

Abstract
Designing multitarget-directed ligands (MTDLs) is considered to be a promising approach to address complex and multifactorial maladies such as Alzheimer’s disease (AD). The concurrent inhibition of the two crucial AD targets, glycogen synthase kinase-3β (GSK-3β) and human acetylcholinesterase (hAChE), might represent a breakthrough in the quest for clinical efficacy. Thus, a novel family of GSK-3β/AChE dual-target inhibitors was designed and synthesized. Among these hybrids, 2f showed the most promising profile as a nanomolar inhibitor on both hAChE (IC50 = 6.5 nM) and hGSK-3β kinase activity (IC50 = 66 nM). It also showed good inhibitory effect on β-amyloid self-aggregation (inhibitory rate = 46%) at 20 μM. Western blot analysis revealed that compound 2f inhibited hyperphosphorylation of tau protein in mouse neuroblastoma N2a-Tau cells. In vivo studies confirmed that 2f significantly ameliorated the cognitive disorders in scopolamine-treated ICR mice and less hepatotoxicity than tacrine. This study provides new leads for assessment of GSK-3β and AChE pathway dual inhibition as a promising strategy for AD treatment.
Keywords: Dual GSK-3β/AChE inhibitor, hybrid, multitarget direct ligands (MTDLs)
Among all the neurodegenerative diseases, Alzheimer’s disease (AD), an irreversible progressive neurodegenerative disorder of the central nervous system (CNS), is described by a progressive loss of cognitive abilities in old age people.1 Due to the very limited number of available drugs and their low efficacy, AD remains incurable. Aberrant protein processing is a distinctive feature of AD in which β-amyloid (Aβ) and abnormally hyperphosphorylated tau protein misfold and self-accumulate in the brains of affected individuals as aggregates, namely, amyloid senile plaques and neurofibrillary tangles (NFTs), respectively.2,3 These assemblies represent the most relevant histopathological hallmarks of AD and have been considered to play crucial roles in its pathogenesis. A series of hypotheses have been proposed, among which tau hypothesis, amyloid cascade hypothesis, and cholinergic hypothesis are of the central importance.4
Tau is a microtubule-associated protein mostly found in neuronal axons in CNS.5 Under normal physiological conditions, tau is associated with microtubules by preventing their dynamic instability, contributing to morphogenesis of neurons.6 Aberrant hyperphosphorylation of the tau protein loses its ability to stabilize microtubules, which leads to accumulation and formation of NFTs.7 As for the tau hypothesis, glycogen synthase kinase-3β (GSK-3β), a multitasking serine/threonine kinase largely expressed in CNS, has been proved to play a significant role in regulating tau phosphorylation mainly at Ser396, Ser199, and Ser413, which causes tau to be detached from the microtubules and precipitates as NFTs under both physiological and pathological conditions.8 Furthermore, increased GSK-3β activity also induces Aβ formation in a unique fashion by regulating γ-secretase and results in toxicity to the cultured neurons.9 A conditional GSK-3β overexpressing transgenic mouse exhibited microtubule destabilization, NFTs formation, and cognitive deficits.10 Both preclinical and early phase clinical studies have validated GSK-3β as a therapeutic target for AD. Recent findings have shown that GSK-3β inhibitors (GSK-3βIs) can be a route to shift the equilibrium from neurodegeneration to neurogenesis both in vitro and in vivo.11 Clearly, hindering generation of Aβ and hyperphosphorylated tau protein by inhibiting GSK-3β activity has been envisaged as a suitable strategy for AD treatment, and thus, the development of selective GSK-3βIs has been popularly pursued (Figure S1).12,13 Up until now, a series of new promising high affinity ATP-competitive GSK-3βI with the basis of a pyridothiazole core has been synthesized.14
However, the decline of acetylcholine (ACh) levels leads to cognitive and memory deficits; thus, recovering cholinergic function is considered to be clinically beneficial.15 Several studies have also indicated that AChE seems to be implicated in AD pathogenesis by promoting the formation of both Aβ fibrils.16 Fortunately, acetylcholinesterase inhibitors (AChEIs) possibly affect metabolic processing of the APP and thus may influence Aβ generation.17 It is worth emphasizing that although many hypotheses and strategies are currently being proposed for the treatment of AD, AChEIs still remain to be a supreme clinical success in AD treatment, which fully demonstrates the value of this target.18 However, the cholinergic deficit is just one of the