

Abstract
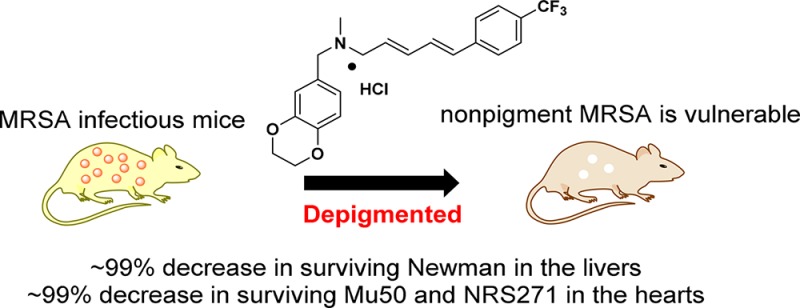
Diapophytoene desaturase (CrtN) is a potential novel target for intervening in the biosynthesis of the virulence factor staphyloxanthin. In this study, 38 1,4-benzodioxan-derived CrtN inhibitors were designed and synthesized to overwhelm the defects of leading compound 4a. Derivative 47 displayed superior pigment inhibitory activity, better hERG inhibitory properties and water solubility, and significantly sensitized MRSA strains to immune clearance in vitro. Notably, 47 displayed excellent efficacy against pigmented S. aureus Newman, Mu50 (vancomycin-intermediate MRSA, VISA), and NRS271 (linezolid-resistant MRSA, LRSA) comparable to that of linezolid and vancomycin in vivo.
Keywords: Virulence factor, pigment, multidrug resistant MRSA, CrtN inhibitor
Staphylococcus aureus (S. aureus), a type of Gram-positive bacteria, causes a series of infections, including skin and soft tissue infections and fatal systemic infections.1,2 Methicillin-resistant S. aureus (MRSA) has been recognized as a major pathogen that mutates rapidly under exposure to antibiotics.3−6 In recent years, more multidrug resistant MRSA strains have been identified, including linezolid-resistant MRSA (LRSA) and vancomycin-intermediate and -resistant MRSA (VISA and VRSA),7 which have almost destroyed the entire “antibio-defense”.8 The World Health Organization (WHO) published its first ever list of antibiotic-resistant “priority pathogens” in 2016, and MRSA, VISA, and VRSA were graded and classified as high priority.9 We urgently need novel strategies against multidrug resistant S. aureus in this postantibiotic era.10
Virulence factors are products of the pathogen, which affect the host–pathogen relationship by damaging the host immune system.11,12 The golden pigment staphyloxanthin (STX) is a carotenoid virulence factor of S. aureus and defends the strains against reactive oxygen substances (ROS) produced by neutrophils.13,14 Therefore, it is a viable avenue to treat MRSA infections by inhibiting STX biosynthesis. The STX biosynthesis genes are organized in the crtOPQMN operon, and biosynthesis begins with the condensation of farnesyl diphosphate catalyzed by dehydrosqualene synthase (CrtM) and a downstream termination region of diapophytoene desaturases (CrtN).15 In 2008, Oldfield and colleagues first reported that cholesterol biosynthesis inhibitor BPH-652 (1, Figure 1) had an inhibitory effect on pigment synthesis by targeting CtrM.16−18 Subsequently, our research group revealed that Naftifine hydrochloride (NTF, 2, Figure 1), an FDA-approved antifungal agent, could block the STX biosynthesis pathway by targeting CrtN, revealing a novel strategy for antivirulence treatments against MRSA infections.19 On the basis of the results above, we further designed and synthesized a series of novel benzofuran and benzocycloalkane CrtN inhibitors, from which the efficient derivatives 3 and 4 were obtained (Figure 1).20,21 Moreover, analogue 4a displayed excellent potency in vitro (pigment inhibition: IC50 = 1.9 nM, Figure 1) but was still limited by its strong inhibition of human Ether-á-go-go Related Gene (hERG), high required dosage, and moderate water solubility in subsequent investigations.21 Therefore, in this study, a novel compound was designed and synthesized to overcome the defects of lead compound 4a. In addition, variable administrations and dosages were introduced for an actual evaluation of the efficiency of our new compounds against multidrug resistant S. aureus in vivo.
