

Abstract
Neurotensin exerts potent analgesic effects following activation of its cognate GPCRs. In this study, we describe a systematic exploration, using structure-based design, of conformationally constraining neurotensin (8–13) with the help of macrocyclization and the resulting impacts on binding affinity, signaling, and proteolytic stability. This exploratory study led to a macrocyclic scaffold with submicromolar binding affinity, agonist activity, and greatly improved plasma stability.
Keywords: Neurotensin, Neurotensin (8−13), macrocycles, GPCR, ring closing metathesis (RCM), conformation, stability
Neurotensin (NT) is a 13-residue peptide N-terminally pyroglutamylated (pGlu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu), which was discovered in 1973 by Carraway et al.1 NT acts as a neurotransmitter and neuromodulator in the central nervous system via two G-protein coupled receptors (GPCRs), NTS1 and NTS2. NTS1 is coupled to several G protein pathways including the Gαq pathway2 and acts as a hormone receptor in the gastrointestinal tract and endothelial cells.3,4 Both receptors, when activated by NT, induce a potent analgesic effect5,6 similar to opioids, yet independently of the opioid receptors.7,8
The neurotensinergic system has been studied to a large extent for its potential in the treatment of pain, and as a result, numerous analogues have been produced to better understand and improve upon the properties of NT.
Structure/activity relationship studies on NT revealed that its C-terminal hexapeptide, NT (8–13), retains all the binding and agonist activity of the full-length peptide.9,10 This shorter peptide has therefore been the scaffold of choice for most studies targeting neurotensin receptors.4
However, NT (8–13) displays the characteristics that often limit the use of peptides as drugs, namely, short in vivo half-life and poor oral bioavailability. Modifications to NT in the form of linear analogues with improved stability and excellent binding affinities have been reported. More specifically, replacement of l-residues by their d-isomers,11 reduction of amide bonds,12N-methylation, or the use of unnatural amino acids13,14 proved to be beneficial when introduced at specific positions in the peptide.
Macrocyclization is a field-tested approach to protect peptides against proteolytic degradation15−18 and has been exploited to successfully improve the pharmacological properties of several peptides.19 In addition to increased proteolytic stability, macrocyclization allows control over peptide conformations20 and fine-tuning of their structural properties, in addition to providing insights on the receptor’s tolerance to ligand conformational changes. To date, only a handful of studies have been reported on the implementation of macrocyclization on NT.21,22 Interestingly, head-to-tail cyclization of NT (8–13) resulted in analogues that display central physiological effects when injected intravenously.23,24
At the outset of this study aimed at identifying suitable sites for macrocyclization on NT, there was evidence that several peptidergic GPCRs recognized turn structures.25,26 In addition, several structural studies based on solution NMR and solid-state NMR, molecular modeling, and mutagenesis suggested that NT was no exception.27,28 At that time, the X-ray structure of NTS1 was not published.
We chose to constrain NT (8–13) via macrocyclization using ring-closing metathesis (RCM). RCM has been widely used to induce turn structure on peptides29 and is particularly suited because of its high tolerance to diversified functional groups.30,31 The required allyl groups are easily integrated into peptides on solid phase (using Fmoc strategy on 2-chlorotrityl chloride resin), usually with the help of commercially available unnatural amino acids such as allylglycine. The macrocyclyzation reaction can be carried out directly on solid support (Scheme 1). Benzoquinone is added along with the catalyst to prevent alkene isomerization,32 and the RCM reaction often benefits from microwave irradiation.33 After simultaneous cleavage of the peptide from the resin and deprotection of the lateral chains under acidic conditions, purification on preparative LC–MS allowed isolation of the desired macrocycle.
Scheme 1. Macrocyclization Reaction.

Reagents and conditions: (a) Hoveyda–Grubbs second generation catalyst, benzoquinone, DCE, 50 °C, microwave, 1 h; (b) TFA/DCM/TIS, 1 h.
