

Abstract
Macrophage activation syndrome (MAS) is an acute episode of overwhelming inflammation characterized by activation and expansion of T lymphocytes and hemophagocytic macrophages. In rheumatology, it occurs most frequently in patients with systemic juvenile idiopathic arthritis (SJIA) and systemic lupus erythematosus. The main clinical manifestations include cytopenias, liver dysfunction, coagulopathy resembling disseminated intravascular coagulation, and extreme hyperferritinemia. Clinically and pathologically, MAS bears strong similarity to hemophagocytic lymphohistiocytosis (HLH), and some authors prefer the term secondary HLH to describe it. Central to its pathogenesis is a cytokine storm, with markedly increased levels of numerous proinflammatory cytokines including IL-1, IL-6, IL-18, TNFα, and IFNγ. Although there is evidence that IFNγ may play a central role in the pathogenesis of MAS, the role of other cytokines is still not clear. There are several reports of SJIA-associated MAS dramatically benefiting from anakinra, a recombinant IL-1 receptor antagonist, but the utility of other biologics in MAS is not clear. The mainstay of treatment remains corticosteroids; other medications, including cyclosporine, are used in patients who fail to respond.
Keywords: hemophagocytic lymphohistiocytosis, systemic juvenile idiopathic arthritis, Still’s disease, hyperferritinemia, cytokine storm
DEFINITIONS
Macrophage activation syndrome (MAS) is a serious, potentially fatal complication of rheumatic diseases caused by excessive activation and expansion of T lymphocytes and macrophages that exhibit hemophagocytic activity (1–4). In MAS, this excessive cellular activation and expansion lead to cytokine overproduction (Figure 1) and a hyperinflammatory state associated with cytopenias, liver dysfunction, and coagulopathy resembling disseminated intravascular coagulation. Extreme hyperferritinemia is another striking laboratory feature of MAS. It is a life-threatening condition, and the reported mortality rates reach 20–30% (5, 6).
Figure 1.
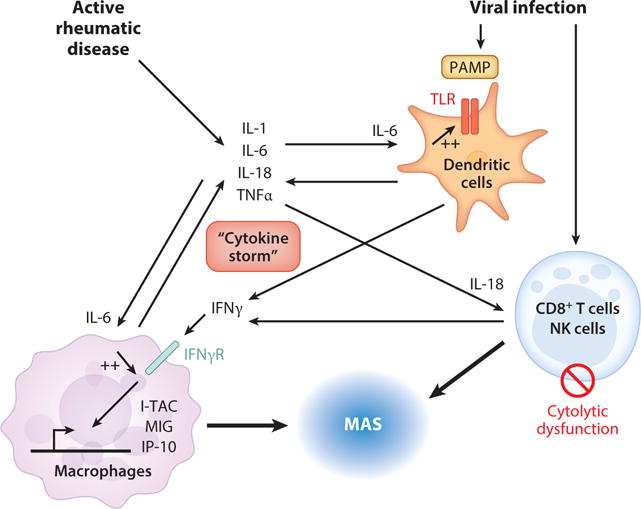
“Cytokine storm” and the development of macrophage activation syndrome (MAS). MAS can develop in the setting of high systemic juvenile idiopathic arthritis (SJIA) disease activity, which is associated with increased levels of cytokines including IL-1, IL-6, IL-18, and TNFα. MAS can also be triggered by viral infections, wherein pathogen-associated molecular patterns (PAMPs) are recognized by toll-like receptors (TLRs) and trigger further secretion of inflammatory cytokines. The proinflammatory environment including elevated IL-6 can enhance signaling through TLRs. Infection also leads to activation and proliferation of CD8+ T cells and NK cells, including secretion of IFNγ. Defects in the cytolytic activity of these lymphocytes also contribute to hyperinflammation. Increased IL-18 levels further drive IFNγ production by these activated lymphocytes. This surge in IFNγ leads to activation of macrophages that acquire a proinflammatory phenotype and generate high levels of chemokines and cytokines. These activated macrophages, along with CD8+ T cells, traffic to tissue including bone marrow and liver and lead to the cytopenias, liver dysfunction, and coagulopathy associated with MAS.
