

Abstract
Ubiquitin specific protease 7 (USP7, HAUSP) has become an attractive target in drug discovery due to the role it plays in modulating Mdm2 levels and consequently p53. Increasing interest in USP7 is emerging due to its potential involvement in oncogenic pathways as well as possible roles in both metabolic and immune disorders in addition to viral infections. Potent, novel, and selective inhibitors of USP7 have been developed using both rational and structure-guided design enabled by high-resolution cocrystallography. Initial hits were identified via fragment-based screening, scaffold-hopping, and hybridization exercises. Two distinct subseries are described along with associated structure–activity relationship trends, as are initial efforts aimed at developing compounds suitable for in vivo experiments. Overall, these discoveries will enable further research into the wider biological role of USP7.
Keywords: USP7, ubiquitination, deubiquitinase, HAUSP, p53, Mdm2, proteasome, ubiquitin
The process of protein ubiquitination is a central tenet of the ubiquitin proteasome system (UPS) and is crucial in many fundamental cellular processes such as proteolysis, cell-cycle control, DNA repair, and apoptosis.1,2 The importance of this pathway in controlling such key cellular processes cannot be understated, and increasing evidence linking the UPS to human diseases such as cancer3 and neurodegenerative disorders4 is emerging. The approved proteasome inhibitor Velcade (bortezimib) has demonstrated that the UPS is a viable target for small molecule therapeutic intervention.5 Targeting the UPS upstream of the proteasome may therefore yield new opportunities for targeted therapeutics with improved specificity and toxicity profiles.
Ubiquitination is a post-translational modification via covalent attachment of the 76 amino acid protein ubiquitin to lysine side chains of target substrates. This elegant and complex molecular “tagging” of target proteins is affected by E1 (activating), E2 (conjugating), and E3 (ligase) enzymes and has multiple functions including targeted substrate degradation, activation for further processing, and cellular localization.6
The process of ubiquitination is reversed by deubiquitinase enzymes (DUBs) of which there are around 100 encoded by human genes.7 Ubiquitin specific proteases (USPs) are cysteine proteases that comprise the largest (>50) subclass of DUBs and are gaining interest as an emerging target class for pharmaceutical intervention.8 There are sparingly few validated small molecule inhibitors reported for USPs, and as such, there is an acute need to develop robust probe compounds for use in deciphering the biological pathways associated with the USP target class.
USP7 represents one of the most studied USPs from a target class that remains largely underexplored and, as such, has gained attention in recent years due to its association with cancer.9,10 USP7 is involved in the regulation of the stability of the tumor suppressor p53 via deubiquitination of the oncoprotein Mdm2.11 USP7 mediated stabilization of Mdm2 reduces cellular p53 and may protect damaged cells from apoptosis. In addition, USP7 has also been implicated in the regulation of several other key signaling proteins linked to tumorigenesis.12−17 Targeting USP7 with small molecules has therefore been of great interest over recent years but has until recently met with limited success due to several factors including poor compound specificity, low potency, and/or poor compound properties.18 Very recent publications have described the characterization of more drug-like USP7 inhibitors that further reinforce the potential druggability of this target class.19−22
Recently, we published the detailed in vitro biological profiling and cocrystal structures of highly potent noncovalent USP7 inhibitors in a variety of biochemical and cellular assays.22 These compounds have proven to be highly valuable tools for interrogating the complex biology of USP7 and will enable further studies aimed at delineating USP7 biology. Herein, we describe the hit-finding and medicinal chemistry efforts toward these tool compounds, summarize the structure–activity relationship (SAR), and outline the identification of a novel subseries of USP7 inhibitors. The binding mode (by way of X-ray cocrystallography) of this subseries is highlighted as is the pharmacokinetic profiling of early leads from both series.