hallmarks of the AD pathology. One of the reasons that the single-target directed drugs have failed to reach clinical trials is the pathological complexity found in AD. An open-label study found that the combination of cholinesterase inhibitors and memantine was well tolerated in dementia therapy.19 As a new paradigm in drug discovery for AD, compared to combination therapies, multifunctional molecules as multitarget direct ligands (MTDL) avoid drug–drug interactions, off-target adverse effects, poor patient compliance, and high development costs.20 Given the fact that AD is a systemic disorder of the central nervous system, multitarget strategies will be more promising. As an AChEI with definite efficacy and binding mode, tacrine is a good scaffold for the design of MTDLs due to its simple structure but high ligand efficiency.21,22 Moreover, tacrine has a good endurance against substantial structural modification while retaining the target-based activity and further provides a sound basis for the design of MTDLs.23,24 In addition, the more potent AChEI 6-Cl-tacrine and the less hepatotoxic 7-MeO-tacrine have provided useful scaffolds to generate new hybrids.25
Given the aforementioned evidence, GSK-3β and AChE, the two main pathways of AD, are ideal candidates for such multitarget approach. Their activities are deeply involved in AD pathogenesis and progression: the cholinergic deficit and NFTs, the two main AD pathological hallmarks. Therefore, the simultaneous modulation of both GSK-3β and AChE, by blocking hyperphosphorylated tau protein and aggregation of Aβ plaques, as well as improving cognition, might serve as a promising strategy for AD treatment. Herein, we designed the first GSK-3β/AChE bifunctional inhibitors by hybridizing the pharmacophores of GSK-3βI with AChEI. The rationale for the MTDL design originated from the cocrystal structure of the selective GSK-3βI 1 with the ATP-binding site of GSK-3β kinase domain. The pyridine carboxamide of 1 acted as the hinge binding head forming hydrogen bonds with the V135 backbone amide and thiazole ring as hinge group, and the carbonyl oxygen of the thiazolyl primary amide formed a critical hydrogen bond with the Lys85 (Figure 1B).14 It should be noted that the thiazolyl methoxy moiety and the primary amide are located in the solvent exposed sites, to be the best position to attach tacrine to compound 1 by alkylenediamine tethers so that critical binding interactions were not disrupted. It is important that the designed hybrids have no significant effect on the binding of tacrine to the catalytic active site (CAS) of AChE and that the pyridoxathiazole fragment acts as a binder for the peripheral anionic site (PAS) and thus may enhance AChE inhibition. Therefore, we incorporated tacrine at the thiazolyl ring of 1 with a proper linker to design a new MTDL (Figure 1A).
Figure 1.

Design strategy of dual GSK-3β/AChE inhibitors. (A) Design of a merged GSK-3β/AChE pharmacophore taking advantage of the solvent exposed sites in GSK-3β and the probable interactions to be gained by a merged tacrine at the entrance to AChE substrate binding pocket. (B) X-ray cocrystal structure of 1 in the kinase domain of GSK-3β. Intermolecular interactions are shown as dotted lines with different colors according to the type of the interaction: green, conventional hydrophobic contact; dark pink, π–π stacked; light pink, π-alkyl contact. The thiazolyl methoxy and the primary amide moiety extend into the solvent-exposed region of the protein (red circle).
The route employed to synthesize target compounds 2a–2k is outlined in Scheme 1. Pyridothiazole ester 3 was obtained from 2-amino-4-cyanopyridine by acylation, addition, and cyclization,26 which was then used in Mitsunobu reaction with N-ethanol tacrine,27 giving compound 2a. Hydrolysis of the ethyl ester in 2a to afford 2b, followed by coupling with NH4Cl afforded compound 2c or methylation with MeI gave out compound 2d. Amination of 4 with the corresponding diamines afforded intermediates 5a–5g. Mitsunobu reaction of 3 with MeOH, followed by hydrolysis to yield carboxyl 4.14 Finally, the carboxylic acid 4 was coupled with tacrine intermediates 5a–5g to produce the target compounds 2e–2k, respectively.
Scheme 1. Preparation of Compounds 2a–2k.

Reagents and conditions: (a) N-ethanol tacrine, PPh3, DIAD, THF, rt, overnight, 52%; (b) 1.5 N LiOH, MeOH/THF/H2O, rt, 5 h, 87%; (c) NH4Cl, HATU, DIPEA, DMF, rt, overnight, 38%; (d) CH3I, K2CO3, DMF, rt, 5 h, 43%; (e) i: anhydrous MeOH, PPh3, DIAD, THF, rt, 18 h; ii: 1 N LiOH, MeOH/THF, rt, 3 h, 80%; (f) HATU, DIPEA, DMF, rt, 10 h, 31–48%.