Expansion of tissue macrophages, or histiocytes, exhibiting hemophagocytic activity is a feature shared by a group of histiocytic disorders collectively known as hemophagocytic lymphohistiocytosis (HLH) (7, 8). In the current classification of histiocytic disorders, HLH is subdivided into primary or familial HLH (FHLH) and secondary or reactive HLH, although clinically they may be difficult to distinguish from each other (7). FHLH is a constellation of rare autosomal recessive immune disorders linked to genetic defects in various genes affecting the cytolytic pathway (see below). The clinical symptoms of FHLH usually become evident within the first months of life. Secondary HLH tends to occur in older children and adults and more often is associated with an identifiable infectious episode, most notably Epstein-Barr virus (EBV) or cytomegalovirus (CMV) infection, as well as malignancy. However, the distinction between primary and secondary HLH is becoming increasingly blurred as the genetic basis for these conditions is further delineated (see below) (8, 9). Given clinical similarities between MAS and HLH, the term reactive HLH has been preferred by some authors to classify patients with MAS occurring in the setting of a rheumatic condition (10, 11).
EPIDEMIOLOGY
In pediatric rheumatology, MAS is seen most frequently in children with systemic juvenile idiopathic arthritis (SJIA) (1–5). However, it is increasingly reported in other rheumatic diseases of childhood, most notably pediatric systemic lupus erythematosus (SLE) (12, 13) and Kawasaki disease (13–15). In these patients, episodes of MAS appear to be most commonly triggered by infections, particularly viral infections, or during periods of high disease activity including disease onset (16, 17). The epidemiologic data on MAS in adults are limited. Based on one survey conducted through the French National Society of Internal Medicine in 1999, of the 26 MAS cases identified, 14 occurred in association with SLE, four with adult-onset Still’s disease (an adult equivalent of SJIA), two with rheumatoid arthritis, and two with polyarteritis nodosa. Infection was identified as a trigger in the majority of the patients (16). The mortality in this group reached 38.5%, most likely owing to the late recognition of the syndrome.
The epidemiologic studies of MAS have been complicated by the lack of defined diagnostic criteria (see below). Most pediatric rheumatologists now agree that approximately 7–17% of patients with SJIA develop overt MAS (6, 18) and that mild, subclinical MAS may be seen in as many as one third of patients with active systemic disease (19, 20). Overt MAS may occur at any stage of the disease, and based on the experience in Cincinnati Children’s Hospital Medical Center, the incidence of overt MAS is 4–6 events per 100 patient-years.
DIAGNOSIS
There are no validated diagnostic criteria for MAS, and early diagnosis is often difficult because of similarities with sepsis-like syndromes. In a febrile patient with an active rheumatologic condition, a fall in platelet count and erythrocyte sedimentation rate (due to decreasing serum fibrinogen) in combination with persistently high C-reactive protein and increasing levels of serum D-dimers should raise suspicion of MAS. Other supportive features include cytopenias involving other cell lines (i.e., decreasing white blood cell count and/or hemoglobin), hyperferritinemia (usually >1,000 ng/ml), liver dysfunction, coagulopathy, decreasing serum fibrinogen, and increasing triglycerides.
Ideally, the diagnosis of MAS should be confirmed by the demonstration of hemophagocytosis in the bone marrow. However, the demonstration of hemophagocytosis may be difficult owing to sampling error, particularly at the early stages of the syndrome. In such cases, additional staining of the bone marrow with anti-CD163 antibodies may be helpful. In the setting of MAS, this usually reveals massive expansion of highly activated histiocytes (19, 20).
In contrast to MAS, the diagnosis of HLH is typically based on the diagnostic criteria developed by the International Histiocyte Society (21). This diagnosis can be made if patients have a known molecular diagnosis consistent with HLH or meet at least 5 of 8 clinical or laboratory criteria (Table 1). Unfortunately, the HLH diagnostic criteria when applied to SJIA patients with suspected MAS are not sufficiently sensitive to diagnose the condition early in its development, when treatment is most successful. Some of the HLH markers, such as lymphadenopathy, splenomegaly, and hyperferritinemia, are common features of active SJIA itself and do not distinguish MAS from a conventional SJIA flare. Other criteria, such as cytopenias and hypofibrinogenemia, become evident only in the later stages of MAS, as SJIA patients often have increased white blood cell and platelet counts as well as elevated serum levels of fibrinogen as a part of the inflammatory response. Finally, other criteria such as natural killer (NK) cell function and bone marrow examination are time consuming and may unnecessarily delay administration of specific therapy.
Table 1.