Figure 1.
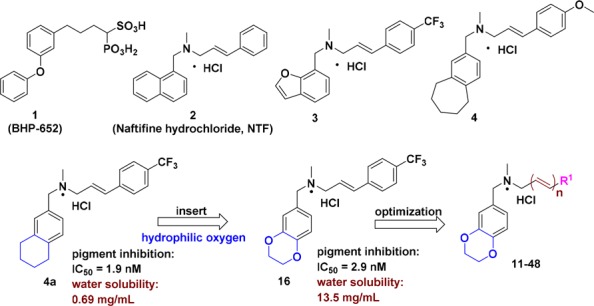
Structures of representative pigment inhibitors reported in the literature and those derived from lead compound 4a.
Our previous study had demonstrated that the strategy of inserting a hydrophilic oxygen into the cycloalkane of lead compound 4a was a feasible means of improving water solubility.22 As an extension to our previous work, we speculated that adding more oxygen atoms into the skeleton of 4a was a rational approach to relieve the stress of solubility brought by possible lipophilic substituents.22 1,4-Benzodioxan derivative 16 was synthesized in accordance with these hypotheses, and it had superior solubility in contrast to 4a (13.5 vs 0.69 mg/mL), along with the maintenance of pigment inhibitory activity (IC50 = 1.9 nM by 4a vs IC50 = 2.9 nM by 16). Building on these data, we next aimed at improving the pharmacologic activity and hERG inhibition properties by rational molecular modification. In addition, our previous work had revealed that the N-methyl group and unsubstituted alkenyl linker were critical for improving pigment inhibitory activity and hERG inhibition properties. Hence, systematic modifications in this study were investigated directly toward diversifying substituents with R1 and inserting more olefins into the linker.
Next, 38 novel 1,4-benzodioxan derivatives 11–48 were synthesized and assayed for pigment inhibitory activities against S. aureus Newman. The results are outlined in Table 1. First, the replacement of phenyl with methyl, cycloalkyls, heteroaryls, or naphthyl groups reduced the pigment inhibitory activity. The results further confirmed that there was no significant relationship between the electron effect and activity, as previously reported.21 Unexpectedly, substitutions at the para-position showed more potency than those at the ortho- or meta-position (12–23 vs 24–29 vs 30–35). Moreover, the suitable bulk of substituents could significantly improve inhibitory activities (12 vs 13 vs 14 and 15 vs 16 vs 17). Among these derivatives, both 15 and 16 displayed excellent pigment inhibitory activities (IC50 = 2.9 ± 0.1 nM, IC50 = 3.2 ± 0.2 nM, respectively), but derivative 16 was selected for further optimization due to its better solubility (∼2-fold higher than 15, Table 2). Next, more unsubstituted multiconjugated polyene groups were introduced into the structure of 16 to yield derivatives 47 and 48, which displayed comparable activities to 16 (IC50 = 2.2 nM by 47 and IC50 = 1.6 nM by 48).
Table 1. Chemical Structures of Derivatives 11–48 and Their Pigment Inhibitory Activities against S. aureus Newman.