Since NT (8–13) is a short peptide, folding it into a turn implies linking the lateral chains of residues situated on both extremities (C- and N-termini). Regardless of the impact on conformation, replacing Ile12 or Leu13 by allylglycine is only a minor structural change from a steric standpoint, and removing one of the side chains of Arg8 or Arg9 has been reported to have a relatively low impact on NTS1-binding affinity.8 Additionally, replacement of Arg by Lys at positions 8 and 9 has only very limited impact on binding and signaling.34
These considerations led to the design and synthesis of macrocycles 1 and 2 (Figure 1). They both form 17-membered rings; however, 1 is cyclized between positions 8 and 12, whereas 2 is cyclized between positions 9 and 13. As a result, 1 bears a C-terminal exocyclic Leu, whereas 2 bears an N-terminal exocyclic Lys residue.
Figure 1.

Structures of neurotensin (8–13) and macrocyclic analogues explored in this study.
The ability of these compounds to bind the NTS1 receptor was determined using a competitive ligand binding assay. Briefly, increasing concentrations of the compounds were incubated with a constant amount of 125I-[Tyr3]NT and NTS1-expressing cell membranes. After filtration, NTS1-bound radioactivity was quantified using a γ-counter, and IC50 values determined are reported in Table 1.
Table 1. Binding affinitiesa.
| code | sequence | IC50 (μM) ± SEM |
|---|---|---|
| NT (8–13) | RRPYIL | 0.0008 ± 0.0003 |
| 1 | [All-KPY-All]L | 17 ± 4 |
| 1-Lb | All-KPY-All-L | 1.7 ± 0.5 |
| 2 | K[All-PYI-All] | >100 |
| 3 | [d-All-KPY-All]L | 10 ± 1.6 |
| 4 | [All-KPY-d-All]-L | 14.3 ± 10.0 |
| 5 | [d-All-KPY-d-All]-L | 19.8 ± 4.9 |
| 6 | [S(All)PYS(All)]L | >100 |
| 7 | KP[Y(All)S(All)L] | 29.8 ± 4.0 |
| 8 | K[All-PY-All]IL | 7.6 ± 2.9 |
| 9 | K[S(All)PY(All)]IL | 10.3 ± 2.6 |
| 10 | [NonKPY(All)]IL | 0.4 ± 0.2 |
| 10-Lb | NonKPY(All)IL | 1.4 ± 0.6 |
IC50 values were determined as described in the Supporting Information. Measurements were performed in triplicate and represent the means ± standard error of the mean (SEM) of at least three independent experiments.
Linear precursors of compounds 1 and 10. All: allylGlycine; Non: nonenoyl.
First, although both compounds have low affinity, compound 1 seems to be preferred (IC50 17 vs >100 μM). However, its linear precursor possesses 10-fold higher affinity than its macrocyclic counterpart (Table 1, entry 1-L, IC50 1.7 μM). This clearly shows that the conformational changes induced by this macrocyclization position alter receptor recognition. This also indicates that simultaneous replacement of Arg8 and Ile12 by allylglycine impairs binding.
Macrocycles 3–5 were then produced using d-allylglycine in N-terminal (3), C-terminal (4), or at both positions (5) as an attempt to elucidate whether these changes in stereochemistry would impact binding.
Although macrocycle 3 shows minor improvement over macrocycle 1 (IC50 10 vs 17 μM, respectively), it remains marginal compared to the reference peptide NT (8–13) (IC50 0.8 nM). Since these macrocycles seemed to be deleterious for binding affinity, we decided to increase the length of the side-chain linker, which was expected theoretically to make the resulting macrocycles more flexible to adapt to the binding pocket.
To accomplish this, we synthesized linker A (Figure 2) by alkylating the lateral chain of Boc-Ser-OH with allyl bromide35 followed by Boc deprotection and subsequent Fmoc protection of the α-amine. This linker was then used during solid phase peptide synthesis to produce the 21-membered macrocycle 6, which possesses lower affinity for NTS1 than its smaller, more constrained congeners.
Figure 2.

Custom linkers A and B.
