

The incidence of diabetes mellitus has increased exponentially over the past 2 decades, and individuals with diabetes mellitus are at increased risk for and have poor prognosis with cardiovascular disease.1 In the myocardium, diabetes mellitus enhances fatty acid metabolism, inhibits glucose oxidation, and modifies intracellular signaling, eventually leading to diabetic cardiomyopathy.2 Diabetic cardiomyopathy is characterized by increased inflammation and accumulation of the extracellular matrix, microangiopathy, as well as impaired cardiac diastolic or systolic function.3 The matricellular proteins are a family of nonstructural extracellular matrix proteins that relay biological, chemical, and mechanical signals through binding to cell surface receptors, growth factors, proteases, and structural matrix proteins.4 At very low expression in the normal heart, most matricelluar proteins are markedly induced in response to injury, indicating roles in the woundhealing response.5
Thrombospondin (TSP)-1, a typical matricellular protein, is a secreted, multimodular, calcium-binding glycoprotein.4 Diabetic db/db mice show upregulated cardiac TSP-1 expression, indicating potential TSP-1 roles in the remodeling diabetic heart. In this issue of Circulation Research, the Frangogiannis laboratory investigated the impact of TSP-1 deletion on remodeling of the left ventricle (LV) in the setting of diabetic cardiomyopathy.6 The db/db TSP-1-null (dbTSP) mice showed weight gain and metabolic function comparable to db/db mice. However, dbTSP animals showed larger LV volume, attenuated LV hypertrophy, and impaired cardiac reserve. The decreased collagen accumulation in dbTSP mice was associated with increased matrix metalloproteinase (MMP)-2 and MMP-9 activities. TSP-1 deletion did not influence the inflammatory response or the transforming growth factor (TGF)-β signaling pathway in db/db mice. In line with in vivo data, in vitro TSP-1 incorporation into collagen pads did not activate TGF-β but suppressed leptin-triggered MMP-2 activation. TSP-1 deletion prevented capillary rarefaction in db/db mice by inhibiting angiopoietin-2 upregulation. In summary, this study illuminated for the first time that key function of TSP-1 in diabetes mellitus–induced cardiomyopathy is to regulate the fibrotic and angiogenic responses (Figure).
Figure. A mechanistic diagram depicting thrombospondin (TSP)-1 regulation of diabetic cardiomyopathy.
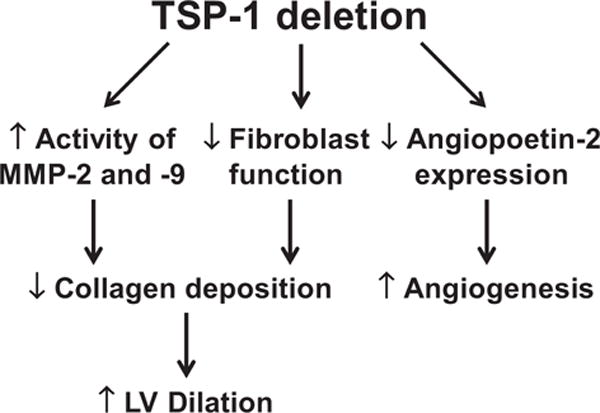
In the diabetic setting, TSP-1 deletion increases matrix metalloproteinase (MMP)-2 and MMP-9 activities to degrade collagen and suppress fibroblast function, thereby inhibiting collagen synthesis. Both these processes reduce collagen deposition but also increase left ventricular dimensions. TSP-1 deletion upregulates angiogenesis through inhibiting angiopoetin-2 expression. In addition, TSP-1 deletion leads to attenuated myocardial hypertrophy and impaired cardiac reserve, but the mechanisms remain to be elucidated.
Inflammation plays a critical role in the initiation and progression of diabetic cardiomyopathy. Activated nuclear factor-κB and increased inflammatory mediators have been reported in clinical patients and experimental animals with diabetic cardiomyopathy.7,8 Unresolved chronic inflammation directly or indirectly triggers myocyte death (both necrosis and apoptosis mechanisms), as well as interstitial and perivascular extracellular matrix deposition, leading to structural and functional impairment.9 Anti-inflammatory therapies targeting nuclear factor-κB and proinflammatory cytokines, such as tumor necrosis factor-α and interleukin-6, have been shown to attenuate cardiac dysfunction in diabetic rodent models.9,10 In this study, the gene expression of proinflammatory cytokines (eg, tumor necrosis factor-α and interleukin-1β) and chemokines (eg, monocyte chemoattractant protein-1 and interferon γ-induced protein 10), however, did not change in diabetic hearts in the presence or absence of TSP-1. This suggests that the role of inflammation in diabetic cardiomyopathy remains incompletely understood.
