the SPSB2-binding domain largely prevented the clearance of iNOS aggregates (Fig. 7B and 7C). This indicated that the interaction with SPSB2 was required for proteasomal clearance of iNOS aggregates. To ensure these findings were not restricted to one particular type of cells, we conducted similar experiments in COS-7 cells. Again, deleting the SPSB2-binding domain abrogated the clearance of iNOS aggregates after Hsp90 inhibition (Fig. 7D and 7E). These results demonstrated the essentiality of SPSB2-iNOS interaction in the UPS clearance of iNOS aggregates in Hsp90-inhibited cells.
Figure 7.
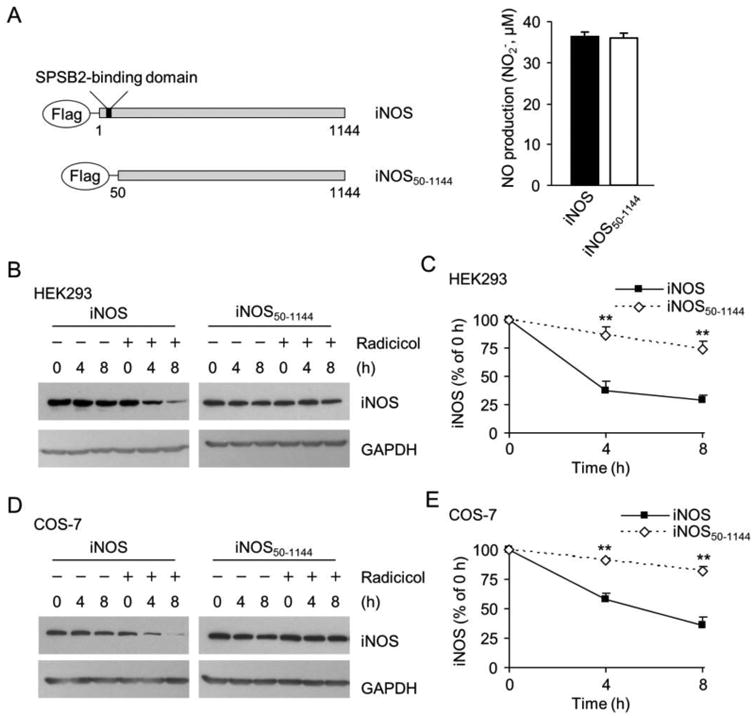
Deletion of SPSB2-interacting domain on iNOS prevents iNOS degradation in Hsp90-inhibited cells. (A) A schematic representation of wild-type iNOS and SPSB2-binding domain deficient mutant (iNOS50-1144). Wild-type and iNOS50-1144 exhibited the same NO-generating activity. Data are mean ± SE, n = 4. (B) Hsp90 inhibition-induced iNOS degradation in HEK 293 cells was significantly decelerated by deleting the SPSB2-binding domain on iNOS. (C) Quantitative analyses of the effects of deleting SPSB2-interacting domain from iNOS on its degradation in Hsp90-inhibited HEK293 cells. **P < 0.01 vs. control, n = 3. (D) Deletion of the SPSB2-binding domain also prevented the degradation of iNOS in COS-7 cells after Hsp90 inhibition (Radicicol, 20 μM). (E) Quantitative analyses of the turnover rate of wild-type and iNOS50-1144 in Hsp90-inhibited COS-7 cells. **P < 0.01 vs. control, n = 3.