| compd | R1 | n | IC50 (nM)a | compd | R1 | n | IC50 (nM)a |
|---|---|---|---|---|---|---|---|
| 11 | phenyl | 1 | 370.9 ± 35.4 | 30 | 3-fluorophenyl | 1 | >1000 |
| 12 | 4-fluorophenyl | 1 | 20.1 ± 0.9 | 31 | 3-chlorophenyl | 1 | 111.6 ± 4.1 |
| 13 | 4-chlorophenyl | 1 | 6.7 ± 0.3 | 32 | 3-methylphenyl | 1 | 43.4 ± 4.8 |
| 14 | 4-bromophenyl | 1 | 8.2 ± 0.3 | 33 | 3-trifluorophenyl | 1 | >1000 |
| 15 | 4-methylphenyl | 1 | 3.2 ± 0.2 | 34 | 3-nitrophenyl | 1 | >1000 |
| 16 | 4-trifluorophenyl | 1 | 2.9 ± 0.1 | 35 | 3-methoxylphenyl | 1 | >1000 |
| 17 | 4-t-butylphenyl | 1 | 156.0 ± 9.3 | 36 | 2,4-dichlorophenyl | 1 | 15.0 ± 3.7 |
| 18 | 4-methoxylphenyl | 1 | 48.6 ± 7.4 | 37 | 2-fluoro-4-trifluorophenyl | 1 | 8.8 ± 0.1 |
| 19 | 4-ethoxylphenyl | 1 | 8.2 ± 0.8 | 38 | 3-fluoro-4-trifluorophenyl | 1 | 13.7 ± 0.1 |
| 20 | 4-nitrophenyl | 1 | 18.9 ± 0.5 | 39 | 2-fluoro-4-methoxylphenyl | 1 | 43.5 ± 1.2 |
| 21 | 4-cyanophenyl | 1 | 19.2 ± 8.0 | 40 | 3-fluoro-4-methoxylphenyl | 1 | 57.9 ± 3.0 |
| 22 | 4-methyl formatephenyl | 1 | 6.5 ± 0.3 | 41 | methyl | 1 | >1000 |
| 23 | 4-biphenyl | 1 | 4.1 ± 0.2 | 42 | cyclopentyl | 1 | 320.8 ± 4.7 |
| 24 | 2-fluorophenyl | 1 | >1000 | 43 | cyclohexyl | 1 | >1000 |
| 25 | 2-chlorophenyl | 1 | 90.3 ± 25.7 | 44 | 1-furyl | 1 | 227.7 ± 4.5 |
| 26 | 2-methylphenyl | 1 | >1000 | 45 | 1-thienyl | 1 | 220.6 ± 8.3 |
| 27 | 2-trifluorophenyl | 1 | >1000 | 46 | 1-naphthyl | 1 | >1000 |
| 28 | 2-nitrophenyl | 1 | >1000 | 47 | 4-trifluorophenyl | 2 | 2.2 ± 0.1 |
| 29 | 2-methoxylphenyl | 1 | >1000 | 48 | 4-trifluorophenyl | 3 | 1.6 ± 0.2 |
| 4 | 4.1 | 4a | 1.9 |
Values given are the IC50 values for pigment inhibition in S. aureus Newman. Values are reported as the average ± SD.
Table 2. Enzyme (CrtN, IC50), Pigment (Mu50, NRS271, USA400 MW2, and USA300 LAC, IC50), hERG, and Water Solubility Results of Representative Derivatives.
| compd | CrtN IC50 (nM)a | Mu50 IC50 (nM)b | NRS271 IC50 (nM)b | USA400 MW2 IC50 (nM)b | USA300 LAC IC50 (nM)b | hERG IC50 (μM)c | water solubility (mg/mL)d |
|---|---|---|---|---|---|---|---|
| 16 | 340.7 ± 65.5 | 1.2 ± 0.3 | 1.1 ± 0.2 | 6.5 ± 0.3 | 5.9 ± 0.2 | 3.9 | 13.5 |
| 47 | 270.4 ± 43.8 | <0.1 | 0.6 ± 0.2 | 6.5 ± 0.1 | 5.5 ± 0.5 | 12.1 | 7.9 |
| 48 | 258.4 ± 57.9 | 1.2 ± 0.5 | 0.4 ± 0.3 | 5.3 ± 0.1 | 3.7 ± 0.2 | 8.7 | 3.3 |
| 4 | 320 | 0.5 | 7.1 | 7.7 | 3.2 | 4.2 | |
| 4a | 1.4 | 0.69 | |||||
| 15 | 4.6 | 6.5 |
Values given are the IC50 values for CrtN enzyme inhibition in S. aureus Newman. The values are reported as the average ± SD, in nM.
Values given are the IC50 values for pigment inhibition in Mu50, NRS271, USA400 MW2, and USA300 LAC, in nM.
Values given are the IC50 values for hERG inhibition, in μM.
Values given are the solubility in water, in mg/mL.