Proposed criteria or features useful in the diagnosis of macrophage activation syndrome
| HLH-2004 criteria (22) | Ravelli criteria (23) | MAS Study Group criteria (24) |
|---|---|---|
| A molecular diagnosis consistent with HLH (i.e., reported mutations in genes encoding either PRF1 or MUNC13-4, STX11, STXBP2, Rab27a, SH2D1A or BIRC4) or At least 5 of the 8 diagnostic criteria for hemophagocytic lymphohistiocytosis listed below: Persistent fever Splenomegaly Cytopenias (affecting ≥2 of 3 lineages in the peripheral blood): hemoglobin <90 g/L, platelets <100 × 109/L, neutrophils <1.0 × 109/L Hypertriglyceridemia (fasting triglycerides ≥3.0 mmol/L) and/or hypofibrinogenemia (≤1.5 g/L) Hemophagocytosis in bone marrow, spleen, or lymph nodes; no evidence of malignancy Serum ferritin ≥500 μg/L Low or absent NK cell activity (according to local laboratory reference) Increased serum sIL-2Rα (according to local laboratory reference) |
Laboratory criteria: Decreased platelet count (≤262 × 109/L) Elevated aspartate aminotransferase (>59 U/L) Decreased white blood count (≤4.0 × 109/L) Hypofibrinogenemia (≤2.5 g/L) Clinical criteria: Central nervous systemic dysfunction (irritability, disorientation, lethargy, headache, seizures, coma) Hemorrhages (purpura, easy bruising, mucosal bleeding) Hepatomegaly (≥3 cm below the costal margin) (Two or more laboratory criteria or any two or more clinical and/or laboratory criteria fulfilled establish the diagnosis) |
Falling platelet count Hyperferritinemia Increased liver enzymes Falling leukocyte count Persistent continuous fever ≥38°C Falling erythrocyte sedimentation rate Hypofibrinogenemia Hypertriglyceridemia Evidence of macrophage hemophagocytosis in the bone marrow (Most frequent features identified by responding clinicians) |
Attempts to modify the HLH criteria to increase sensitivity and specificity for the diagnosis of MAS in rheumatic conditions including SJIA have been initiated (22, 23). The first proposed criteria for diagnosis of MAS in patients with SJIA were by Ravelli and colleagues (22) (Table 1). However, these criteria have not been further validated in a large cohort of patients, nor examined in patients who have MAS without SJIA.
Recently, an international collaborative project was started to develop a new and robust set of diagnostic criteria for MAS in patients with SJIA (23). As a first step, this project used the Delphi survey technique to identify the clinical features most pertinent to the diagnosis of MAS. The nine features ranked highest by the 232 respondent physicians are shown in Table 1. These candidate clinical variables are currently being evaluated for their performance in the early recognition of MAS with the goal of defining cut-off numerical values to achieve the highest sensitivity and specificity.
PATHOPHYSIOLOGY
The main pathophysiologic feature of MAS is excessive activation and expansion of T lymphocytes (mainly cytotoxic CD8+T cells) and macrophages (3, 13). These activated immune cells produce large amounts of proinflammatory cytokines, creating a “cytokine storm.”
In clinically similar primary HLH, the uncontrolled expansion of T cells and macrophages has been linked to decreased NK cell and cytotoxic T cell functions (8). In about 30% of FHLH patients, this is due to mutations in the gene encoding perforin (24). Perforin is a protein that cytolytic cells utilize to induce apoptosis of target cells such as tumor cells or cells infected by viruses. In ~10% of patients with primary HLH, the disease is caused by mutations in another gene, MUNC13-4 (25). The protein encoded by this gene is involved in the release of perforin into the immune synapse with a target cell. Although the cytolytic cells of patients with MUNC13-4 mutations produce sufficient amounts of perforin, their ability to kill target cells is greatly diminished. More recently, mutations in two other genes have been linked to the development of primary HLH, both encoding proteins that facilitate granule fusion in intracellular trafficking events leading to the release of perforin: syntaxin 11, a member of the SNARE protein family (26), and syntaxin binding protein 2 (STXBP2, known as MUNC18-2) (27, 28). Finally, several immunodeficiency syndromes that predispose to HLH are caused by mutations in gene products involved in the function of lytic granules. These include Griscelli syndrome type II (Rab27a) and Chediak Higashi syndrome (Lyst) (29). Although familial cases of MAS in SJIA have not been reported, SJIA/MAS patients have functional defects in the exosome degranulation pathway (30), and these abnormalities are associated with the presence of rare variants in the genes encoding MUNC13-4 (31, 32), STXBP2, STX11, and PRF1 (33).
Normally, cytotoxic cells induce apoptosis of cells infected with intracellular microbes or cells undergoing malignant transformation. In some circumstances, cytotoxic cells may also be directly involved in induction of apoptosis of activated macrophages and T cells during the contraction stage of the immune response. It has been proposed that in both HLH and MAS, failure to induce apoptosis due to cytotoxic dysfunction leads to prolonged expansion of T cells and macrophages and escalating production of proinflammatory cytokines.