4. Discussion
The key finding of this study is that Hsp90 is essential for maintaining the protein stability of iNOS. Unlike eNOS or nNOS whose activation is largely driven by intracellular Ca2+ transient, NO production from iNOS is dependent on the levels of iNOS proteins. Historically, intracellular iNOS levels have been thought to be determined by the magnitude of its gene transcription. Thus, most studies in this field focused on deciphering the mechanisms of iNOS gene transactivation (34-36). Little attention was paid to the potential roles of changed protein stability or turnover in iNOS biology. Recently, studies on the changes in iNOS protein stability and the impacts of these changes on iNOS function began to emerge (16-18, 21, 27). The findings in current study underscore the importance of protein aggregation in affecting iNOS function, and identify Hsp90 as a critical component in controlling iNOS protein stability. Hsp90 inhibition causes iNOS aggregation and deactivation. NO production in the macrophages exposed to inflammatory stimuli is dependent on Hsp90. Under Hsp90 inhibition, NO production is largely diminished because of iNOS aggregation. Hence in addition to the previously reported roles in regulating iNOS function and expression (14, 15), Hsp90 is also essential in keeping iNOS proteins stable inside cells.
The details of iNOS aggregation following Hsp90 inhibition remain to be determined. Nevertheless, our data suggest that the aggregation process may begin with Hsp90 dissociation from iNOS. A comparison of the time-courses of Hsp90 dissociation and iNOS aggregation showed that Hsp90-iNOS dissociation occurred prior to iNOS aggregation. This suggests a model that Hsp90 inhibition cause Hsp90-iNOS dissociation and Hsp90-uncoupled iNOS subsequently forms aggregates (Fig. 8). Further studies are needed to verify such a model and decipher why Hsp90-uncoupled iNOS is prone to aggregation. As a chaperon protein, Hsp90 is known to play crucial roles in maintaining the conformation of its client proteins. It is conceivable that lacking the chaperon of Hsp90 may render iNOS into an unstable conformation that favors protein aggregation.
Figure 8.
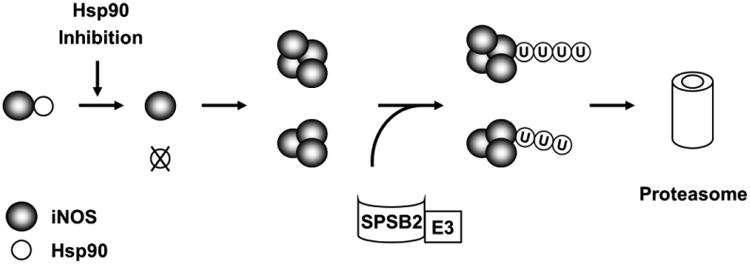
Schematic diagram depicting iNOS aggregation and degradation in Hsp90-inhibited cells. Hsp90 inhibition triggers the dissociation of Hsp90 from iNOS and subsequent iNOS aggregation. SPSB2 interacts with iNOS and recruits E3 ligase to ubiquitinate aggregated iNOS. Ubiquitinated iNOS aggregates are degraded by the proteasome.
Another finding in this study is the elucidation on how iNOS aggregates are coped with in cells. Our results show that iNOS aggregates are cleared via the UPS. Aggregation not only causes proteins to lose their functionalities, aggregated proteins often inflame additional harm to cells. In fact, abnormal accumulations of protein aggregates are a mechanism of many diseases. Whether or not iNOS aggregates further cause detrimental consequences remains to be seen. Nonetheless, cells appear to take prompt actions to eliminate iNOS aggregates. Thus, when UPS function is normal, iNOS aggregation can be effectively dealt with except for the cells to lose NO-generating capability. However, when UPS function is hampered, as occurred in various diseases, iNOS aggregates will retain in cells. Not only these aggregated iNOS stops producing NO, they may also pose a new burden to cells. It will be interesting to investigate whether or not iNOS aggregates induce detrimental effects in disease conditions in the future.
Prior to proteasomal degradation, target proteins must be ubiquitinated and this is mainly accomplished by a cascade of reactions catalyzed by a group of enzymes including E1, E2, and E3 (37). Among them, the E3 ligase is responsible for target specificity and thus plays determining role in directing proteins to the proteasome for degradation. The E3 ligase CHIP was previously reported to facilitate iNOS ubiquitination and proteasomal degradation in transfected cells (27, 28). A recent study, however, shows that CHIP was not responsible for iNOS ubiquitination and lifetime in macrophages (38). Interestingly, CHIP is an Hsp90 dependent enzyme. Our findings that Hsp90 inhibition promotes iNOS degradation suggest that CHIP is dispensable for proteasomal clearance of iNOS. Indeed, CHIP knockdown has no effect on iNOS degradation in Hsp90-inhibited cells. These results do not exclude the possible role of CHIP in iNOS turnover under certain circumstances. But they demonstrate that when CHIP is inhibited or absent, there are other E3 ligases that account for iNOS ubiquitination and degradation.