Further in vitro investigation results are shown in Table 2. First, three potency derivatives, 16, 47, and 48 were, bioassayed for CrtN enzymatic inhibitory activity, and all of the selected derivatives had hundred-nanomolar inhibitory activity ranges, which were comparable with 4. Next, two multidrug resistant MRSA strains (Mu50 as VISA and NRS271 as LRSA)23,24 and two community-acquired MRSA strains (USA300 LAC and USA400 MW)25,26 were introduced to test the pigment inhibitory activities of selected derivatives. All of the compounds displayed potent inhibitory activities in the single-nanomolar range against the four MRSAs above (IC50 = 0.1–6.5 nM, Table 2). In this study, both hERG profiles and solubility were important properties to investigate with regards to the drugability of compounds. Regrettably, we found conflicts in these derivatives relating to their solubility and hERG inhibition, as shown in Table 2. Appropriate olefins significantly ameliorated the hERG inhibitory activities of 47 and 48, but decreased their solubility. In light of the balance between hERG safety profiles and solubility, derivative 47 was selected to further survey, due to its significant improvement of hERG inhibition (IC50 = 12.1 μM by 47 vs 1.4 μM by 4a, ∼8-fold increase) and good water solubility (7.9 mg/mL). In contrast with traditional antibiotics, derivative 47, which originated from antifungal agent 2, not only did not affect the growth of MRSAs at 0.2 mM (Newman and three MRSA strains, Figure S1, Supporting Information) but also lost its antifungal capacity (Table S1, Supporting Information). Furthermore, the identification of CrtN targeting demonstrated that derivative 47 indeed inhibited the function of CrtN (Figure S2, Supporting Information).
STX could protect S. aureus from the oxidative stress by ROS, and nonpigmented MRSAs were vulnerable to immune clearance.19−22 Based on this, a hydrogen peroxide (H2O2) assay and human whole blood killing assay were used to imitate ROS in vitro to verify the efficiency of our compounds. As shown in Figure 2, three groups, including mock-treated (yellow), 47-treated (blue), and N-acetylcysteine (NAC, known as an antioxidant substance)-treated (red), were incubated with S. aureus Newman or the other three MRSAs (Mu50, USA300 LAC, and USA400 MW). In the H2O2 killing assay, the survival of all 47-treated groups was obviously lower than that of the mock-treated and NAC-treated groups, which demonstrated that the depigmented MRSAs were vulnerable to H2O2 after treatment with our compound. In addition, antioxidant NAC-treated groups gave the worst performance due to counteracting H2O2. Similarly, in the whole blood killing model, 47-treated S. aureus groups (in blue) were more susceptible to being killed by blood (survival, <0.5% in general).
Figure 2.
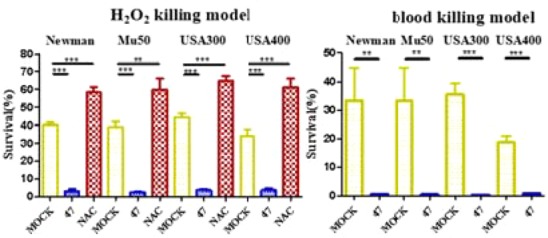
Effects of derivative 47 on susceptibility of S. aureus Newman, Mu50, USA300 LAC, and USA400 MW2 to hydrogen peroxide and human whole blood killing assays in vitro.
The results above indicated that candidate 47 could sensitize MRSAs to immune clearance in vitro. Next, murine abscess formation models were established to assess the bioactivity of 47 in vivo. Candidate 47 with two administrations timings (normal treatment or pretreatment) at two dosages (0.4 mg b.i.d. or 0.1 mg b.i.d.) were used to imitate authentic infections, along with vancomycin (VAN) and linezolid (LZD) as positive controls. Bacterial survival was then measured in mice hearts and livers. First, as shown in Figure 3, in contrast with the nontreatment groups by a 6.34 log10 CFU in hearts or by a 6.41 log10 CFU in livers, 47-treatment groups displayed significant activities against Newman strains (at least a 0.96 log10 CFU reduction in the hearts, an 89.01% decrease in surviving bacteria, and 0.87 log10 CFU reduction in the livers, an 86.70% decrease in surviving bacteria). Although 47-treatment appeared less efficacious than the two positive controls in the hearts, it was still comparable with the positive controls in the livers. In particular, the dosage of 0.1 mg/bid exhibited an excellent performance in the livers of the normal treatment groups (3.31 log10 CFU, a ∼99% decrease in surviving bacteria), even better than positive controls.