In 2012, White and co-workers reported the X-ray structure of the rat NTS1 receptor (PDB ID 4grv).36 Remarkably, this was the first time that the structure of a GPCR was obtained by cocrystallizing with a peptidergic agonist, in this case, the unmodified ligand NT (8–13). This allowed us to assess the previous results in a new context. Essentially, the newly reported structure demonstrated that the peptide NT (8–13) actually adopts a linear conformation inside the binding pocket, which was congruent with several structural studies on neurotensin.9,37−40
This perspective granted us with the opportunity to get an in-depth understanding of the first-generation of macrocycles and support the design of new improved analogues using the Molecular Operating Environment (MOE) package,41 which was instrumental to superimpose macrocycles 1, 2, and 6 to the receptor-bound conformation of NT (8–13) (Figure 3A,B,C, respectively). Toward this end, the macrocycle underwent a flexible alignment onto the fixed NT (8–13) structure to get the best possible fit of the features of both structures. It showed, as expected, that 1 and 2 adopted a turn structure that is too compact in comparison with the bound linear peptide, and although 6 displayed improved structural overlap, the added linkers are likely too bulky and would not fit into the receptor’s pocket, possibly because of unfavorable interactions with Asn127 or His132.
Figure 3.
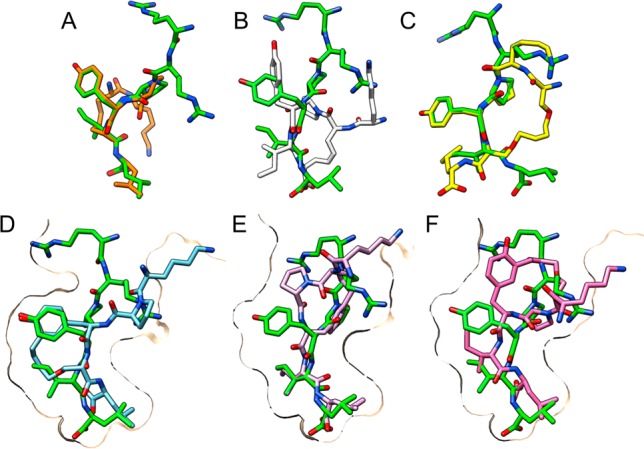
Top row: Macrocycles 1, 2, and 6 (A, B, and C, respectively) aligned with the conformation of NT (8–13) within the crystal structure of NTS1 (green). Bottom row: Macrocycles 7, 8, and 9 (D, E, and F, respectively) docked into NTS1. NT (8–13) (green) is shown for comparison. Molecular graphics were performed with the UCSF Chimera package.43
In light of the above, it became apparent that the design of a linear-shaped, low steric bulk macrocycle implied that the extremities could not be bound together, be it by a short or a long linker. On such a short peptide, new options appeared in the form of side-chain-to-C-terminal or side-chain-to-N-terminal macrocyclization. Knowing that existing SAR on NT (8–13) had demonstrated that an aromatic residue is vital in position 11 in order to bind to NTS1, the replacement of Tyr by a linker such as allylglycine or serine(O-allyl) was therefore not a viable avenue.9,27,34,40,42
To overcome this, ortho-allylated tyrosine linker B (Figure 2) was synthesized via a Claisen rearrangement of the commercially available Fmoc-Tyr(O-allyl)-OH in the presence of diethylaluminum chloride as a Lewis acid.44 To prevent an unfavorable interaction of the free phenol group with the ruthenium catalyst during the RCM step, which would decrease macrocyclization yield,45 phenol was acetylated immediately prior to the RCM. The acetyl group was removed before the final deprotection step by treatment with piperidine similar to Fmoc deprotection.
Using this strategy, macrocycle 7, 8, and 9 were produced, featuring side-chain-to-C-terminal link between Tyr11 and linker A in position 12 and side-chain-to-N-terminal link between Tyr11 and allylGly or linker A in position 9, respectively. In addition to affinity measurements, we used molecular modeling to dock these compounds into a homology model of human NTS1 based on the crystal structure of rat NTS1.
Although docking results (Figure 3, bottom row) indicate a good overlap with the reference peptide, the measured affinities of these compounds are still very low (7.6–29.8 μM). Taken together, these results show that the receptor is rather intolerant to minor changes in the ligand’s conformation in the core of the binding pocket.
Closer examination of the structure of NT (8–13) in the binding pocket of NTS1 revealed a certain proximity between residues Tyr11 and Arg8 (7.4 Å) as well as the same orientation with an apparently empty space between them (Figure 4A). In order to exploit this feature, macrocycle 10 was designed, where Arg8 was replaced with nonenoic acid, which possesses the appropriate length to reach the Tyr11. This compound gave very promising docking results (Figure 4B,C) and turned out to be the best binding macrocycle of this series with an IC50 of 0.4 μM. Interestingly, 10 possesses a three-fold improved affinity compared to its linear precursor (10-L, IC50 1.4 μM), which suggests that the conformational constraint imposed by cyclization favorably impacts interaction with the receptor. Supplementary Figure S1 shows that 10 is involved in fewer interactions with the receptor than NT (8–13) is.