Our studies show that SPSB2-associated E3 ligase is responsible for iNOS ubiquitination and degradation in Hsp90-inhibited cells. SPSB2 belongs to a group of SOCS box-containing proteins that regulate other proteins via a SOCS box associated E3 ligase (39, 40). Nicholson and colleagues reported that SPSB2 interacts with iNOS via an iNOS N-terminal DINNN motif. SPSB2 couples with the elongin B/C-cullin5-SPRY protein complex to form an E3 ligase to ubiquitinate iNOS (32, 33). This report addressed the role of SPSB2 in controlling the lifetime of normal iNOS. Important question remains regarding the role of SPSB2 in the turnover of other forms of iNOS proteins. We now show that SPSB2 is also essential for proteasomal clearance of aggregated iNOS. Knockdown of SPSB2 markedly reduced the ubiquitination of iNOS aggregates in Hsp90-inhibted cells. SPSB2 knockdown or deleting the SPSB2-interating domain in iNOS prevented the degradation of iNOS aggregates. Together with previous reports (32, 33, 38), our findings highlight the crucial role of SPSB2 in controlling iNOS ubiquitination and turnover in both physiological and pathophysiological settings (Fig. 8).
Aggregation and proteasomal clearance are efficient means to remove active iNOS in cells. As iNOS levels determine the NO formation quantity; in principle, NO production could be modulated through Hsp90 control of iNOS stability and turnover. Known as a molecular chaperon, Hsp90 is often thought to function as a supporter rather than an initiator in cell signaling. For example, Hsp90 is known to serve as a structural scaffold to bring kinases and their substrates together. This process may not need a change of Hsp90 function. However, recent progresses demonstrate that Hsp90 is able to actively participate in molecular regulation. Hsp90 function can be modulated by protein phosphorylation and protein-protein interactions (41-43). Therefore, down-regulation of Hsp90 function would be expected to lead to iNOS aggregation and removal. This could serve as a counter mechanism to gene expression so that the level of iNOS proteins can be precisely tuned to accommodate the biological need. It will be interesting to study if intrinsic pathways exist to modulate NO production in host defense via Hsp90 control of iNOS protein stability. Exploring this issue may give rise to novel mechanisms in iNOS regulation.
In summary, in the third report of a serial studies on Hsp90 regulation of iNOS, we show that the interaction with Hsp90 is crucial for iNOS protein stability. Together with previous reports (14, 15), we have demonstrated a comprehensive role of Hsp90 in modulating iNOS gene transcription, catalytic function, and protein stability. Lack of Hsp90 interaction results in iNOS aggregation and loss of catalytic function. iNOS aggregates are cleared by the UPS and this is mediated by SPSB2. The identified Hsp90 control of iNOS protein stability and turnover suggest a novel approach to modulate NO production from iNOS in cells.
Highlights.
Hsp90 is essential for iNOS protein stability.
Loss of Hsp90 function renders iNOS aggregation.
iNOS aggregates are cleared by UPS
Acknowledgments
We thank the members of the Xia lab for reading our manuscript. This work was supported by National Institutes of Health Grant HL86965, the National Program on Key Basic Research Project (973 Program, 2014CB542400), the National Natural Science foundation of China grants (81170112, 81270210), an American Diabetes Association grant (1-15-BS-200) and an OSU faculty development fund.