Figure 3.
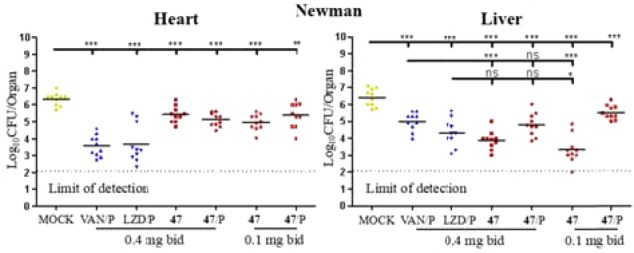
Effects of derivative 47 (in red) on S. aureus bacteria survival in the hearts and livers of mice (n = 10 for each group) challenged with 2.2 × 107 CFU of Newman. P = pretreatment, drugs or compounds were intraperitoneally administered 24 h before infection in contrast to normal treatment (in which drugs or compounds were administered 6 h after infection). Each symbol represents the value for an individual mouse. Horizontal bars indicate the observation means, and dashed lines mark the limits of detection. Statistical significance was determined by the Mann–Whitney test (two-tailed): *p < 0.05, **p < 0.01, ***p < 0.001, and ns indicates no significant difference.
We had demonstrated that CrtN inhibitor 47 could efficaciously ameliorate the infections of Newman strains. Next, two multidrug resistant MRSAs, Mu50 (VISA) and NRS271 (LRSA), were introduced for evaluating the inhibitory effects of 47 against multidrug resistant MRSA infections. In general, most of the 47-treated cases in either Mu50 or NRS271 showed significant efficacy in contrast with the mock treatments, as shown in Figure 4. On one hand, as shown in the heart section in Mu50, the staphylococcal loads of groups that received the normal treatment of compound 47 were decreased significantly, as measured by 3.81 log10 CFU with 0.4 mg/b.i.d. and 3.67 log10 CFU with 0.1 mg/b.i.d., corresponding to a ∼99% decrease in surviving bacteria, compared with positive controls, while the two pretreatment groups produced less benefit. In parallel, as shown in the liver section in Mu50, most of the 47-treated groups displayed good resistance against encroachments, which was comparable with positive control groups. On the other hand, as shown in the NRS271 infectious model in Figure 4, in general, compared with the VAN-treatment groups and most of the 47-treatment groups in the hearts and livers, both mock and LZD-treatment groups displayed inferior inhibitory activities. Furthermore, the surviving bacteria of normal administration of 47-treatment groups decreased over 99.0%, comparable to that of the VAN-treated groups in the hearts. However, although most of the 47-treated groups were reduced by at least 0.75 log10 CFU in the livers, these groups were inferior to the VAN group. Consequently, all of the pharmacodynamic results showed that (1) most 47-treatment groups had more beneficial effects on lowering abscess formation than mock controls; (2) 47 in the low-dosage by normal treatment displayed comparable effects with the two positive groups (VAN and LZD) in the hearts in Mu50 and NRS271 and was even more effective than the two positive groups in the livers in Newman. In view of this anomaly, we suggest a preliminary hypothesis: (i) as shown in liver microsomes in Table 3, a moderate stability (t1/2 = 7.9 min) of 47 caused the pretreatment to fail to proactively exploit its advantage against MRSA infections; (ii) as a superactive CrtN inhibitor, derivative 47 still had enough ability to be efficacious against MRSAs at the low dosage. However, a potential cytotoxicity of 47 possibly reduced the immunity (Table 3), which constrained the potency of the high dosage groups.
Figure 4.