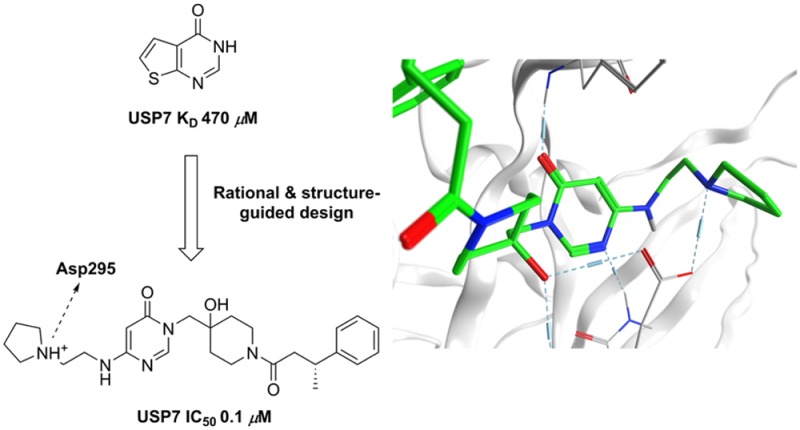
Our USP7 hit-finding strategy involved initial fragment screening using surface plasmon resonance (SPR) coupled with the parallel in vitro benchmarking of published USP7 inhibitors. Briefly, SPR screening of 1.9k fragments versus immobilized USP7 catalytic domain afforded a range of primary USP7 binding fragments including thieno-pyrimidinone 1 (Figure 1).
Figure 1.
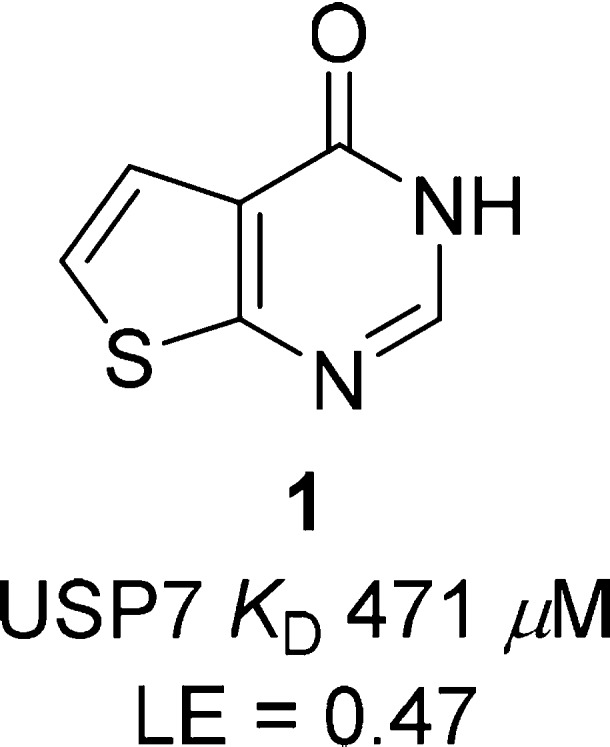
Primary USP7 SPR binding fragment 1.
Compound 1 was found to be a high ligand efficiency USP7 binder (LE = 0.47) with an SPR equilibrium binding constant (KD) of 471 μM (Figure S1, SI). Binding of compound 1 to USP7 was subsequently confirmed by orthogonal techniques (ligand observed STD, CPMG, and WaterLOGSY NMR experiments) (Figure S2, SI). Further profiling revealed that 1 had excellent aqueous kinetic solubility (>200 μM) and was free from redox cycling activity, a liability that has the potential to lead to false positive readouts in biochemical assays. Compound 1 and analogues thereof were then incorporated into our wider USP7 medicinal chemistry program, which involved scaffold-hopping as well as hybridization with known literature USP binding motifs (e.g., 4-hydroxypiperidines23). From this program of work, compounds 2 and 3 were synthesized and found to have modest USP7 biochemical activity (Table 1).
Table 1. Early USP7 Hits and Associated Physicochemical Properties.

Compounds 2 and 3 were deemed suitable for further SAR studies due to their reasonable ligand efficiencies (LE) and favorable physicochemical properties. A range of analogues derived by scaffold-hopping to other fused five-membered ring pyrimidinones were explored in parallel to substitution on the bicyclic pyrimidinone core (Table 2). Furano, pyrazolo, and thiazolo-pyrimidinone analogues 4–8 were largely equipotent with thieno-pyrimidinones 2 and 3. Substitution of a lipophilic bromine atom at either the C-5 or C-6 positions of the thiophene ring in compounds 9 and 10 did not lead to marked USP7 potency gains, whereas substitution at the C-7 position led to an appreciable (ca. 5-fold) potency enhancement as demonstrated by compound 11. Pleasingly, substitution at this position with other lipophilic groups such as cyclopropyl 12, alkynyl 13, and phenyl 14 also afforded noticeable potency gains versus the nonsubstituted analogue 3. Substitution at the 2-position of the pyrimidinone ring with a methyl substituent was not tolerated (compound 15).