The GSK-3β enzymatic inhibition of all the target compounds was analyzed using human recombinant GSK-3βusing a luminescence method.28 Compounds 2a-2k displayed potent GSK-3β kinase inhibitory activities. The IC50 values ranged from 18 to 270 nM, similar to compound 1 (Table 1). These results suggested that the GSK-3β inhibitory effect was durable by introducing tacrine moiety at the solvent exposure zone of 1. The inhibitory potency was significantly reduced when the amino group (1) was replaced by ethoxy group (2a), carboxyl group (2b) and methoxy group (2d). This indicated that the amino group was crucial for the enzymatic inhibition of 1, probably because the NH of the amide bond forms an intramolecular hydrogen bond with the oxygen atom of the thiazole ring to stabilize the orientation of the amide bond. However, 2c did not show potency against hAChE activity. Given the importance of the amino group on GSK-3β inhibitory activity, tacrine was introduced in the position of the amide group by alkyl chain, resulting in compounds 2e-2k. As shown in Table 1, compound 2e showed potent inhibitory activity against GSK-3β, with IC50 value of 68 nM, while 6-chlorotacrine and 7-methoxytacrine hybrids (2h-2k) showed similar GSK-3β inhibitory effects to 2e, with activities ranging from 63 to 98 nM. These results suggested that GSK-3β inhibition could rarely be affected by changing the length of the side chain, or by addition of the tacrine moiety. However, 2h-2k exhibited potent antiproliferative activities against human neuroblastoma SH-SY5Y cell lines (IC50 values ranging from 3.1 to 8.9 μM), the improved cytotoxicity might limit the further cell-based evaluation. It is noteworthy that 2f and 2g, with an n-propane or an n-hexane linker, displayed potent GSK-3β inhibition with IC50 of 66 and 18 nM, respectively. In addition, 2g exhibited an IC50 of 30 μM against SH-SY5Y cells while 2f exhibited a slightly lower IC50 value of 18 μM. According to their single- or double-digit nanomolar activities on ChEs and GSK-3β, these two compounds were thought to own the best safety among all the target molecules to exert their biological effects as dual GSK-3β/AChE inhibitors. Here we chose 2f as the representative for further investigations.
Table 1. Inhibition of hGSK-3β, hAChE and hBuChE, Aβ1-42 Self-Aggregation, and Anti-Proliferative Activitiesa.
| compd | R | R′ | n | hGSK-3β (IC50 nM)a | hAChE (IC50 nM)a | hBuChE (IC50 nM)a | SIc | Aβ1–42 IR (%)d | SH-SY5Y (IC50 μM)e |
|---|---|---|---|---|---|---|---|---|---|
| 2a | OEt | 180 ± 20 | 310 ± 20 | 610 ± 50 | 2 | 30 ± 2.9 | 7.1 ± 0.6 | ||
| 2b | OH | 270 ± 30 | 580 ± 50 | 450 ± 50 | 0.8 | 25 ± 2.1 | 21 ± 1.8 | ||
| 2c | NH2 | 34 ± 3.1 | 300 ± 10 | 150 ± 10 | 0.5 | 15 ± 1.8 | 19 ± 2.5 | ||
| 2d | OMe | 99 ± 10 | 130 ± 7.1 | 28 ± 2.3 | 0.2 | 47 ± 5.1 | 13 ± 0.9 | ||
| 2e | H | 1 | 68 ± 5.3 | 6.3 ± 0.2 | 51 ± 6.1 | 9 | 40 ± 4.3 | 6.3 ± 0.5 | |
| 2f | H | 2 | 66 ± 6.2 | 6.4 ± 0.3 | 260 ± 32 | 43 | 46 ± 4.1 | 18 ± 1.2 | |
| 2g | H | 5 | 18 ± 1.4 | 22 ± 2.1 | 100 ± 7.9 | 5 | 39 ± 4.1 | 30 ± 2.8 | |
| 2h | 6-Cl | 1 | 63 ± 4.9 | 2.1 ± 0.9 | 410 ± 39b | 190 | 40 ± 3.7 | 2.3 ± 0.3 | |
| 2i | 6-Cl | 2 | 98 ± 9.3 | 3.6 ± 0.3 | 290 ± 23b | 80 | 42 ± 4.1 | 3.1 ± 0.2 | |
| 2j | 7-OMe | 1 | 83 ± 6.5 | 23 ± 1.7 | 2100 ± 180b | 91 | 47 ± 4.5 | 8.9 ± 0.9 | |
| 2k | 7-OMe | 2 | 65 ± 4.7 | 38 ± 4.2 | 2300 ± 170b | 60 | 45 ± 4.2 | 2.9 ± 0.2 | |
| Tac. | 230 ± 31 | 40 ± 3.7 | 0.17 | <5 | 120 ± 15 |
Recombinant human AChE, BuChE, and GSK-3β were used. Data are the mean of at least three independent determinations.