Clues from Animal Models
Some clues to the pathophysiology of MAS are provided by animal models of HLH. First, Jordan et al. demonstrated that perforin-deficient mice infected with lymphochoriomeningitic virus (LCMV) developed fevers, splenomegaly, pancytopenia, extreme hyperferritinemia, and hypercytokinemia, as well as histologic features including hemophagocytosis characteristic of HLH (34). More importantly, these clinical features were prevented by the administration of anti-CD8 antibodies or neutralization of interferon gamma (IFNγ), whereas antibodies against CD4 and the neutralization of other inflammatory cytokines including tumor necrosis factor alpha (TNFα) had no effect. These results suggest that, at least in this model, IFNγ-producing CD8+ T cells are central in the pathogenesis of the hemphagocytic syndrome (34). As a consequence of continuous stimulation with IFNγ and other cytokines derived from these CD8+ cells, macrophages expand and acquire a distinct phenotype associated with hemophagocytic activity. Similar results have been obtained in mice deficient in other HLH-associated gene products including Munc13-4 and Rab27a (35, 36). These animals also developed HLH-like clinical features upon infection with LCMV in an IFNγ-dependent manner. Several lines of evidence suggest that IFNγ-producing CD8+ cells are important in patients with MAS as well. A recent study of liver biopsies from patients with MAS revealed extensive periportal infiltration with hemophagocytic macrophages secreting TNFα and interleukin (IL)–6, as well as IFNγ-producing CD8+ T cells (37). Furthermore, MAS patients usually have very high levels of soluble IL2 receptor α (sIL-2Rα) chains, most likely originating from overly activated T cells (19). In addition, MAS features usually improve in response to treatment with cyclosporine A, a drug that acts predominantly on T cells rather than macrophages (2).
In all the above-mentioned animal models, however, the HLH-like clinical features emerge only in response to viral infections. Although a viral illness is a common trigger of hemophagocytic syndromes, many FHLH patients develop the first symptom of the disease spontaneously without an identifiable infection (8). Similarly, MAS is often associated with a flare of underlying SJIA rather than infection. These considerations prompted a search for other animal models that would not be dependent on an infectious trigger. Recent reports showing the critical need for the toll-like receptor (TLR) signaling adaptor MyD88 in the development of HLH-like disease in LCMV-infected MUNC13-4–deficient mice (38), combined with evidence of persistently activated TLR/IL-1R signaling pathways in SJIA (39, 40), provided a rationale for repeated activation of TLR to replicate the environment that would allow MAS to develop in a genetically predisposed host. Indeed, wild-type mice given repeated TLR9 stimulation develop some MAS features, including hepatic dysfunction and cytopenias (41). Interestingly, this model appears to be only partially IFNγ dependent, and in contrast to the models of primary HLH, IFNγ in these animals appears to be produced mainly by dendritic cells and NK cells but not by CD8+ T lymphocytes. Furthermore, in this model, many clinical features including hemophagocytosis do not appear to depend on IFNγ. Although the findings in this model are intriguing, their relevance to the disease in humans still needs to be elucidated.
Cytokine Storm
Whatever the upstream events that trigger MAS, the end product is escalating production of cytokines. Indeed, in both MAS and HLH, strikingly high levels of circulating cytokines including IFNγ, IL-2, macrophage colony stimulating factor (M-CSF), IL-1, IL-6, IL-18, and TNFα, as well as natural cytokine inhibitors such as soluble TNF receptors and IL-1R antagonists, have been reported (42–44). The interpretation of these data, however, is not straightforward, as even if cytokine levels are high and strongly correlate with disease activity, those observations do not necessarily establish causality. Below, we further discuss several cytokines that are increased in MAS and examine the effects of biologics blocking these cytokines on MAS risk and clinical presentation.
IL-1β
IL-1β is a proinflammatory cytokine produced primarily by monocytes and macrophages. It is present as an inactive form, pro-IL-1β; however, upon activation of cells, it is cleaved by caspase-1 to the biologically active form. IL-1β signals through its receptor and causes leukocyte and endothelial cell activation as well as production of other inflammatory cytokines including IL-6. IL-1β is believed to be central to the pathogenesis of SJIA (39, 45–48). Newly diagnosed SJIA patients show an IL-1-drive gene expression profile (40, 45), and serum from patients with active SJIA triggers the induction of IL-1-related genes in monocytes from healthy donors (39). Indeed, large series (45–47) as well as phase III randomized trials (49, 50) have shown that IL-1 blockade could induce long-lasting clinical remission in >50% of SJIA patients.