Figure 4.

Comparison of the crystallized NT (8–13) conformation with 10. (A) NTS1 crystal. Blue arrow indicates available space between Tyr11 and Arg8. (B) Superimposition of NT (8–13) from the crystal and macrocycle 10 (docked). (C) Macrocycle 10 docked into a homology model of human NTS1 based on the crystal. Molecular graphics were performed with the UCSF Chimera package.43
The ability of 10 to activate the receptor was confirmed using BRET-based biosensors assay to measure activation of the Gαq pathway and the recruitment of β-arrestin 2 (see Supplementary Figure S2). Indeed, compound 10 was able to induce full Gαq activation (EC50 166.3 nM) and to stimulate β-arrestin 2 recruitment at NTS1 (EC50 1370 nM).
Furthermore, the sensitivity of 10 to proteolytic degradation was assessed by incubation with rat plasma followed by UPLC–MS quantification (see Supporting Information for full experimental procedures). Under these conditions, 10 possessed a half-life of 12 h, which is far superior to NT (8–13), which possesses a half-life around 3 min (Figure 5). Such extended stability is a significant advantage of these compounds as pharmacological tools. Further work is under way to decipher the SAR of this new lead series and use it to better understand the pharmacology of the neurotensinergic system.
Figure 5.

Plasma stability of 10 compared to NT (8–13).
In conclusion, we identified a novel macrocyclic scaffold that possesses good binding affinity for the NTS1 receptor, behaves as a full agonist, and displays excellent in vitro stability. We believe macrocycle 10 is a promising starting point for the development of new neurotensin macrocyclic analogues. Subsequent analogues in this series will be reported in due course.
Acknowledgments
Funding from the Réseau Québécois de Recherche sur le Médicament (RQRM), the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian institutes of Health Research (CIHR), and the Canadian Foundation for Innovation (CFI) is gratefully acknowledged. E.M. is a member of the RQRM and Proteo networks. P.S. is the holder of the Canada Research Chair Tier 1 in the Neurophysiopharmacology of chronic pain. The authors wish to thank Dr. Pierre-Luc Boudreault for help with the MOE package.
Glossary
ABBREVIATIONS
- NT
neurotensin
- NT (8–13)
neurotensin (8–13)
- NTS1
neurotensin receptor 1
- NTS2
neurotensin receptor 2
- GPCR
G-protein coupled receptor
- RCM
ring-closing metathesis
- NMR
nuclear magnetic resonance
- BRET
bioluminescence resonance energy transfer
- UPLC
ultra performance liquid chromatography
- MS
mass spectroscopy
- HRMS
high resolution mass spectroscopy
- All
allylGlycine
- DCE
dichloroethane
- TFA
trifluoroacetic acid
- DCM
dichloromethane
- TIS
triisopropylsilane
- DMF
dimethylformamide
- THF
tetrahydrofuran
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00500.