BuChE (EC 3.1.1.8) from horse serum.
Selectivity index (SI) = IC50hBuChE/IC50hAChE.
Inhibition of Aβ1–42 self-aggregation investigated by the thioflavin-T fluorescence assay. Assays were carried out in the presence of 20 μM inhibitor and 50 μM Aβ1–42.
SH-SY5Y cells exposing for 48 h to compound.
The inhibitory potency of compounds 2a–2k on hAChE and BuChE from human serum (hBuChE) was tested by Ellman’s method (Table 1).29 All tested derivatives showed an IC50 against hAChE in nanomolar range, which were consistent with the data on the inhibition of cholinesterase from the electric eel (see Table S1). We retained the amino group of the GSK-3βI 1 or introduced other groups, including carboxylic acid, methyl ester and ethyl ester, at the formyl side chain. Hybrids exhibited slight hAChE inhibitory activities from 130 to 580 nM. Concerning the effect of the spacer chain length on the anti-AChE activity, we introduced various lengths of alkyl chains as linker to the side chain based on the GSK-3βI 1. As shown in Table 1, the compounds with various linkers exhibited potent the anti-AChE activities (from 2.1 to 280 nM), which verified the flexibility of the side chain in AChE inhibitory activity. It should be noted that 2h–2k, bearing 6-Cl-tacrine or 7-MeO-tacrine fragments, were superior AChEIs to tacrine. Besides, these four compounds exhibited much higher inhibitory potency on AChE than on BuChE, indicating that such modifications could help to design highly selective hAChE inhibitors. The most potent hAChE inhibitor was 2h (IC50 = 2.1 nM), which also had the best selectivity toward AChE with 190-fold better than BuChE. Unfortunately, these hybrids possessed a strong antiproliferative activity against SH-SY5Y cell lines, giving IC50 values spanning from 2.3 to 8.9 μM. The optimal tethers were two–three carbon chains (2e, 2f) for the thiazolylpyridine hybrids bearing tacrine unit, showing IC50 values of 6.4 and 6.5 nM, respectively.
Considering that roles of AChE and BuChE in the modulation of the central cholinergic tone vary with the progression of the disease, it is conceivable that selective AChEIs are more effective in the early stages of AD. The best selectivity for hAChE was demonstrated by 2h, which was 190-fold more active toward hAChE versus BuChE. Targeting BuChE has become a viable alternative for the treatment of patients with mild-to-late stages of AD. BuChE is primarily expressed and secreted by glial cells, and it is thought to play a compensatory role in response to the decrease of AChE activity as AD progresses. Selective inhibition of BuChE thus represents a useful strategy to ameliorate the cholinergic deficit and improve the cognitive performance of patients in the late stages of AD. Hybrid 2d was the most potent and selective hBuChEI (IC50 = 28 nM) of this series, with a 4.6-fold better inhibition. The accumulation of oligomeric aggregates of Aβ is considered to play an important role in the pathogenesis of AD. Thus, the inhibition of Aβ oligomerization has been investigated as an attractive therapeutic strategy for AD treatment. All compounds were evaluated for their inhibitory capacity on self-induced Aβ1–42 aggregation based on a thioflavin T-based fluorometric assay. Most of the hybrids showed moderate inhibition on Aβ1–42 self-aggregation (ranging from 15 to 47%, Table 1), while tacrine was not able to inhibit Aβ1–42 self-aggregation. Considering its better inhibition activity of AChE, Aβ1–42 self-aggregation, and smaller molecular weight compared to 2g, we assumed that the most promising achievement of the present study is 2f, which showed a dual low-nanomolar inhibition profile in in vitro GSK-3β/AChE (IC50 values of 66 and 6.5 nM, respectively).