The exact role of IL-1 in the development of MAS remains unclear. Because MAS episodes are often triggered by SJIA flare, it is reasonable to expect at least some response to IL-1 inhibition due to better control of the underlying disease. Indeed, there are several reports of SJIA-associated MAS dramatically benefiting from anakinra, a recombinant IL-1R antagonist, after inadequate response to corticosteroids and cyclosporine (51–53). Successful treatment of refractory adult onset Still’s disease (AOSD) complicated by MAS with IL-1 blockade has been reported as well (53). In contrast, in two reports summarizing the experience with the use of anakinra in SJIA in several pediatric rheumatology centers, anakinra was a suspected MAS trigger in several children at doses of 1–2 mg/kg/day (46). However, cause and effect were difficult to establish in these patients, and permanent discontinuation of anakinra was unnecessary for any of them. In fact, in some of these patients, MAS features improved after the dose of anakinra was increased.
Canakinumab, a monoclonal antibody directed against IL-1β, is another IL-1-blocking biologic agent (50). Although canakinumab is an effective treatment in SJIA, it does not appear to have a significant effect on reducing the risk of MAS, even in patients whose underlying SJIA is well controlled (54). Infections were the most prevalent trigger for MAS in this group. Furthermore, the overall clinical features of MAS in patients treated with canakinumab do not appear to be modified by treatment.
IL-6
IL-6 is a pleotropic cytokine produced in the early stages of inflammation and is central in driving the acute-phase response. Patients with SJIA also demonstrate increased levels of IL-6, which correlate with disease activity (55). However, like IL-1β, the role of IL-6 in the pathogenesis of MAS remains unknown. One study of patients with MAS demonstrated the presence of IL-6-producing activated macrophages obtained from liver biopsies (37). In addition, IL-6 is elevated in patients with HLH, although not as markedly as in syndromes such as sepsis (42, 56). Interestingly, one study of IL-6-overexpressing transgenic mice found that prolonged exposure to IL-6 in vivo led to an exaggerated inflammatory response to TLR ligands, with some clinical features reminiscent of MAS, including cytopenias and hyperferritinemia (57). These findings suggest that IL-6 may amplify the inflammatory response to infections and contribute to a cytokine storm. There are no reports of MAS treated specifically with tocilizumab. In contrast, in a phase III clinical trial in SJIA, MAS was observed in three patients receiving IL-6 blockade with tocilizumab. This corresponded to 1.5 MAS cases per 100 patient-years. Intriguingly, the underlying SJIA in these patients responded well to the treatment (58). Another recent report from Japan described a patient with severe AOSD who showed a very good initial response to tocilizumab but then rapidly progressed to develop MAS (59). Furthermore, it has been suggested that treatment with tocilizumab may mask some MAS features. Shimizu and colleagues described several SJIA patients who developed MAS while on tocilizumab; their CRP levels remained normal, and the increase in levels of ferritin was relatively modest (60). This is not surprising given that IL-6 is a strong inducer of acute-phase proteins, including CRP and ferritin.
TNFα
TNFα is a pleomorphic cytokine implicated in the pathogenesis of several inflammatory diseases. It is produced largely by monocytes and macrophages. The role of TNFα in SJIA appears to be rather limited, and the introduction of TNFα-inhibiting agents in clinical practice had no major impact on the SJIA outcome. Similarly, the role of TNFα in MAS and other hemophagocytic syndromes is unclear. Several animal models of HLH have shown increased levels of TNFα (34, 36, 61). However, there are conflicting results as to the pathologic role of increased TNFα; one report showed this cytokine may mediate tissue damage (62) whereas another found no effect of neutralizing this cytokine (34). TNFα similarly does not appear to be central to the pathogenesis of MAS in patients with SJIA. There are occasional reports of MAS that showed rapid improvement with TNFα blockade (63–65). However, there are numerous cases described in the literature of MAS in the setting of TNFα therapy, as well as reports of patients whose MAS worsened upon initiation of therapy (66). Taken together, these findings may suggest that elevated TNFα levels reflect the degree of underlying cellular activation rather than playing a causative role in MAS.
IL-18
IL-18 is a unique cytokine in the IL-1 family, and in a precursor form it is constitutively present in keratinocytes, epithelial cells, and blood monocytes (67). Because the IL-18 precursor is inactive, caspase-1 is required for processing and secretion of the active cytokine. IL-18 induces production of IFNγ by NK cells and T cells as well as TNFα and chemokine secretion by macrophages (68). IL-18 activity may be counterbalanced by a naturally occurring, high-affinity binding protein termed the IL-18 binding protein (IL-18BP).