Table 2. Early SAR of Five-membered Heterocyclic Pyrimidinones.

With potency gains of ca. 5–10-fold attainable via modification of the bicyclic thienopyrimidinone ring, our attention turned to SAR analysis of the phenethylamide moiety. During the course of our studies a key breakthrough was the observation that methyl substitution at the benzylic position of compounds such as 11 led to a significant (>40-fold) increase in USP7 potency. This large increase in potency was fully dependent on the stereochemistry of the newly installed chiral methyl group as exemplified by compounds 16 and ent-16 (Figure 2).
Figure 2.

Effects of benzylic substitution.
Interestingly, the (R)-enantiomer 16 was found to be >150 times more potent than the corresponding (S)-enantiomer ent-16. With potent USP7 compounds such as 16 now in hand, we initiated cocrystallization studies with the catalytic domain of USP7. We subsequently obtained a 2.3 Å resolution X-ray cocrystal structure of compound 16 and USP7 that revealed the ligand bound in the catalytic cleft between the β-sheet of the palm subdomain and the helices of the thumb subdomain.22 Removal of key ligand hydrogen-bonding heteroatoms (such as the amide carbonyl, tertiary alcohol, or pyrimidinone N-1 nitrogen from the bicyclic core) was highly deleterious to USP7 binding (data not shown) highlighting the importance of each individual interaction.
Aided by the X-ray cocrystal structure, we were able to use structure-based design to further optimize our USP7 inhibitors with the aim of enhancing target affinity (while retaining favorable physicochemical properties). Our initial focus centered on the thiophene ring of compound 16, which contains a bromine substituent at C-7 that is orientated toward the protein surface with a high degree of solvent exposure and was thus viewed as a promising vector for new analogue design and optimization. Encouragingly, scaffold-hopping from the thieno-pyrimidinone core of compound 16 to the more drug-like N-methyl pyrazolo-pyrimidine core (compound 17, Table 3) not only lowered overall lipophilicity (logD7.4 1.5 vs 2.3) but was also not detrimental to USP7 activity. Given this result, we decided to concentrate our efforts on the pyrazolo-pyrimidinone core, and a summary of the SAR is presented in Table 3.
Table 3. USP7 Biochemical Potencies of N-Methyl Pyrazolo-pyrimidinones.


Our strategy (guided by docking studies using the cocrystal structure) was centered on targeting both hydrogen-bond and hydrophobic interactions via selective substitution at the pyrazole C-3 position. Incorporation of alkynyl, alkenyl, or isopropyl substituents at the C-3 position (compounds 18–21) did not prove fruitful; however, when pyrazole or phenyl groups were incorporated at this position a ca. 10-fold increase in USP7 potency was observed (e.g., compound 23, IC50 0.03 μM). This marked increase in potency may involve a CH−π interaction between the pendant pyrazole or phenyl groups of 22 or 23 and Gln351 as postulated in the published cocrystal structure.22 Addition of an ortho-fluoro group to the pendant phenyl in compound 23 led to a slight decrease in potency (compound 24, IC50 0.09 μM) in contrast to ortho-anilino substituted compound 25, which was around 40-fold less potent than 23, possibly due to the larger ortho-NH2 substituent increasing the dihedral angle between the phenyl ring and the core, causing an unfavorable steric clash between the phenyl ring and the protein surface. Addition of hydrogen-bond acceptors and donors at the meta and para positions on the pendant phenyl group (compounds 26-–30) largely maintained potency in relation to 23 with the exception of compound 31, which showed a marked decrease in potency, most likely due to the added steric bulk of the morpholine group. Interestingly, the para-carboxamide or benzylic alcohol groups in compounds 32 and 33 did not lead to potency increases, whereas the para-benzylic amine moiety in compound 34 (IC50 = 6 nM) provided a ca. 5-fold increase in potency over compound 23. The (S)-Me enantiomer of 34 was found to have an IC50 of 2.4 μM, representing a 400-fold decrease in USP7 potency.22
In addition to identifying the promising pyrazolo-pyrimidinones described above, we also investigated how truncation of the bicyclic core would affect USP7 binding. Hence, a series of monocyclic pyrimidinones such as compound 35 was synthesized (Table 4). Although only moderately active, compound 35 (IC50 = 23 μM) represented a promising and ligand efficient (LE = 0.25) starting point for further analogue work with respect to its low molecular weight and logD7.4 (355 and 0.9, respectively). A range of analogues were subsequently prepared substituted at the C-6 position of the pyrimidinone ring with representative examples shown in Table 4.