To investigate the binding pattern of 2f with hAChE, molecular docking studies were performed using Discovery Studio (DS). As shown in Figure 2, 2f bound to AChE in a dual-site manner by occupying both CAS and PAS. The location and orientation of 2f in AChE active-site gorge was consistent with the critical role of tacrine fragments, which binds at the anionic site by stacking against the aromatic rings of Trp86 and Tyr124. The endocyclic nitrogen of 2f was hydrogen bonded to the main-chain carbonyl oxygen of the catalytic residue His447, indicating that the catalytic triad was disrupted. It was noticeable that pyridothiazole moiety was located at the PAS of hAChE binding groove, which formed π–π stacking contacts with the side chain of Trp286. The Lineweaver–Burk plots for AchE showed the point of intersection was in the third quadrant, but very close to the X axis. It is a mixed but noncompetitive combination, suggesting that 2f should combine the PAS site as a noncompetitive inhibitor, similarly to donepezil.
Figure 2.
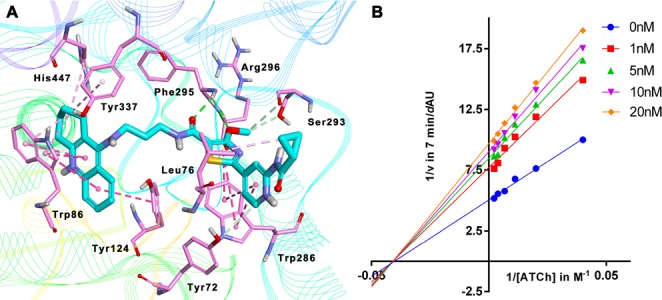
Binding mode prediction of 2f with hAChE (PDB id: 4EY7). (A) Compound 2f was shown in stick mode colored in green. Key residues were labeled as thin stick mode colored in orange. Intermolecular interactions were shown with different colored dotted lines: light green, hydrophobic contact; yellow, π-cation; purple, π-alkyl contact. (B) Lineweaver–Burk plots resulting from subvelocity curves of AChE activity with different substrate concentrations (25–450 μM) in the absence and presence of 1, 5, 10, and 20 nM 2f.
It is widely believed that elevated levels of hyperphosphorylated tau (p-tau) protein are causal in the process of forming the neurofibrillatory tangles, which are one of the pathologic features in the brains of AD patients.30 Encouraged by the potent inhibitory activity of 2f against GSK-3β, Western blot assays were conducted to further confirm the effect of the dual-action targeting of GSK-3β and AChE on p-tau levels. We treated mouse neuroblastoma N2a-Tau cells with increasing concentrations of 2f for 6 h, and the lysates were subjected to Western blot analyses. The results in Figure 3B showed that the phosphorylation of tau protein at Ser396 site decreased after treatment with 10 μM 2f. Meanwhile, the total tau protein level was not changed resulting in an attenuation of p-tau/total tau. These data suggested that 2f could significantly inhibit tau protein hyperphosphorylation, thus offering promises for reducing NFTs in the development of thiazolylpyridine-based multitarget drug candidates for AD treatment.
Figure 3.
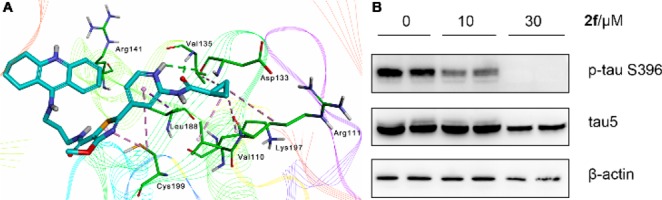
(A) Binding mode prediction of 2f with hGSK-3β (PDB id: 4PTC). Compound 2f was shown in stick mode colored in green. (B) Compound 2f effectively reduce tau protein hyperphosphorylation in mouse neuroblastoma N2a-Tau cells. Protein expression was detected by immunoblot analysis with a specific antibody. The effect of 2f on phosphorylation status of tau.
Cognition-improving potency is the most important profile of anti-AD agents. Given the interesting multipotent activity profile of 2d and 2f, their ability to ameliorate scopolamine-induced cognition impairment in ICR mice was investigated in a behavioral study by a Morris water maze test. Scopolamine-induced cognition-impaired adult mice were used as an animal model to measure the cognitive improvement effects of 2d and 2f, compared with tacrine (20 μmol/kg body weight). Five days of memory training and learning and a probe trial on the sixth day were performed in the test. The mean escape latency values and distance to target of all the groups on the sixth day are shown in Figure 4 (the mean escape latency values for each training day are provided in Table S2 and the trajectories of the mice in each group are shown in Figure S5). Compared to the control group, scopolamine-induced mice exhibited a prominent delay of mean escape latency compared to the control group (41 s vs 8.6 s). Meanwhile, the 2f-treated group had better performance than the tacrine-treated group, displaying the superior latency to target (17 s) to the tacrine group (26 s). Furthermore, 2d significantly reduced the latency to target (11 s), closed to the control group. The trend of distance to target was basically consistent with the latency to target (Figure 5B). The results suggested 2d and 2f considerably ameliorated the cognitive impairment of the treated mice and was much better than tacrine, and also suggested that 2d and 2f may penetrate the blood–brain barrier and target the central nervous system.