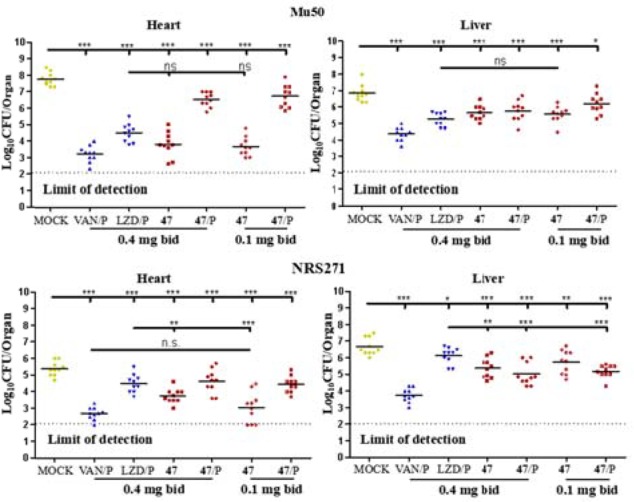
Effects of derivative 47 (in red) on S. aureus bacteria survival in the hearts and livers of mice (n = 10 for each group), challenged with 1.1 × 109 CFU of Mu50 and 1.8 × 108 CFU of NRS271 in vivo section, respectively. P = pretreatment, drugs or compounds were intraperitoneally administered 24 h before infection in contrast with the normal treatment (in which drugs or compounds were administered 6 h after infection). Each symbol represents the value for an individual mouse. Horizontal bars indicate the observation means, and dashed lines mark the limits of detection. Statistical significance was determined by the Mann–Whitney test (two-tailed): *p < 0.05, **p < 0.01, ***p < 0.001, and ns indicates no significant difference.
Table 3. Liver Microsomes and Cytotoxicity of Derivative 47.
| liver
microsomes |
cytotoxicity CC50 (μM) |
|||
|---|---|---|---|---|
| compd | k | t1/2 (min) | HEK-293T | HepG2 |
| 47 | 0.089 | 7.9 | 50.33 ± 2.7 | 67.76 ± 3.5 |
| Midazolam | 0.165 | 4.2 | ||
| Amphotericin B | 27.46 ± 5.2 | 37.81 ± 4.6 | ||
In summary, to improve the water solubility, hERG inhibition, and dosage of leading compound 4a, 38 derivatives were synthesized for a systematic study of structure and activity relationships, along with an investigation of pigment inhibitory activities against S. aureus Newman in vitro. Candidate 47 was selected for further evaluation of its potency in vivo against multidrug resistant MRSA infections, including S. aureus Newman, Mu50 (VISA/MRSA), and NRS271 (LRSA/MRSA). Fortunately, normal 47-treatment in the low-dosage group had a comparable effect to the positive groups (VAN and LZD). Altogether, these results demonstrate that 47 is a good candidate for combating MRSA, VISA, and LRSA infections.
Glossary
ABBREVIATIONS
- S. aureus
Staphylococcus aureus
- MRSA
methicillin-resistant Staphylococcus aureus
- STX
staphyloxanthin
- NTF
naftifine hydrochloride
- CrtM
dehydrosqualene synthase
- CrtN
diapophytoene desaturases
- ROS
reactive oxygen species
- SAR
structure–activity relationship
- IC50
half maximal inhibitory concentration
- CFU
colony-forming unit
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00501.
Purity, chemical synthesis, structural characterization, and solubility of the target compounds; protocols of biological assays (PDF)
Author Contributions
§ These authors contributed equally to this work.