Complete experimental procedures, supplementary figures, compound characterization, UPLC-MS, HRMS, and binding curves (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Carraway R.; Leeman S. E. The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. S. E. J. Biol. Chem. 1973, 248 (19), 6854–6861. [PubMed] [Google Scholar]
- Besserer-Offroy É.; Brouillette R. L.; Lavenus S.; Froehlich U.; Brumwell A.; Murza A.; Longpré J. M.; Marsault É.; Grandbois M.; Sarret P.; Leduc R. The signaling signature of the neurotensin type 1 receptor with endogenous ligands. Eur. J. Pharmacol. 2017, 805 (1), 1–13. 10.1016/j.ejphar.2017.03.046. [DOI] [PubMed] [Google Scholar]
- Vincent J. P.; Mazella J.; Kitabgi P. Neurotensin and neurotensin receptors. Trends Pharmacol. Sci. 1999, 20 (7), 302–309. 10.1016/S0165-6147(99)01357-7. [DOI] [PubMed] [Google Scholar]
- Kleczkowska P.; Lipkowski A. Neurotensin and neurotensin receptors: characteristic, structure-activity relationship and pain modulation -A review. W. Eur. J. Pharmacol. 2013, 716, 1–7. 10.1016/j.ejphar.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Roussy G.; Dansereau M.-A.; Doré-Savard L.; Belleville K.; Beaudet N.; Richelson E.; Sarret P. J. J.; Spinal NTS1 receptors regulate nociceptive signaling in a rat formalin tonic pain model. J. Neurochem. 2008, 105 (4), 1100–1114. 10.1111/j.1471-4159.2007.05205.x. [DOI] [PubMed] [Google Scholar]
- Tétreault P.; Beaudet N.; Perron A.; Belleville K.; René A.; Cavelier F.; Martinez J.; Stroh T.; Jacobi A. M.; Rose S. D.; Behlke M. A.; Sarret P. Spinal NTS2 receptor activation reverses signs of neuropathic pain. FASEB J. 2013, 27 (9), 3741–3752. 10.1096/fj.12-225540. [DOI] [PubMed] [Google Scholar]
- Clineschmidt B. V.; McGuffin Neurotensin administered intracisternally inhibits responsiveness of mice to noxious stimuli. Eur. J. Pharmacol. 1977, 46 (4), 395–396. 10.1016/0014-2999(77)90236-9. [DOI] [PubMed] [Google Scholar]
- Clineschmidt B. V.; McGuffin J. C.; Bunting P. B. Neurotensin: Antinocisponsive action in rodents. Eur. J. Pharmacol. 1979, 54 (1–2), 129–139. 10.1016/0014-2999(79)90415-1. [DOI] [PubMed] [Google Scholar]
- Barroso S.; Richard F.; Nicolas-Ethève D.; Reversat J. L.; Bernassau J. M.; Kitabgi P.; Labbé-Jullié C. Identification of residues involved in neurotensin binding and modeling of the agonist binding site in neurotensin receptor 1. J. Biol. Chem. 2000, 275 (1), 328–336. 10.1074/jbc.275.1.328. [DOI] [PubMed] [Google Scholar]
- Glimcher P. W.; Margolin D. H.; Giovino a a; Hoebel B. G. Neurotensin: a new ’reward peptide. Brain Res. 1984, 291 (1), 119–124. 10.1016/0006-8993(84)90657-7. [DOI] [PubMed] [Google Scholar]
- Checler F.; Vincent J. P.; Kitabgi P. Neurotensin analogs [D-TYR11] and [D-PHE11]neurotensin resist degradation by brain peptidases in vitro and in vivo. J. Pharmacol. Exp. Ther. 1983, 227 (3), 743–748. [PubMed] [Google Scholar]
- Wustrow D. J.; Davis M. D.; Akunne H. C.; Corbin A. E.; Wiley J. N.; Wise L. D.; Heffner T. G. Reduced amide bond neurotensin 8–13 mimetics with potent in vivo activity. Bioorg. Med. Chem. Lett. 1995, 5 (9), 997–1002. 10.1016/0960-894X(95)00155-M. [DOI] [Google Scholar]
- Smith K. E.; Boules M.; Williams K.; Richelson E. NTS1 and NTS2 mediate analgesia following neurotensin analog treatment in a mouse model for visceral pain. Behav. Brain Res. 2012, 232 (1), 93–97. 10.1016/j.bbr.2012.03.044. [DOI] [PubMed] [Google Scholar]
- Fanelli R.; Besserer-Offroy É.; René A.; Côté J.; Tétreault P.; Collerette-Tremblay J.; Longpré J.-M.; Leduc R.; Martinez J.; Sarret P.; Cavelier F. Synthesis and Characterization in Vitro and in Vivo of (l)-(Trimethylsilyl)alanine Containing Neurotensin Analogues. J. Med. Chem. 2015, 58 (19), 7785–7795. 10.1021/acs.jmedchem.5b00841. [DOI] [PubMed] [Google Scholar]