Figure 4.

Effects of oral administration of 2d (15 mg/kg), 2g (15 mg/kg), 2f (15 mg/kg), and tacrine (15 mg/kg) on scopolamine-induced cognitive impairment in ICR mice determined by the Morris water maze test. (A) Latency to target. (B) Distance to target. Data are presented as the mean ± SEM (n = 6; ***p < 0.001, ****p < 0.0001 vs scopolamine group).
Figure 5.
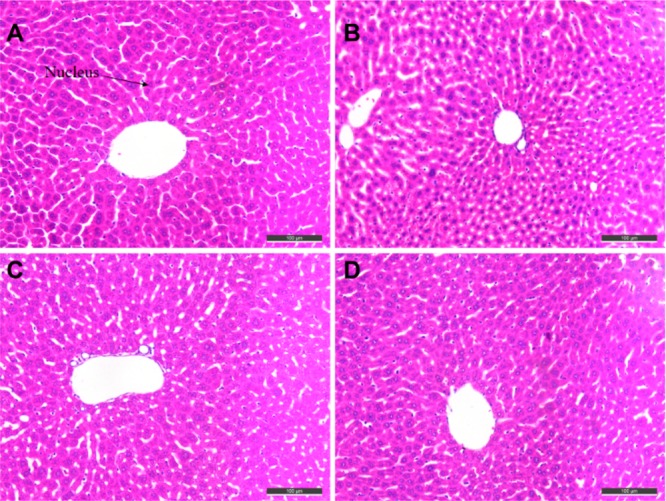
Histomorphological appearance of livers of male mice after treatment with the solvent only (control, A) or 22 h after administration of tacrine (B), 2d (C), or 2f (D). HE staining, original magnification × 200.
Given the serious hepatotoxicity of tacrine for its long-term use, the possible drug-induced hepatotoxicity associated with the presence of the tetrahydroacridine nucleus in 2d and 2f was investigated by comparing to tacrine. Heparinized serum was collected 8, 22, and 36 h after the administration of tacrine, 2d, and 2f, and the levels of alanine aminotransferase (ALT) and aspartate aminotranferase (AST) were evaluated (Table 2). ALT is primarily distributed in liver, and damaged hepatocytes release ALT and AST into the extracellular space. Thus, the serum levels of AST and ALT are directly related to the extent of the liver damage. Eight hours after tacrine administration, the levels of ALT and AST were slightly induced. However, 2d and 2f did not show any hepatotoxicity at all time-points, and the level of ALT and AST even slightly reduced at 8 h, compared to those of the tacrine group. To confirm the hepatotoxicity of 2d and 2f ulteriorly, morphologic studies by immunohistochemical staining were performed. Treatment of tacrine (Figure 5B), 2d (Figure 5C), or 2d (Figure 5D) did not result in remarkable morphologic changes, such as hepatic cell gap and number in liver compared to the control group (Figure 5A). Cytotoxicity profile was also demonstrated in human hepatocellular HepG2 cells, where 2f showed low impairment of the cell viability (IC50 = 35 μM) (see Figure S4). In general, 2f showed hepatic safety profile, ensuring its further development.
Table 2. ALT and ASAT Activity after the Administration of 2d, 2f, and Reference Compound Tacrine (30 mg/kg)a.
| ALT (U/L) |
AST (U/L) |
|||||
|---|---|---|---|---|---|---|
| group | 8 h | 22 h | 36 h | 8 h | 22 h | 36 h |
| control | 35 ± 8.4 | 39 ± 9.4 | 30 ± 8 | 120 ± 20 | 120 ± 20 | 120 ± 13 |
| tacrine | 32 ± 4.9 | 25 ± 7 | 25 ± 7 | 130 ± 22 | 120 ± 30 | 110 ± 28 |
| 2d | 25 ± 7.9 | 31 ± 12 | 30 ± 4 | 110 ± 24 | 120 ± 19 | 130 ± 32 |
| 2f | 28 ± 7.6 | 29 ± 7.9 | 29 ± 7 | 110 ± 24 | 120 ± 24 | 120 ± 21 |
Values were expressed as the mean ± SD (n = 6).