In MAS, serum IL-18 levels are elevated out of proportion compared with other cytokines (69, 70). This is in distinct contrast to IL-18 levels in other diseases, such as rheumatoid arthritis (71), sepsis (72), or SLE (73, 74), where only moderate elevation is seen. In SJIA and AOSD, the levels of free IL-18 strongly correlate with underlying disease activity (69–71) and slowly decrease with clinical remission. Notably, serum IL-18 levels in patients with MAS were only slightly higher than those in patients with active SJIA (75).
In one study of patients with MAS, serum IL-18 levels were highly increased, whereas the levels of IL-18BP were only moderately elevated, resulting in a high level of biologically active free IL-18 (69). Free IL-18, but not other cytokines, significantly correlated with overall clinical status as well as many specific features of MAS, including anemia, hypertriglyceridemia, hyperferritinemia, and soluble IL-2Rα chains, along with elevated levels of IFN-γ. Interestingly, IL-18 is a strong stimulator of NK cell activity, but despite high IL-18 levels, in vitro NK cell cytolytic activity is severely impaired in MAS patients. This impairment is due to both NK cell lymphopenia and intrinsic NK cell functional deficiency (76). Thus, a severe IL-18/IL-18BP imbalance may result in T lymphocyte and macrophage activation, which escapes immunoregulatory control by NK cell cytotoxicity. In patients with underlying inflammatory diseases, this creates conditions favoring the development of secondary MAS. However, one recent study suggests that IL-18 may have a more limited role in MAS. Chiossone and colleagues examined perforin-deficient mice infected with murine CMV, which leads to uncontrolled viral replication along with pancytopenia, hepatic dysfunction, hemophagocytosis, and death (61). Administration of synthetic IL-18BP ameliorated liver damage in the perforin-deficient mice. However, the animals still produced substantial proinflammatory cytokines, and there was no change in overall survival. Further work is needed to better characterize the role of IL-18 in other model systems.
IFNγ
IFNγ is a proinflammatory cytokine produced by NK cells and T lymphocytes when activated by antigen-presenting cells. IFNγ is an important activator of monocytes and macrophages (77). Activated macrophages are divided into several general classes based on stimuli and their resulting polarization, with M1 macrophages driven by IFNγ into a classical proinflammatory phenotype characterized by increased microbicidal capacity, heightened responses to TLR ligands, and upregulated antigen processing and presentation. These cells are potent producers of proinflammatory cytokines, including IL-6, IL-12, and IL-23, as well as the chemokines IP-10, monokine induced by IFNγ (MIG), and IFN-inducible T cell alpha-chemoattractant (I-TAC), which recruit polarized Th1 cells as well as NK cells (78, 79). There is also evidence that continuous stimulation with IFNγ may be a critical driver of hemophagocytosis by these activated macrophages (80). Macrophages can also be “alternatively activated” by other stimuli including IL-4 to exhibit an anti-inflammatory, wound-healing phenotype (81). These macrophages are likely a heterogeneous population that encompasses distinct cellular phenotypes involved in activating polarized Th2 cells, immunoregulation, and wound healing (79).
The role of IFNγ in MAS in SJIA has not been fully characterized. However, IFNγ does not appear to play a central role in the pathogenesis of SJIA without MAS. Patients with both active and inactive SJIA do not exhibit increased serum IFNγ (45, 82). In addition, three independent gene expression studies have failed to find a prominent INFγ-induced signature in the peripheral blood monocytes of children who do not exhibit MAS clinical features (39, 40, 83).
However, multiple lines of evidence suggest an essential role for IFNγ in the pathogenesis of MAS. IFNγ appears central to the pathogenesis of FHLH. First, IFNγ expression is highly elevated in these patients, out of proportion to other proinflammatory cytokines such as TNFα and IL-6, and rapidly returns to normal upon effective treatment (42, 84, 85). These children also exhibit increased levels of the IFNγ-induced chemokines IP-10 and MIG (86). Second, multiple independent animal models of FHLH have also demonstrated increased levels of IFNγ, and neutralization of this cytokine dramatically improves survival (34, 36, 41). Taken together, these findings support IFNγ blockade as a novel therapy for HLH, and a clinical trial is currently in progress (87). There is also increasing evidence for the role of IFNγ in SJIA patients who develop MAS. MAS episodes are frequently triggered by viral infections, which are known activators of IFNγ-induced pathways (88). Patients with MAS also show significant proliferation of IFNγ-producing T cells in tissue (37). The precise molecular phenotype of macrophages in patients with MAS has not been defined. However, children with MAS do exhibit increased levels of neopterin, a product of IFNγ-activated macrophages (89). In addition, levels of neopterin could distinguish patients with MAS from those with active SJIA without MAS clinical features (75). Combined, these observations suggest that IFNγ-induced pathways are important in the pathogenesis of MAS in patients with SJIA.