- Marsault É.; Peterson M. L.. Practical Medicinal Chemistry with Macrocycles: Design, Synthesis, and Case Studies; Marsault É., Peterson M. L., Eds.; Wiley, 2017. [Google Scholar]
- Marsault E.; Peterson M. L. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 2011, 54 (7), 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- Yudin A. K. Macrocycles: lessons from the distant past, recent developments, and future directions. Chem. Sci. 2015, 6 (1), 30–49. 10.1039/C4SC03089C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. The exploration of macrocycles for drug discovery--an underexploited structural class. Nat. Rev. Drug Discovery 2008, 7 (7), 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- Beck J. G.; Chatterjee J.; Laufer B.; Kiran M. U.; Frank A. O.; Neubauer S.; Ovadia O.; Greenberg S.; Gilon C.; Hoffman A.; Kessler H. Intestinal permeability of cyclic peptides: common key backbone motifs identified. J. Am. Chem. Soc. 2012, 134 (29), 12125–12133. 10.1021/ja303200d. [DOI] [PubMed] [Google Scholar]
- Glas A.; Wamhoff E.-C.; Krüger D. M.; Rademacher C.; Grossmann T. N. Increased Conformational Flexibility of a Macrocycle-Receptor Complex Contributes to Reduced Dissociation Rates. Chem. - Eur. J. 2017, 23 (64), 16157–16161. 10.1002/chem.201702776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akunne H. C.; Darling S.; Zoski K.; Sefler a M.; He J. X.; Sawyer T. K.; Pugsley T. a; Cody W. L. Functional activity of new C-terminal cyclic-neurotensin fragment analogs. Neuropeptides 1996, 30 (3), 213–218. 10.1016/S0143-4179(96)90066-9. [DOI] [PubMed] [Google Scholar]
- Lundquist J. T.; Dix T. A. Preparation and receptor binding affinities of cyclic C-terminal neurotensin (8–13) and (9–13) analogues. Bioorg. Med. Chem. Lett. 1999, 9 (17), 2579–2582. 10.1016/S0960-894X(99)00420-5. [DOI] [PubMed] [Google Scholar]
- Van Kemmel F. M.; Dubuc I.; Bourdel E.; Fehrentz J. a; Martinez J.; Costentin J. A C-terminal cyclic 8–13 neurotensin fragment analog appears less exposed to neprilysin when it crosses the blood-brain barrier than the cerebrospinal fluid-brain barrier in mice. Neurosci. Lett. 1996, 217 (1), 58–60. 10.1016/0304-3940(96)13074-3. [DOI] [PubMed] [Google Scholar]
- Bredeloux P.; Cavelier F.; Dubuc I.; Vivet B.; Costentin J.; Martinez J. Synthesis and biological effects of c(Lys-Lys-Pro-Tyr-Ile-Leu-Lys-Lys-Pro-Tyr-Ile-Leu) (JMV2012), a new analogue of neurotensin that crosses the blood-brain barrier. J. Med. Chem. 2008, 51 (6), 1610–1616. 10.1021/jm700925k. [DOI] [PubMed] [Google Scholar]
- Tyndall J. D. a; Pfeiffer B.; Abbenante G.; Fairlie D. P. Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem. Rev. 2005, 105 (3), 793–826. 10.1021/cr040689g. [DOI] [PubMed] [Google Scholar]
- Ruiz-Gómez G.; Tyndall J. D. a; Pfeiffer B.; Abbenante G.; Fairlie D. P. Update 1 of: Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem. Rev. 2010, 110 (4), PR1–41. 10.1021/cr900344w. [DOI] [PubMed] [Google Scholar]
- Pang Y. P.; Cusack B.; Groshan K.; Richelson E. Proposed ligand binding site of the transmembrane receptor for neurotensin(8–13). J. Biol. Chem. 1996, 271 (25), 15060–15068. 10.1074/jbc.271.25.15060. [DOI] [PubMed] [Google Scholar]
- Luca S.; White J. F.; Sohal A. K.; Filippov D. V.; van Boom J. H.; Grisshammer R.; Baldus M. The conformation of neurotensin bound to its G protein-coupled receptor. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (19), 10706–10711. 10.1073/pnas.1834523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell H. E.; Sadowsky J. D.; Howard R. J.; Sampson J. N.; Chao J. A.; Steinmetz W. E.; O’Leary D. J.; Grubbs R. H. Ring-closing metathesis of olefinic peptides: Design, synthesis, and structural characterization of macrocyclic helical peptides. J. Org. Chem. 2001, 66 (16), 5291–5302. 10.1021/jo015533k. [DOI] [PubMed] [Google Scholar]