Interstitial and perivascular fibrosis are pathological hallmarks of diabetic cardiomyopathy, reflective of increased collagen deposition and crosslinking.2 Excessive collagen accumulation promotes myocardial stiffness and diastolic dysfunction. The TGF-β/Smad signaling pathway is activated in experimental models of diabetes mellitus and contributes to cardiac fibrosis by stimulating cardiac fibroblast proliferation and secretion of extracellular matrix, particularly collagen I.11 TSP-1 can activate TGF-β1 by preventing its latency-associated peptide from silencing the mature domain of TGF-β.12 As such, this links TSP-1 to diabetes mellitus-associated myocardial fibrosis. Consistent with TGF-β1 activation, Smad2/3 signaling was activated and collagen accumulated in the diabetic db/db myocardium. TSP-1 deletion, however, did not affect TGF-β1 signaling, implying that matrix-preserving actions of TSP-1 in the remodeling diabetic heart are not because of TGF-β1 activation. In contrast, dbTSP mice had lower collagen content and higher MMP-2 and MMP-9 activities compared with db/db counterparts, which explained the larger LV volume in the absence of TSP-1. In vitro studies using isolated cardiac fibroblasts validated these findings. However, the underlying mechanisms whereby TSP-1 regulates MMP-2 and MMP-9 activity remain to be elucidated.
Microangiopathy develops in the myocardium of diabetic patients and animals and presents as a thickening of capillary basement membrane, medial thickening of the arteriole, perivascular fibrosis, and decreased capillary density.13,14 TSP-1 is an endogenous angiostatic factor that inhibits angiogenesis by reducing endothelial cell migration and proliferation, decreasing survival by stimulating apoptosis, and inhibiting nitric oxide and vascular endothelial growth factor signaling.15,16 In this study, the authors revealed that capillary rarefaction occurred in diabetic db/db mice, an effect abrogated in the absence of TSP-1. The authors demonstrated that the TSP-1 induced angiostatic mechanism involved upregulation of angiopoietin-2 but not vascular endothelial growth factor. Angiopoietin-2 facilitates vessel regression by interrupting Tie2 signaling and pericyte recruitment.17
The findings of this study are novel and reveal critical roles of TSP-1 in diabetic cardiomyopathy. The following concepts may further elucidate molecular mechanisms of diabetic cardiomyopathy and help in the search for potential therapeutic targets. First, the dbTSP mouse is a systemic deletion model. The impact of TSP-1 deletion on other glucose-metabolizing organs (eg, liver and pancreas) needs to be considered when interpreting the data. The authors showed higher plasma cholesterol levels in dbTSP mice than in db/db counterparts. TSP-1 is overexpressed in hypercholesterolemic arteries, suggesting a close interaction between cholesterol and TSP-1.18 Whether the increased cholesterol in dbTSP mice contributes to TSP-1 roles in diabetic cardiomyopathy remains to be determined. A discovery proteomics approach would also be useful for providing novel targets affected by TSP-1.19
Second, the molecular mechanisms of TSP-1 regulation of diabetic cardiomyopathy are not totally understood. How TSP-1 deficiency enhances MMP-2 and MMP-9 activities and downregulates angiopoietin-2 expression need to be investigated further. Additional experiments examining TSP-1 effects to inhibit MMP functions may bring new insights and potential therapeutic approaches.20 The mechanisms whereby TSP-1 deletion attenuates LV hypertrophy and compromises cardiac reserve need to be clarified. Measuring the expression of fetal genes, such as atrial or B-type natriuretic peptides, β-myosin heavy chain, and α-skeletal actin, may help further identify mechanisms.
Third, TSP-1 roles involve multiple components. In the setting of diabetes mellitus, TSP-1 deletion attenuates LV fibrosis, LV hypertrophy, and microvessel rarefaction, and thus is beneficial. At the same time, TSP-1 deletion impairs cardiac reserve and increases LV dimensions, and thus is detrimental. TSP-1-null mice fed a high-fat diet show attenuated adipose inflammation and improved insulin sensitivity.21 Endogenous TSP-1 protects the myocardium from myocardial infarction-induced and pressure overload-induced cardiac remodeling.22,23 Because diabetes mellitus predisposes humans to cardiovascular morbidity and mortality, having a better understanding of how TSP-1 in the diabetic setting responds to stressed conditions (eg, myocardial infarction and hypertension) is warranted.