Financial support for this research was provided by the National Key R&D Program of China (Grant 2017YFB0202600), the National Natural Science Foundation of China (Grant 21672064), the “Shu Guang” project supported by the Shanghai Municipal Education Commission, and Shanghai Education Development Foundation (Grant 14SG28). The Fundamental Research Funds for the Central Universities are gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- McColl K.; Murray L.; El-Omar E.; Dickson A.; El-Nujumi A.; Wirz A.; Kelman A.; Penny C.; Knill-Jones R.; Hilditch T. Symptomatic benefit from eradicating Helicobacter pyloriInfection in patients with nonulcer dyspepsia. N. Engl. J. Med. 1998, 339 (26), 1869–1874. 10.1056/NEJM199812243392601. [DOI] [PubMed] [Google Scholar]
- Kyaw M. H.; Kern D. M.; Zhou S.; Tunceli O.; Jafri H. S.; Falloon J. Healthcare utilization and costs associated with S. aureus and P. aeruginosa pneumonia in the intensive care unit: a retrospective observational cohort study in a US claims database. BMC Health Serv. Res. 2015, 15 (1), 241–252. 10.1186/s12913-015-0917-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Reisman I.; Ronin I.; Gefen O.; Braniss I.; Shoresh N.; Balaban N. Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–839. 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- Fridman O.; Goldberg A.; Ronin I.; Shoresh N.; Balaban N. Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 2014, 473, 418–421. 10.1038/nature13469. [DOI] [PubMed] [Google Scholar]
- Mechler L.; Herbig A.; Paprotka K.; Fraunholz M.; Nieselt K.; Bertram R. A novel point mutation promotes growth phase-dependent daptomycin tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59 (9), 5366–5376. 10.1128/AAC.00643-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh B.; Michiels J. E.; Wenseleers T.; Windels E. M.; Boer P. V.; Kestemont D.; De Meester L.; Verstrepen K. J.; Verstraeten N.; Fauvart M.; Michiels J. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 2016, 1 (9), 16020–16027. 10.1038/nmicrobiol.2016.20. [DOI] [PubMed] [Google Scholar]
- Russo A.; Campanile F.; Falcone M.; Tascini C.; Bassetti M.; Goldoni P.; Trancassini M.; Siega P. D.; Menichetti F.; Stefani S.; Venditti M. Linezolid-resistant staphylococcal bacteraemia: A multicentre case-case-control study in Italy. Int. J. Antimicrob. Agents 2015, 45 (3), 255–261. 10.1016/j.ijantimicag.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Rossi F.; Diaz L.; Wollam A.; Panesso D.; Zhou Y.; Rincon S.; Narechania A.; Xing G.; Di-Gioia T. S.; Doi A.; Tran T. T.; Reyes J.; Munita J. M.; Carvajal L. P.; Hernandez-Roldan A.; Brandão D.; van der Heijden I. M.; Murray B. E.; Planet P. J.; Weinstock G. M.; Arias C. A. Transferable vancomycin resistance in a community-associated MRSA lineage. N. Engl. J. Med. 2014, 370 (16), 1524–1531. 10.1056/NEJMoa1303359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/ (accessed February 27, 2016). [Google Scholar]
- Dickey S. W.; Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discovery 2017, 16 (7), 457–471. 10.1038/nrd.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A.; Pirofski L. A. Host-Pathogen Interactions: Redefining the Basic Concepts of Virulenlence and Pathogenicity. Infect. Immun. 1999, 67 (8), 3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauditz A.; Resch A.; Wieland K. P.; Peschel A.; Götz F. Staphyloxanthin Plays a Role in the Fitness of Staphylococcus aureus and Its Ability to Cope with Oxidative Stress. Infect. Immun. 2006, 74 (8), 4950–4953. 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodade V. S.; Sharath Chandra M.; Banerjee A.; Lahiri S.; Pulipeta M.; Rangarajan R.; Chakrapani H. Bioreductively Activated Reactive Oxygen Species (ROS) Generators as MRSA Inhibitors. ACS Med. Chem. Lett. 2014, 5 (7), 777–781. 10.1021/ml5001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield E.; Feng X. Resistance-resistant antibiotics. Trends Pharmacol. Sci. 2014, 35 (12), 664–674. 10.1016/j.tips.