- Pérez de Vega M. J.; García-Aranda M. I.; González-Muñiz R. A role for ring-closing metathesis in medicinal chemistry: Mimicking secondary architectures in bioactive peptides. Med. Res. Rev. 2010, 31 (5), 677–715. 10.1002/med.20199. [DOI] [PubMed] [Google Scholar]
- Brik A. Metathesis in Peptides and Peptidomimetics. Adv. Synth. Catal. 2008, 350 (11–12), 1661–1675. 10.1002/adsc.200800149. [DOI] [Google Scholar]
- Hong S. H.; Sanders D. P.; Lee C. W.; Grubbs R. H. Prevention of undesirable isomerization during olefin metathesis. J. Am. Chem. Soc. 2005, 127 (49), 17160–17161. 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- Patgiri A.; Menzenski M. Z.; Mahon A. B.; Arora P. S. Solid-phase synthesis of short α-helices stabilized by the hydrogen bond surrogate approach. Nat. Protoc. 2010, 5 (11), 1857–1865. 10.1038/nprot.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier C.; van Rietschoten J.; Kitabgi P.; Poustis C.; Freychet P. Synthesis and characterization of neurotensin analogues for structure/activity relationship studies. Acetyl-neurotensin-(8--13) is the shortest analogue with full binding and pharmacological activities. Eur. J. Biochem. 1982, 124 (1), 117–124. 10.1111/j.1432-1033.1982.tb05913.x. [DOI] [PubMed] [Google Scholar]
- Boal A. K.; Guryanov I.; Moretto A.; Crisma M.; Lanni E. L.; Toniolo C.; Grubbs R. H.; O’Leary D. J. Facile and E-selective intramolecular ring-closing metathesis reactions in 310-helical peptides: A 3D structural study. J. Am. Chem. Soc. 2007, 129 (22), 6986–6987. 10.1021/ja071148m. [DOI] [PubMed] [Google Scholar]
- White J. F.; Noinaj N.; Shibata Y.; Love J.; Kloss B.; Xu F.; Gvozdenovic-Jeremic J.; Shah P.; Shiloach J.; Tate C. G.; Grisshammer R. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490 (7421), 508–513. 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutant J.; Curmi P. a; Toma F.; Monti J.-P. NMR solution structure of neurotensin in membrane-mimetic environments: molecular basis for neurotensin receptor recognition. Biochemistry 2007, 46 (19), 5656–5663. 10.1021/bi602567p. [DOI] [PubMed] [Google Scholar]
- Bittermann H.; Einsiedel J.; Hübner H.; Gmeiner P. Evaluation of lactam-bridged neurotensin analogues adjusting psi(Pro10) close to the experimentally derived bioactive conformation of NT(8–13). J. Med. Chem. 2004, 47 (22), 5587–5590. 10.1021/jm049644y. [DOI] [PubMed] [Google Scholar]
- Härterich S.; Koschatzky S.; Einsiedel J.; Gmeiner P. Novel insights into GPCR-peptide interactions: mutations in extracellular loop 1, ligand backbone methylations and molecular modeling of neurotensin receptor 1. Bioorg. Med. Chem. 2008, 16 (20), 9359–9368. 10.1016/j.bmc.2008.08.051. [DOI] [PubMed] [Google Scholar]
- Einsiedel J.; Hübner H.; Hervet M.; Härterich S.; Koschatzky S.; Gmeiner P. Peptide backbone modifications on the C-terminal hexapeptide of neurotensin. Bioorg. Med. Chem. Lett. 2008, 18 (6), 2013–2018. 10.1016/j.bmcl.2008.01.110. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2016.08; Chemical Computing Group ULC: Montreal, Canada, 2017. [Google Scholar]
- Da Costa G.; Bondon A.; Coutant J.; Curmi P.; Monti J.-P. Intermolecular interactions between the neurotensin and the third extracellular loop of human neurotensin 1 receptor. J. Biomol. Struct. Dyn. 2013, 31 (12), 1381–1392. 10.1080/07391102.2012.736776. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–12. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- The Claisen Rearrangement: Methods and Applications; Hiersemann M., Nubbemeyer U., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Macrocyclizations undergone without prior acetylation gave significantly lower yields (unpublished results).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.