TREATMENT
Standard Nonbiologic Treatments
MAS is a life-threatening condition associated with high mortality rates. Therefore, early recognition and immediate therapeutic intervention to produce a rapid response are critical. Most clinicians start with intravenous methylprednisolone pulse therapy, e.g., 30 mg/kg for three consecutive days, followed by 2–3 mg/kg/day in 2–4 divided doses. If response to steroids is not immediately evident, parenteral administration of cyclosporine A (CsA) (2–7 mg/kg/day) is usually initiated (2, 4, 90). Although high intravenous doses of CsA have been associated with increased risk for posterior reversible encephalopathy syndrome, in most MAS patients, addition of CsA not only provides rapid control of symptoms but also avoids excessive use of steroids (5). Patients in whom MAS remains active, despite the use of corticosteroids and CsA, present a serious challenge. In these patients, the HLH-2004 treatment protocol developed by the International Histiocyte Society might be considered (21). This protocol, in addition to steroids and CsA, includes etoposide (or VP16), a podophyllatoxin derivative that inhibits DNA synthesis by forming a complex with topoisomerease II and DNA. Although successful use of etoposide in MAS has been reported, potential toxicity of the drug is a major concern, particularly in patients with hepatic impairment. Deaths associated with the use of etoposide, caused by severe bone marrow suppression and overwhelming infection, have been reported (91, 92). Recently, it has been suggested that in patients unresponsive to the combination of steroids and cyclosporine A, particularly in those with renal and hepatic impairment, antithymocyte globulin (ATG) might be a safer alternative to etoposide (93, 94). ATG depletes both CD4+ and CD8+ T cells through complement-dependent cell lysis. Mild depletion of monocytes is noted in some patients as well. Although in the reported cases this treatment was tolerated well, one must remember infusion reactions are frequently reported with the use of ATG, and adequate laboratory and supportive medical resources must be readily available if this treatment is used.
Biologic Agents
The utility of biologic drugs in MAS treatment remains unclear. Although TNFα-inhibiting agents have been reported to be effective in a few MAS patients, other reports describe patients in whom MAS developed while they were on TNFα-inhibiting agents. Because, at least in SJIA, MAS episodes are often triggered by disease flare, biologics that neutralize IL-1, a cytokine that plays a pivotal role in SJIA pathogenesis, have been tried. There are several reports of SJIA-associated MAS dramatically benefiting from anakinra after inadequate response to corticosteroids and CsA (51–53). Intravenous immune globulin treatment has been a successful treatment in virus-associated reactive HLH (95, 96). Rituximab, an anti-CD20 antibody that depletes B lymphocytes, has been successfully used in EBV-induced lymphoproliferative disease (97, 98) and could be considered in EBV-driven MAS. In children with treatment-refractory HLH, alemtuzumab, an anti-CD52 antibody that depletes lymphocytes as well as mononuclear cells, has been used successfully as salvage therapy prior to hematopoietic stem cell transplant (99). There is also a single report of a patient with SLE who developed reactive HLH/MAS that was treated successfully with alemtuzumab (100).
SUMMARY
In summary, the mainstay of treatment of MAS presenting as a complication of rheumatic diseases remains high-dose corticosteroids, cyclosporine, and in more difficult instances, etoposide. Understanding the balance of cytokines in the cytokine storm of MAS is crucial to developing and utilizing the available cytokine-targeted biologic therapies. In systemic JIA patients, even those whose systemic JIA is well controlled, continuous treatment with canakinumab or tocilizumab does not provide full protection against MAS. Anakinra may be beneficial at least in some MAS patients. Findings in patients with FHLH support the IFNγ blockade as a novel therapy for FHLH, and a phase II clinical trial of NI-0501, an anti-IFNγ monoclonal antibody, is currently in progress.
Acknowledgments
Dr. Schulert is supported by the Amgen Fellowship Training Award from the Rheumatology Research Foundation. Dr. Grom is supported by NIH grants NIH RO1-AR059049 and NIH PO1-AR048929.
Glossary
- FHLH
familial hemophagocytic lymphohistiocytosis
- SLE
systemic lupus erythematosus
Footnotes
DISCLOSURE STATEMENT
Dr. Grom has received consulting fees from Novartis and Roche and has research collaborations with NovImmune and Novartis.