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland B.; Feil C.; Gloria-Maercker E.; Thumm G.; Lechner M.; Bravo J. M.; Poralla K.; Götz F. Genetic and biochemical analyses of the biosynthesis of the yellow carotenoid 4, 4′-diaponeurosporene of Staphylococcus aureus. J. Bacteriol. 1994, 176 (24), 7719–7726. 10.1128/jb.176.24.7719-7726.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. I.; Liu G. Y.; Song Y.; Yin F.; Hensler M. E.; Jeng W. Y.; Nizet V.; Wang A. H.; Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Lin F. Y.; Yin F.; Hensler M.; Rodrígues Poveda C. A.; Mukkamala D.; Cao R.; Wang H.; Morita C. T.; Gonzalez Pacanowska D.; Nizet V.; Oldfield E. Phosphonosulfonates are potent, selective inhibitors of dehydrosqualene synthase and staphyloxanthin biosynthesis in Staphylococcus aureus. J. Med. Chem. 2009, 52 (4), 976–988. 10.1021/jm801023u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Liu C. I.; Lin F. Y.; No J. H.; Hensler M.; Liu Y. L.; Jeng W. Y.; Low J.; Liu G. Y.; Nizet V.; Wang A. H. J.; Oldfield E. Inhibition of staphyloxanthin virulence factor biosynthesis in Staphylococcus aureus: in vitro, in vivo, and crystallographic results. J. Med. Chem. 2009, 52 (13), 3869–3880. 10.1021/jm9001764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Di H.; Wang Y.; Cao Q.; Xu B.; Zhang X.; Yang N.; Liu G.; Yang C. G.; Xu Y.; Jiang H.; Lian F.; Zhang N.; Li J.; Lan L. Small molecule targeting of a diapophytoene desaturase inhibits S. aureus virulence. Nat. Chem. Biol. 2016, 12 (3), 174–179. 10.1038/nchembio.2003. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chen F.; Di H.; Xu Y.; Xiao Q.; Wang X.; Wei H.; Lu Y.; Zhang L.; Zhu J.; Sheng C.; Lan L.; Li J. Discovery of Potent Benzofuran-Derived Diapophytoene Desaturase (CrtN) Inhibitors with Enhanced Oral Bioavailability for the Treatment of Methicillin-Resistant Staphylococcus aureus (MRSA) Infections. J. Med. Chem. 2016, 59 (7), 3215–3230. 10.1021/acs.jmedchem.5b01984. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Di H.; Chen F.; Xu Y.; Xiao Q.; Wang X.; Wei H.; Lu Y.; Zhang L.; Zhu J.; Lan L.; Li J. Discovery of Benzocycloalkane Derivatives Efficiently Blocking Bacterial Virulence for the Treatment of Methicillin-Resistant S. aureus (MRSA) Infections by Targeting Diapophytoene Desaturase (CrtN). J. Med. Chem. 2016, 59 (10), 4831–4747. 10.1021/acs.jmedchem.6b00122. [DOI] [PubMed] [Google Scholar]
- Ni S.; Wei H.; Li B.; Chen F.; Liu Y.; Chen W.; Xu Y.; Qiu X.; Li X.; Lu Y.; Liu W.; Hu L.; Lin D.; Wang M.; Zheng X.; Mao F.; Zhu J.; Lan L.; Li J. Novel Inhibitors of Staphyloxanthin Virulence Factor in Comparison with Linezolid and Vancomycin versus Methicillin-Resistant, Linezolid-Resistant, and Vancomycin-Intermediate Staphylococcus aureus Infections in Vivo. J. Med. Chem. 2017, 60 (19), 8145–8159. 10.1021/acs.jmedchem.7b00949. [DOI] [PubMed] [Google Scholar]
- Kuroda M.; Ohta T.; Uchiyama I.; Baba T.; Yuzawa H.; Kobayashi I.; Cui L.; Oguchi A.; Aoki K. I.; Nagai Y.; Lian J.; Ito T.; Kanamori M.; Matsumaru H.; Maruyama A.; Murakami H.; Hosoyama A.; Mizutani-Ui Y.; Takahashi N. K.; Sawano T.; Inoue R. i.; Kaito C.; Sekimizu K.; Hirakawa H.; Kuhara S.; Goto S.; Yabuzaki J.; Kanehisa M.; Yamashita A.; Oshima K.; Furuya K.; Yoshino C.; Shiba T.; Hattori M.; Ogasawara N.; Hayashi H.; Hiramatsu K. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- Wilson P.; Andrews J. A.; Charlesworth R.; Walesby R.; Singer M.; Farrel D. J.; Robbins M. Linezolid resistance in clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 2003, 51 (1), 186–188. 10.1093/jac/dkg104. [DOI] [PubMed] [Google Scholar]
- David M. Z.; Daum R. S. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23 (3), 616–687. 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Matos P. D.; de Oliveira T. L.; Cavalcante F. S.; Ferreira D. C.; Iorio N. L.; Pereira E. M.; Chamon R. C.; Dos Santos K. R. Molecular markers of antimicrobial resistance in methicillin-resistant Staphylococcus aureus SCCmec IV presenting different genetic backgrounds. Microb. Drug Resist. 2016, 22, 700. 10.1089/mdr.2015.0255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.