In conclusion, dual GSK-3β/AChE inhibitors were successfully designed, which showed nanomolar potency on both targets. To the best of our knowledge, it is the first attempt to design GSK-3β/AChE dual inhibitor as anti-AD agents. The representative compound 2f significantly inhibited tau protein hyperphosphorylation, while it also reduced the self-aggregation of Aβ1–42. Furthermore, ICR male mice treated with scopolamine exhibited significantly ameliorated memory performance in a Morris water maze test when 2f was administrated. Additionally, 2f exhibited preliminary safety in hepatotoxicity studies, without improving the level of ALT and AST. Such biological properties highlight 2f as a very interesting prototype in the search for new disease-modifying drugs in the treatment of AD.
Acknowledgments
We gratefully thank the support from the grants of the National Natural Science Foundation of China (Nos. 81402851, 81373956, and 81573281) and Natural Science Foundation of Jiangsu Province (BK20140957). We also thank the support from Fundamental Research Funds for the Central Universities (2015ZD009), Jiangsu Qing Lan Project.
Glossary
ABBREVIATIONS
- MTDLs
multitarget-directed ligands
- AD
Alzheimer’s disease
- GSK-3β
glycogen synthase kinase-3β
- h-AChE
human acetylcholinesterase
- CAS
catalytic active site
- PAS
peripheral anionic site
- Aβ
β-amyloid
- NFTs
neurofibrillary tangles
- CNS
central nervous system
- BuChE
butyrylcholinesterase
- PAS
peripheral anionic site
- CPCCI
cyclopropanecarbonyl chloride
- TFA
trifluoroacetic acid
- DIAD
diisopropyl azodiformate
- DIPEA
N,N-diisopropylethylamine
- ALT
alanine aminotransferase
- AST
aspartate aminotranferase
Biographies
Dr. Feng Feng graduated in Chemistry from Shaanxi Normal University in 1991. He received his Ph.D. in Medicinal Chemistry from China Pharmaceutical University in 2001, and his thesis focused on anti-tumor natural products. He worked as a visiting scholar at the University of California, Irvine, in 2005. In 2010, he was promoted to Professor of Natural Medicinal Chemistry at China Pharmaceutical University. He has published more than 70 papers in journals indexed by the Science Citation Index. His major research interests include extraction and isolation of chemical constituents from natural medicines, structural modification, and analysis of active compositions.
Dr. Haopeng Sun graduated in Pharmacy from the China Pharmaceutical University in 2006. He received his Ph.D. in 2011 in Medicinal Chemistry, with a thesis on the structural optimization and mechanism study of the natural product. In 2014, he was promoted to Associate Professor of Medicinal Chemistry at China Pharmaceutical University. So far, he has published more than 70 papers in peer-reviewed journals indexed by Science Citation Index. His major research interests include the design and development of small molecule bioactive compounds targeting neurodegenerative diseases, inflammation modulation, and cancer prevention.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00463.
Synthetic procedures, characterization data, binding mode prediction of 2f with eeAChE and hGSK-3β, and biological assay procedures (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Lazarov O.; Marr R. A. Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol. 2010, 223, 267–281. 10.1016/j.expneurol.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth H. W.; LaFerla F. M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Kepp K. P. Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 2012, 112, 5193–5239. 10.1021/cr300009x. [DOI] [PubMed] [Google Scholar]
- Dolan P. J.; Johnson G. V. The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Disc. 2010, 13, 595–603. [PMC free article] [PubMed] [Google Scholar]
- Weingarten M. D.; Lockwood A. H.; Hwo S. Y.; Kirschner M. W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 1858–1862. 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. The relationship between amyloid and tau. J. Mol. Neurosci. 2003, 20, 203–206. 10.1385/JMN:20:2:203. [DOI] [PubMed] [Google Scholar]