Abbreviations
- Hsp90
heat shock protein 90
- NO
nitric oxide
- iNOS
inducible NO synthase
- SPSB2
SPRY domain-containing SOCS box protein 2
- CHIP
carboxyl terminus of Hsc70 interacting protein
- UPS
ubiquitin-proteasome system
- GA
geldanamycin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 2.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Kojda G, Kottenberg K. Regulation of basal myocardial function by NO. Cardiovasc Res. 1999;41:514–523. doi: 10.1016/s0008-6363(98)00314-9. [DOI] [PubMed] [Google Scholar]
- 4.Lowenstein CJ, Snyder SH. Nitric oxide, a novel biologic messenger. Cell. 1992;70:705–707. doi: 10.1016/0092-8674(92)90301-r. [DOI] [PubMed] [Google Scholar]
- 5.Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298(Pt 2):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 7.Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 8.Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 9.Masters BS, McMillan K, Sheta EA, Nishimura JS, Roman LJ, Martasek P. Neuronal nitric oxide synthase, a modular enzyme formed by convergent evolution: structure studies of a cysteine thiolate-liganded heme protein that hydroxylates L-arginine to produce NO. as a cellular signal. FASEB J. 1996;10:552–558. doi: 10.1096/fasebj.10.5.8621055. [DOI] [PubMed] [Google Scholar]
- 10.Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 12.Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Nathan C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J Exp Med. 1992;176:599–604. doi: 10.1084/jem.176.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marletta MA. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida M, Xia Y. Heat shock protein 90 as an endogenous protein enhancer of inducible nitric-oxide synthase. J Biol Chem. 2003;278:36953–36958. doi: 10.1074/jbc.M305214200. [DOI] [PubMed] [Google Scholar]
- 15.Luo S, Wang T, Qin H, Lei H, Xia Y. Obligatory role of heat shock protein 90 in iNOS induction. Am J Physiol Cell Physiol. 2011;301:C227–33. doi: 10.1152/ajpcell.00493.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musial A, Eissa NT. Inducible nitric-oxide synthase is regulated by the proteasome degradation pathway. J Biol Chem. 2001;276:24268–24273. doi: 10.1074/jbc.M100725200. [DOI] [PubMed] [Google Scholar]
- 17.Kolodziejska KE, Burns AR, Moore RH, Stenoien DL, Eissa NT. Regulation of inducible nitric oxide synthase by aggresome formation. Proc Natl Acad Sci USA. 2005;102:4854–4859. doi: 10.1073/pnas.0500485102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolodziejski PJ, Koo JS, Eissa NT. Regulation of inducible nitric oxide synthase by rapid cellular turnover and cotranslational down-regulation by dimerization inhibitors. Proc Natl Acad Sci USA. 2004;101:18141–18146. doi: 10.1073/pnas.0406711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Q, Xia Y. Proteasome inhibition down-regulates endothelial nitric-oxide synthase phosphorylation and function. J Biol Chem. 2006;281:21652–21659. doi: 10.1074/jbc.M602105200. [DOI] [PubMed] [Google Scholar]
- 20.Blagosklonny MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002;16:455–462. doi: 10.1038/sj.leu.2402415. [DOI] [PubMed] [Google Scholar]
- 21.Pandit L, Kolodziejska KE, Zeng S, Eissa NT. The physiologic aggresome mediates cellular inactivation of iNOS. Proc Natl Acad Sci USA. 2009;106:1211–1215. doi: 10.1073/pnas.0810968106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- 23.Magzoub M, Miranker AD. Protein aggregation: p53 succumbs to peer pressure. Nat Chem Biol. 2011;7:248–249. doi: 10.1038/nchembio.569. [DOI] [PubMed] [Google Scholar]