Table 4. USP7 Biochemical Potencies of Monocyclic Pyrimidinones.

Overall SAR at this position suggested that substitution was largely beneficial for USP7 potency. Substitution with simple amines such as methylamine or aniline (compounds 36 and 37) increased potency by 5- to 10-fold versus the unsubstituted analogue 35. Switching the −NH linker in compound 37 to −CH2 in compound 38 resulted in a significant USP7 potency drop-off (IC50 = 1.9 μM versus 90 μM). Potency was regained when the methylene phenyl linker in compound 38 was switched to an alkynyl linker group (compound 39, IC50 = 2.8 μM). Ethylenediamine analogue 40 (IC50 = 0.12 μM) represented a >190-fold improvement in USP7 biochemical potency over the simple analogue 35, suggestive of new hydrogen-bonding interactions. Indeed, close analogues of compound 40 in which the hydrogen-bonding interaction potential of the ethylenediamine side-chain was modified led to decreases in potency (compounds 41–45). Pyrrolidine analogue 46 represented the first sub-100 nM compound from this series and suggested that the basic pyrrolidine nitrogen was making a critical hydrogen-bond with USP7.
Intrigued by the observed SAR in which the precise nature of the side-chain linker and the pKa of the amine both seemed crucial, molecular modeling suggested that a close interaction between the protonated amine of 46 and Asp295 was potentially achievable. This was subsequently confirmed via a high-resolution (2.2 Å) X-ray cocrystal structure of compound 46 bound to USP7, which demonstrates a similar binding mode to that of compound 16 reported previously (Figure 3A,B).22
Figure 3.
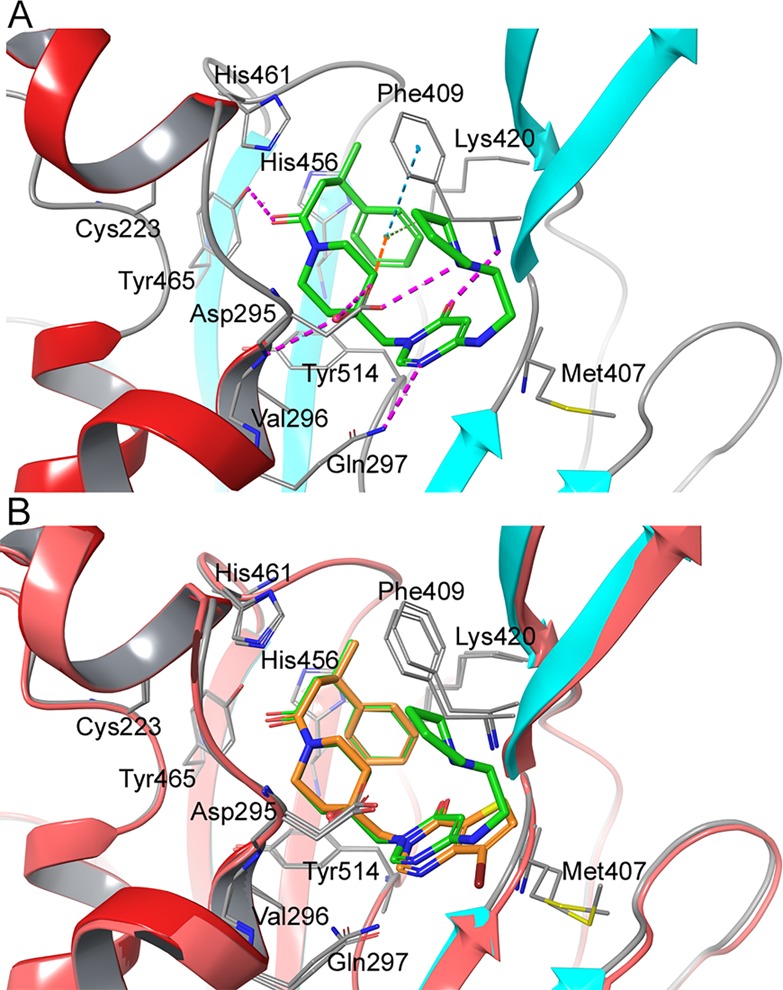
(A) High-resolution X-ray cocrystal structure of USP7 in complex with 46 (PDB code: 6F5H). (B) Overlay of compounds 46 (green) and 16(22) (orange, PDB code: 5N9R) bound to USP7.