In conclusion, the Frangogiannis team has revealed direct and indirect effects of TSP-1 on cardiac changes that occur in the setting of diabetes mellitus. This report serves as a foundation for future studies to define the underlying mechanisms of TSP-1 in regulating diabetic cardiomyopathy.
Acknowledgments
Sources of Funding
We acknowledge support from NIH/NHLBI HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center, from R01 HL075360 and HL051971, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505 (to M.L.L.).
Footnotes
The opinions expressed in this editorial are not necessarily those of the editors or of the American Heart Association.
Disclosures
None.
References
- 1.Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, Rao M, Ali MK, Riley LM, Robinson CA, Ezzati M, Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Blood Glucose) National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 27 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 2.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev. 2013;18:149–166. doi: 10.1007/s10741-012-9313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. 2008;121:748–757. doi: 10.1016/j.amjmed.2008.03.046. [DOI] [PubMed] [Google Scholar]
- 4.Ma Y, Halade GV, Lindsey ML. Extracellular matrix and fibroblast communication following myocardial infarction. J Cardiovasc Transl Res. 2012;5:848–857. doi: 10.1007/s12265-012-9398-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, Frunza O, Dobaczewski M, Shinde A, Frangogiannis NG. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res. 2013;113:1331–1344. doi: 10.1161/CIRCRESAHA.113.302593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falcão-Pires I, Leite-Moreira AF. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail Rev. 2012;17:325–344. doi: 10.1007/s10741-011-9257-z. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 9.Wen HL, Liang ZS, Zhang R, Yang K. Anti-inflammatory effects of triptolide improve left ventricular function in a rat model of diabetic cardiomyopathy. Cardiovasc Diabetol. 2013(12):50. doi: 10.1186/1475-2840-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y, Zhang L, Qiao Y, et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS One. 2013;8:e75927. doi: 10.1371/journal.pone.0075927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan HY. Transforming growth factor-β/Smad signalling in diabetic nephropathy. Clin Exp Pharmacol Physiol. 2012;39:731–738. doi: 10.1111/j.1440-1681.2011.05663.x. [DOI] [PubMed] [Google Scholar]
- 12.Schultz-Cherry S, Ribeiro S, Gentry L, Murphy-Ullrich JE. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J Biol Chem. 1994;269:26775–26782. [PubMed] [Google Scholar]
- 13.Factor SM, Minase T, Sonnenblick EH. Clinical and morphological features of human hypertensive-diabetic cardiomyopathy. Am Heart J. 1980;99:446–458. doi: 10.1016/0002-8703(80)90379-8. [DOI] [PubMed] [Google Scholar]
- 14.Factor SM, Minase T, Cho S, Fein F, Capasso JM, Sonnenblick EH. Coronary microvascular abnormalities in the hypertensive-diabetic rat. A primary cause of cardiomyopathy? Am J Pathol. 1984;116:9–20. [PMC free article] [PubMed] [Google Scholar]
- 15.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2:a006627. doi: 10.1101/cshperspect.a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: implications for antiangiogenic therapies. Nat Rev Cancer. 2009;9:182–194. doi: 10.1038/nrc2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerald D, Chintharlapalli S, Augustin HG, Benjamin LE. Angiopoietin-2: an attractive target for improved antiangiogenic tumor therapy. Cancer Res. 2013;73:1649–1657. doi: 10.1158/0008-5472.CAN-12-4697. [DOI] [PubMed] [Google Scholar]
- 18.Roth JJ, Gahtan V, Brown JL, Gerhard C, Swami VK, Rothman VL, Tulenko TN, Tuszynski GP. Thrombospondin-1 is elevated with both intimal hyperplasia and hypercholesterolemia. J Surg Res. 1998;74:11–16. doi: 10.1006/jsre.1997.5209. [DOI] [PubMed] [Google Scholar]
- 19.de Castro Brás LE, Ramirez TA, DeLeon-Pennell KY, Chiao YA, Ma Y, Dai Q, Halade GV, Hakala K, Weintraub ST, Lindsey ML. Texas 3-step decellularization protocol: looking at the cardiac extracellular matrix. J Proteomics. 2013;86:43–52. doi: 10.1016/j.jprot.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yabluchanskiy A, Li Y, Chilton RJ, Lindsey ML. Matrix metalloproteinases: drug targets for myocardial infarction. Curr Drug Targets. 2013;14:276–286. [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PLoS One. 2011;6:e26656. doi: 10.1371/journal.pone.0026656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 23.Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, Lee DW, Frangogiannis NG. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension. 2011;58:902–911. doi: 10.1161/HYPERTENSIONAHA.111.175323. [DOI] [PMC free article] [PubMed] [Google Scholar]