- Avila J.; Wandosell F.; Hernández F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 2010, 10, 703–710. 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- Phiel C. J.; Wilson C. A.; Lee V. M.; Klein P. S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Hernández F.; Borrell J.; Guaza C.; Avila J.; Lucas J. J. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3β in the brain but do not form tau filaments. J. Neurochem. 2002, 83, 1529–1533. 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- Morales-Garcia J. A.; Luna-Medina R.; Alonso-Gil S.; Sanz-SanCristobal M.; Palomo V.; Gil C.; Santos A.; Martinez A.; Perez-Castillo A. Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem. Neurosci. 2012, 3, 963–971. 10.1021/cn300110c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maqbool M.; Mobashir M.; Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. 10.1016/j.ejmech.2015.10.018. [DOI] [PubMed] [Google Scholar]
- Giacobini E.; Gold G. Alzheimer disease therapy—moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686. 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- Sivaprakasam P.; Han X.; Civiello R. L.; Jacutin-Porte S.; Kish K.; Pokross M.; Lewis H. A.; Ahmed N.; Newitt J. A.; Baldwin E. T. Discovery of new acylaminopyridines as GSK-3 inhibitors by a structure guided in-depth exploration of chemical space around a pyrrolopyridinone core. Bioorg. Med. Chem. Lett. 2015, 25, 1856–1863. 10.1016/j.bmcl.2015.03.046. [DOI] [PubMed] [Google Scholar]
- Esquivias-Pérez M.; Maalej E.; Romero A.; Chabchoub F.; Samadi A.; Marco-Contelles J.; Oset-Gasque M. J. Nontoxic and neuroprotective β-naphthotacrines for Alzheimer’s disease. Chem. Res. Toxicol. 2013, 26, 986–992. 10.1021/tx400138s. [DOI] [PubMed] [Google Scholar]
- Inestrosa N. C.; Dinamarca M. C.; Alvarez A. Amyloidcholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. 10.1111/j.1742-4658.2007.06238.x. [DOI] [PubMed] [Google Scholar]
- Nitsch R. M.; Slack B. E.; Wurtman R. J.; Growdon J. H. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 1992, 258, 304–307. 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Zemek F.; Drtinova L.; Nepovimova E.; Sepsova V.; Korabecny J.; Klimes J.; Kuca K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–74. [DOI] [PubMed] [Google Scholar]
- Hartmann S.; Möbius H. J. Tolerability of memantine in combination with cholinesterase inhibitors in dementia therapy. Int. Clin. Psychopharm. 2003, 18, 81–85. 10.1097/00004850-200303000-00003. [DOI] [PubMed] [Google Scholar]
- Leon R.; Garcia A. G.; Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. 10.1002/med.20248. [DOI] [PubMed] [Google Scholar]
- Spilovska K.; Korabecny J.; Nepovimova E.; Dolezal R.; Mezeiova E.; Soukup O.; Kuca K. Multitarget tacrine hybrids with neuroprotective properties to confront Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 1006–1026. 10.2174/1568026605666160927152728. [DOI] [PubMed] [Google Scholar]
- Romero A.; Cacabelos R.; Oset-Gasque M. J.; Samadi A.; Marco-Contelles J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. 10.1016/j.bmcl.2013.02.017. [DOI] [PubMed] [Google Scholar]
- Nepovimova E.; Korabecny J.; Dolezal R.; Babkova K.; Ondrejicek A.; Jun D.; Kuca K. Tacrine-trolox hybrids: a novel class of centrally active, nonhepatotoxic multi-target-directed ligands exerting anticholinesterase and antioxidant activities with low in vivo toxicity. J. Med. Chem. 2015, 58, 8985–9003. 10.1021/acs.jmedchem.5b01325. [DOI] [PubMed] [Google Scholar]
- Nepovimova E.; Uliassi E.; Korabecny J.; Peña-Altamira L. E.; Samez S.; Pesaresi A.; Bolognesi M. L. Multitarget drug design strategy: quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–89. 10.1021/jm5010804. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kaur M.; Chadha N.; Silakari O. Hybrids: a new paradigm to treat Alzheimer’s disease. Mol. Diversity 2016, 20, 271–297. 10.1007/s11030-015-9628-9. [DOI] [PubMed] [Google Scholar]
- Reichelt A.; Bailis J. M.; Bartberger M. D.; Yao G.; Shu H.; Kaller M. R.; Allen J. G.; Weidner M. F.; Keegan K. S.; Dao J. H. Synthesis and structure–activity relationship of trisubstituted thiazoles as Cdc7 kinase inhibitors. Eur. J. Med. Chem. 2014, 80, 364–382. 10.1016/j.ejmech.2014.04.013. [DOI] [PubMed] [Google Scholar]
- But T. Y. S.; Toy P. H. Organocatalytic mitsunobu reactions. J. Am. Chem. Soc. 2006, 128, 9636–9637. 10.1021/ja063141v. [DOI] [PubMed] [Google Scholar]
- Baki A.; Bielik A.; Molnar L.; Szendrei G.; Keseru G. M. A high throughput luminescent assay for glycogen synthase kinase-3beta inhibitors. Assay Drug Dev. Technol. 2007, 5, 75–84. 10.1089/adt.2006.029. [DOI] [PubMed] [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V. Jr.; Feather-Stone R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Cai Z. Y.; Zhao Y.; Zhao B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 864–879. 10.2174/156720512802455386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.