In addition to the important hydrogen bond interaction network observed previously with 16, we also observed the postulated additional hydrogen bond between the protonated nitrogen of the pyrrolidine side-chain of 46 and Asp295, demonstrating a unique bidentate binding interaction pattern with Asp295, which had not been previously reported. This extra interaction appears to be crucial for improving affinity within the monocyclic series. As with compound 16, the amide carbonyl in 46 interacts with the Tyr465 hydroxyl group and the ligand tertiary alcohol and forms hydrogen bonds with both Asp295 and Val296. With regards to the pyrimidinone ring of 46, the carbonyl oxygen atom forms a hydrogen bond with the backbone NH of Phe409, whereas the N-4 ring atom forms a hydrogen bond with Gln297 effectively stabilizing the monocyclic ring with interactions above and below the ring system (Figure 3A). The folded bioactive conformation of the phenethylamide side chain of 46 may partly be induced by allylic 1,3-strain between the benzylic CH and the phenyl ring as well as stabilization via a CH−π intramolecular interaction between the piperidine C-3 axial hydrogen and the phenyl ring. These intramolecular conformational drivers may in part be responsible for inducing binding site side-chain movements that create the overall binding pocket that accommodates this portion of the ligand. The resulting conformation is stabilized by a cation−π interaction between Lys420 and the ligand phenyl ring in addition to an edge-to-face π–π interaction with Phe409. Additional contacts between the phenethylamide methylene hydrogen atoms of 46 and the π system of His461 may also contribute to overall binding efficiency.
With highly potent USP7 inhibitors (from two distinct subseries) such as 34 and 46 in hand, we performed extensive in vitro profiling and demonstrated that 34 shows potent target engagement of endogenous USP7 in cells as well as excellent selectivity for USP7 in panels of deubiquitinases, proteases, and kinases.22 In addition, we also identified cancer cell lines that are hypersensitive to our USP7 inhibitors. Similar to compound 34, monocyclic analogue 46 shows excellent selectivity versus a panel of USPs (n = 21) when screened at a fixed concentration of 10 μM as well as potent intracellular USP7 target engagement (EC50 = 0.32 μM) in cells (Figure 4A,B).
Figure 4.

(A) Selectivity profile of 46 against a panel of 21 USPs. Screening was performed at a fixed concentration of 10 μM (Ubiquigent). (B) USP7 target engagement of 46 in HCT116 cells. See Supporting Information for assay conditions.
In parallel to the in-depth cellular profiling outlined above, potent compounds were assessed in a range of in vitro assays in order to determine their suitability for in vivo studies. Generally, nonbasic analogues were unstable in both human and mouse liver microsomes and had poor Caco-2 permeability if they contained >1 hydrogen bond donor (HBD), thus precluding them from in vivo PK studies (e.g., compounds 23 and 33, Table 5). Likewise, benzylic amine 34 was found to have high in vitro metabolic turnover in both human and mouse liver microsomes (with predicted hepatic clearances of 18 and 62 mL/min/kg, respectively), whereas monocycle 46 had moderate metabolic stability in human and mouse liver microsomes (11 and 34 mL/min/kg, respectively). The aqueous kinetic solubility of both 34 and 46 was high (Ksol >190 μM), but the Caco-2 A/B permeability of each at pH 6.5 was found to be low (Papp < 0.3 × 10–6 cm/s), limiting their potential for oral dosing. The low predicted in vivo hepatic stability of the highly potent benzylic amine 34 encouraged us to carry out further chemical optimization in order to identify compounds with improved metabolic stability to facilitate in vivo proof-of-concept studies. As part of the chemistry program aimed at improving metabolic stability of compound 34, trifluoromethyl analogue 47 (Figure S3, SI) was prepared via an asymmetric hydrogenation route described previously.22
Table 5. Biochemical and ADME Profiling of Compounds 23, 33, 34, 46, and 47a.