- 24.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 25.Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, Neckers LM, Toft DO. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 26.Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 27.Sha Y, Pandit L, Zeng S, Eissa NT. A critical role for CHIP in the aggresome pathway. Mol Cell Biol. 2009;29:116–128. doi: 10.1128/MCB.00829-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, Kong X, Fu J, Xu Y, Fang S, Hua P, Luo L, Yin Z. CHIP facilitates ubiquitination of inducible nitric oxide synthase and promotes its proteasomal degradation. Cell Immunol. 2009;258:38–43. doi: 10.1016/j.cellimm.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Kolodziejski PJ, Musial A, Koo JS, Eissa NT. Ubiquitination of inducible nitric oxide synthase is required for its degradation. Proc Natl Acad Sci USA. 2002;99:12315–12320. doi: 10.1073/pnas.192345199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int J Biochem Cell Biol. 2003;35:572–578. doi: 10.1016/s1357-2725(02)00394-1. [DOI] [PubMed] [Google Scholar]
- 31.Jiang J, Cyr D, Babbitt RW, Sessa WC, Patterson C. Chaperone-dependent regulation of endothelial nitric-oxide synthase intracellular trafficking by the co-chaperone/ubiquitin ligase CHIP. J Biol Chem. 2003;278:49332–49341. doi: 10.1074/jbc.M304738200. [DOI] [PubMed] [Google Scholar]
- 32.Kuang Z, Lewis RS, Curtis JM, Zhan Y, Saunders BM, Babon JJ, Kolesnik TB, Low A, Masters SL, Willson TA, Kedzierski L, Yao S, Handman E, Norton RS, Nicholson SE. The SPRY domain-containing SOCS box protein SPSB2 targets iNOS for proteasomal degradation. J Cell Biol. 2010;190:129–141. doi: 10.1083/jcb.200912087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishiya T, Matsumoto K, Maekawa S, Kajita E, Horinouchi T, Fujimuro M, Ogasawara K, Uehara T, Miwa S. Regulation of inducible nitric-oxide synthase by the SPRY domain- and SOCS box-containing proteins. J Biol Chem. 2011;286:9009–9019. doi: 10.1074/jbc.M110.190678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nathan C, Xie QW. Regulation of biosynthesis of nitric oxide. J Biol Chem. 1994;269:13725–13728. [PubMed] [Google Scholar]
- 35.Xie Q, Nathan C. The high-output nitric oxide pathway: role and regulation. J Leukoc Biol. 1994;56:576–582. doi: 10.1002/jlb.56.5.576. [DOI] [PubMed] [Google Scholar]
- 36.Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 37.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto K, Nishiya T, Maekawa S, Horinouchi T, Ogasawara K, Uehara T, Miwa S. The ECS(SPSB) E3 ubiquitin ligase is the master regulator of the lifetime of inducible nitric-oxide synthase. Biochem Biophys Res Commun. 2011;409:46–51. doi: 10.1016/j.bbrc.2011.04.103. [DOI] [PubMed] [Google Scholar]
- 39.Kamura T, Sato S, Haque D, Liu L, Kaelin WG, Jr, Conaway RC, Conaway JW. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang JG, Metcalf D, Rakar S, Asimakis M, Greenhalgh CJ, Willson TA, Starr R, Nicholson SE, Carter W, Alexander WS, Hilton DJ, Nicola NA. The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc Natl Acad Sci USA. 2001;98:13261–13265. doi: 10.1073/pnas.231486498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao YG, Gilmore R, Leone G, Coffey MC, Weber B, Lee PW. Hsp90 phosphorylation is linked to its chaperoning function. Assembly of the reovirus cell attachment protein. J Biol Chem. 2001;276:32822–32827. doi: 10.1074/jbc.M105562200. [DOI] [PubMed] [Google Scholar]
- 42.Zuehlke AD, Johnson JL. Chaperoning the Chaperone: A Role for the Co-chaperone Cpr7 in Modulating Hsp90 Function in Saccharomyces cerevisiae. Genetics. 2012;191:805–814. doi: 10.1534/genetics.112.140319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mollapour M, Tsutsumi S, Truman AW, Xu W, Vaughan CK, Beebe K, Konstantinova A, Vourganti S, Panaretou B, Piper PW, Trepel JB, Prodromou C, Pearl LH, Neckers L. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol Cell. 2011;41:672–681. doi: 10.1016/j.molcel.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]