| Cmpd | USP7 IC50 (μM) | HLM/MLM CLhep | logD7.4/Ksol | Caco-2 Papp A/B |
|---|---|---|---|---|
| 23 | 0.030 | 19/84 | 2.3/175 | 1.93 |
| 33 | 0.040 | 19/86 | 1.7/200 | 0.15 |
| 34 | 0.006 | 18/62 | –0.1/191 | 0.28 |
| 46 | 0.087 | 11/34 | 0/200 | 0.26 |
| 47 | 0.022 | 7/28 | 0.1/179 | 0.14 |
See Supporting Information for assay conditions. HLM/MLM units: mL/min/kg. Ksol units: μM. Caco-2 units: 10–6 cm/s
Compound 47 was found to have improved in vitro microsomal stability in both human and mouse microsomes compared to compound 34 (HLM CLhep 7 vs 18 mL/min/kg and MLM CLhep 28 vs. 62 mL/min/kg) (Table 5). Furthermore, 47 largely maintained USP7 biochemical potency when contrasted with methyl analogue 34 (IC50 = 22 vs 6 nM).
The pharmacokinetic profiles of compounds 46 and 47 were subsequently assessed in male CD-1 mice (Table 6). As expected from their low Caco-2 permeabilities (A/B Papp < 0.3 × 10–6 cm/s), both compounds exhibited poor oral bioavailability (F < 1%). However, when dosed intraperitoneally (i.p.) both compounds exhibited reasonable bioavailability (F = 44% and 64% for 46 and 47, respectively). Volume of distribution for each compound was low (Vss ≤ 1 L/kg), in line with their low lipophilicities (logD7.4 ≤ 0.1). Compound 46 demonstrated low plasma clearance (CL = 13 mL/min/kg), whereas compound 47 had moderate clearance (CL = 32 mL/min/kg). Further optimization studies aimed at improving PK profiles based on these encouraging preliminary in vivo results are underway and will be reported in due course.
Table 6. Pharmacokinetic Profile of Compounds 46 and 47 in Male CD-1 Mice.
| Routea | Cmpd | Cmaxb | AUCallc | t1/2d | CLe | Vssf |
|---|---|---|---|---|---|---|
| i.v. | 46 | 4035 | 1700 | 1.1 | 13 | 0.4 |
| 47 | 2808 | 516 | 1.0 | 32 | 0.9 | |
| i.p | 46 | 3430 | 3002 | 1.1 | - | - |
| 47 | 2876 | 1904 | 0.8 | - | - |
i.v., 46 1.3 mg/kg, 47 0.9 mg/kg in 2% DMSO in 20% aq. 2-hydroxypropyl-β-cyclodextrin; i.p., 46 6.2 mg/kg, 47 5.7 mg/kg in saline.
ng/mL.
ng·h/mL.
hr.
mL/min/kg.
L/kg.
In conclusion, we have identified and optimized highly potent USP7 inhibitors based on two different core chemotypes. Key compounds have a well understood mode of binding as evidenced by the high-resolution cocrystal structures obtained. These USP7 inhibitors have been highly valuable in validating the druggability of USP7 as well as enabling studies toward a deeper understanding of the underlying biology of USP7 and its potential as a therapeutic target. Efforts are continuing toward the further development of these inhibitors into compounds suitable for in vivo proof-of-concept studies.
Acknowledgments
This study was supported by the Almac Group and the European Regional Development Fund and Invest Northern Ireland (Grant RD1010668).
Glossary
ABBREVIATIONS
- CPMG
Carr–Purcell–Meiboom–Gill sequence NMR experiment
- DUB
deubiquitinase
- ee
enantiomeric excess
- HAC
heavy atom count
- HLM
human liver microsomes
- KSol
kinetic solubility
- LE
ligand efficiency (−1.4 pKD or pIC50/HAC)
- MLM
mouse liver microsomes
- PK
pharmacokinetics
- SAR
structure–activity relationship
- STD
saturation transfer difference
- UPS
ubiquitin proteasome system
- USP
ubiquitin specific protease
- WaterLOGSY
water-ligand observed via gradient spectroscopy
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00512.
Experimental details and characterization data for all compounds, biochemical, cellular, and selectivity assay protocols, crystallization conditions and methodology, in vitro ADME assay protocols, in vivo procedures, computational methods, and supplementary figures (PDF)
Author Present Address
§ Randox Laboratories, 55 Diamond Road, Crumlin, County Antrim BT29 4QY, U.K.
Author Present Address
∥ UCB Celltech, 208 Bath Road, Slough SL1 3WE, U.K.
Author Present Address
⊥ The European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Cambridgeshire CB10 1SD, U.K.
Author Present Address
# Medivir AB, 141 22 Huddinge, Sweden.
Author Present Address
∇ Redag Crop Protection, BioHub, Alderley Park, Mereside, Macclesfield SK10 4TG, U.K.
Author Contributions
T.H. conceived the concept and directed the research. C.O.D. and G.G. helped conceive and develop the concept and designed and supervised medicinal chemistry and biology experiments. M.H., E.A., J.F., H.M., J.S.S.R., and L.J. carried out the design, synthesis, and characterization of compounds. N.P. performed SPR experiments. E.R., C.H., K.M., E.O., E.C., and N.P. carried out compound screening, target validation, and biochemical and cellular profiling studies. O.B. carried out computational modeling and structural analysis. S.F.B. carried out ADME profiling. S.D. performed NMR experiments. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Hershko A.; Ciechanover A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Clague M. J.; Urbé S. Ubiquitin: Same Molecule, Different Degradation Pathways. Cell 2010, 143, 682–685. 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Hoeller D.; Hecker C. M.; Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Zhang L.; Zhou Y.; Luo H.; Wang X.; Huang T.; Xu H. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 1–10. 10.3389/fnagi.2016.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson P. G.; Barlogie B.; Berenson J.; Singhal S.; Jagannath S.; Irwin D.; Rajkumar S. V.; Srkalovic G.; Alsina M.; Alexanian R.; Siegel D.; Orlowski R. Z.; Kuter D.; Limentani S. A.; Lee S.; Hideshima T.; Esseltine D. L.; Kauffman M.; Adams J.; Schenkein D. P.; Anderson K. C. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- Komander D.; Rape M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Komander D.; Clague M. J.; Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Pal A.; Young M. A.; Donato N. J. Emerging Potential of Therapeutic Targeting of Ubiquitin-Specific Proteases in the Treatment of Cancer. Cancer Res. 2014, 74, 4955–4966. 10.1158/0008-5472.CAN-14-1211. [DOI] [PubMed] [Google Scholar]
- Nicholson B.; Suresh Kumar K. G. The Multifaceted Roles of USP7: New Therapeutic Opportunities. Cell Biochem. Biophys. 2011, 60, 61–68. 10.1007/s12013-011-9185-5. [DOI] [PubMed] [Google Scholar]
- Wu J.; Kumar S.; Wang F.; Wang H.; Chen L.; Arsenault P.; Mattern M.; Weinstock J. Chemical Approaches to Intervening in Ubiquitin Specific Protease 7 (USP7) Function for Oncology and Immune Oncology Therapies. J. Med. Chem. 2018, 61, 422–443. 10.1021/acs.jmedchem.7b00498. [DOI] [PubMed] [Google Scholar]
- Cummins J. M.; Rago C.; Kohli M.; Kinzler K. W.; Lengauer C.; Vogelstein B. Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature 2004, 428, 1–2. 10.1038/nature02501. [DOI] [PubMed] [Google Scholar]
- Song M. S.; Salmena L.; Carracedo A.; Egia A.; Lo-Coco F.; Teruya-Feldstein J.; Pandolfi P. P. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008, 455, 813–817. 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.-T.; Kuo Y.-C.; Hung J.-J.; Huang C.-H.; Chen W.-Y.; Chou T.-Y.; Chen Y.; Chen Y.-J.; Chen Y.-J.; Cheng W.-C.; Teng S.-C.; Wu K.-J. K63-polyubiquitinated HAUSP deubiquitinates HIF-1α and dictates H3K56 acetylation promoting hypoxia-induced tumour progression. Nat. Commun. 2016, 7, 13644. 10.1038/ncomms13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavana O.; Li D.; Dai C.; Lopez G.; Banerjee D.; Kon N.; Chen C.; Califano A.; Yamashiro D.; Sun H.; Gu W. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat. Med. 2016, 22, 1180–1186. 10.1038/nm.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Ma S.; Song N.; Li X.; Liu L.; Yang S.; Ding X.; Shan L.; Zhou X.; Su D.; Wang Y.; Zhang Q.; Liu X.; Yu N.; Zhang K.; Shang Y.; Yao Z.; Shi L. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Invest. 2016, 126, 2205–2220. 10.1172/JCI85747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Yao X.; Li S.; Xiong Y.; Dong X.; Zhao Y.; Jiang J.; Zhang Q. Deubiquitination of Ci/Gli by Usp7/HAUSP Regulates Hedgehog Signaling. Dev. Cell 2015, 34, 58–72. 10.1016/j.devcel.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst A.; de Vries-Smits A. M.; Brenkman A. B.; van Triest M. H.; van den Broek N.; Colland F.; Maurice M. M.; Burgering B. M. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 2006, 8, 1064–1073. 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- Kemp M. Recent Advances in the Discovery of Deubiquitinating Enzyme Inhibitors. Prog. Med. Chem. 2016, 55, 149–192. 10.1016/bs.pmch.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kategaya L.; Di Lello P.; Rougé L.; Pastor R.; Clark K. R.; Drummond J.; Kleinheinz T.; Lin E.; Upton J. P.; Prakash S.; Heideker J.; McCleland M.; Ritorto M. S.; Alessi D. R.; Trost M.; Bainbridge T. W.; Kwok M. C. M.; Ma T. P.; Stiffler Z.; Brasher B.; Tang Y.; Jaishankar P.; Hearn B. R.; Renslo A. R.; Arkin M. R.; Cohen F.; Yu K.; Peale F.; Gnad F.; Chang M. T.; Klijn C.; Blackwood E.; Martin S. E.; Forrest W. F.; Ernst J. A.; Ndubaku C.; Wang X.; Beresini M. H.; Tsui V.; Schwerdtfeger C.; Blake R. A.; Murray J.; Maurer T.; Wertz I. E. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550, 534–538. 10.1038/nature24006. [DOI] [PubMed] [Google Scholar]
- Turnbull A. P.; Ioannidis S.; Krajewski W. W.; Pinto-Fernandez A.; Heride C.; Martin A. C. L.; Tonkin L. M.; Townsend E. C.; Buker S. M.; Lancia D. R.; Caravella J. A.; Toms A. V.; Charlton T. M.; Lahdenranta J.; Wilker E.; Follows B. C.; Evans N. J.; Stead L.; Alli C.; Zarayskiy V. V.; Talbot A. C.; Buckmelter A. J.; Wang M.; McKinnon C. L.; Saab F.; McGouran J. F.; Century H.; Gersch M.; Pittman M. S.; Marshall C. G.; Raynham T. M.; Simcox M.; Stewart L. M. D.; McLoughlin S. B.; Escobedo J. A.; Bair K. W.; Dinsmore C. J.; Hammonds T. R.; Kim S.; Urbé S.; Clague M. J.; Kessler B. M.; Komander D. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. 10.1038/nature24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto I.; Liu X.; Seo H. S.; Schauer N. J.; Iacob R. E.; Hu W.; Das D.; Mikhailova T.; Weisberg E. L.; Engen J. R.; Anderson K. C.; Chauhan D.; Dhe-Paganon S.; Buhrlage S. J. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem. Biol. 2017, 24, 1490–1500. 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavory G.; O’Dowd C. R.; Helm M. D.; Flasz J.; Arkoudis E.; Dossang A.; Hughes C.; Cassidy E.; McClelland K.; Odrzywol E.; Page N.; Barker O.; Miel H.; Harrison T. Discovery and Characterization of Highly Potent and Selective Allosteric Inhibitors of USP7. Nat. Chem. Biol. 2018, 14, 118–125. 10.1038/nchembio.2528. [DOI] [PubMed] [Google Scholar]
- Selective and reversible inhibitors of ubiquitin specific protease 7. WO2013030218.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.