LITERATURE CITED
- 1.Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106:561–66. doi: 10.1016/s0022-3476(85)80072-x. [DOI] [PubMed] [Google Scholar]
- 2.Mouy R, Stephan JL, Pillet P, et al. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996;129:750–54. doi: 10.1016/s0022-3476(96)70160-9. [DOI] [PubMed] [Google Scholar]
- 3.Grom AA, Passo M. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis. J Pediatr. 1996;129:630–32. doi: 10.1016/s0022-3476(96)70140-3. [DOI] [PubMed] [Google Scholar]
- 4.Ravelli A, De Benedetti F, Viola S, Martini A. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine. J Pediatr. 1996;128:275–78. doi: 10.1016/s0022-3476(96)70408-0. [DOI] [PubMed] [Google Scholar]
- 5.Stephan JL, Kone-Paut I, Galambrun C, et al. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40:1285–92. doi: 10.1093/rheumatology/40.11.1285. [DOI] [PubMed] [Google Scholar]
- 6.Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Childhood. 2001;85:421–26. doi: 10.1136/adc.85.5.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol. 1997;29:157–66. doi: 10.1002/(sici)1096-911x(199709)29:3<157::aid-mpo1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 8.Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–52. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang KJ, Jordan MB, Marsh R, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118:5794–98. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Athreya BH. Is macrophage activation syndrome a new entity? Clin Exp Rheumatol. 2002;20:121–23. [PubMed] [Google Scholar]
- 11.Ramanan AV, Schneider R. Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003;30:401–3. [PubMed] [Google Scholar]
- 12.Parodi A, Davi S, Pringe AB, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum. 2009;60:3388–99. doi: 10.1002/art.24883. [DOI] [PubMed] [Google Scholar]
- 13.Avcin T, Tse SM, Schneider R, et al. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148:683–86. doi: 10.1016/j.jpeds.2005.12.070. [DOI] [PubMed] [Google Scholar]
- 14.Latino GA, Manlhiot C, Yeung RS, et al. Macrophage activation syndrome in the acute phase of Kawasaki disease. J Pediatr Hematol/Oncol. 2010;32:527–31. doi: 10.1097/MPH.0b013e3181dccbf4. [DOI] [PubMed] [Google Scholar]
- 15.Simonini G, Pagnini I, Innocenti L, et al. Macrophage activation syndrome/hemophagocytic lymphohistiocytosis and Kawasaki disease. Pediatr Blood Cancer. 2010;55:592. doi: 10.1002/pbc.22630. [DOI] [PubMed] [Google Scholar]
- 16.Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633–39. doi: 10.1002/art.11368. [DOI] [PubMed] [Google Scholar]
- 17.Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31:1223–30. doi: 10.1007/s10067-012-1998-0. [DOI] [PubMed] [Google Scholar]
- 18.Moradinejad MH, Ziaee V. The incidence of macrophage activation syndrome in children with rheumatic disorders. Minerva Pediatr. 2011;63:459–66. [PubMed] [Google Scholar]
- 19.Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:965–71. doi: 10.1002/art.22416. [DOI] [PubMed] [Google Scholar]
- 20.Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34:1133–38. [PubMed] [Google Scholar]
- 21.Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 22.Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146:598–604. doi: 10.1016/j.jpeds.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 23.Davi S, Consolaro A, Guseinova D, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38:764–68. doi: 10.3899/jrheum.100996. [DOI] [PubMed] [Google Scholar]
- 24.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–59. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 25.Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–73. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 26.zur Stadt U, Schmidt S, Diler AS, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14:827–34. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 27.Crozat K, Hoebe K, Ugolini S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med. 2007;204:853–63. doi: 10.1084/jem.20062447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to Syntaxin 11. Am J Hum Genet. 2009;85:482–92. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bode SF, Lehmberg K, Maul-Pavicic A, et al. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther. 2012;14:213–25. doi: 10.1186/ar3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grom AA, Villanueva J, Lee S, et al. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142:292–96. doi: 10.1067/mpd.2003.110. [DOI] [PubMed] [Google Scholar]
- 31.Kaufman KM, Linghu B, Szustakowski JD, et al. Whole exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheum. 2014 doi: 10.1002/art.38793. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang K, Biroschak J, Glass DN, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. 2008;58:2892–96. doi: 10.1002/art.23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology. 2010;49:441–49. doi: 10.1093/rheumatology/kep418. [DOI] [PubMed] [Google Scholar]
- 34.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–43. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 35.Pachlopnik Schmid J, Ho CH, Chretien F, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1:112–24. doi: 10.1002/emmm.200900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pachlopnik Schmid J, Ho CH, Diana J, et al. A Griscelli syndrome type 2 murine model of hemophagocytic lymphohistiocytosis (HLH) Eur J Immunol. 2008;38:3219–25. doi: 10.1002/eji.200838488. [DOI] [PubMed] [Google Scholar]
- 37.Billiau AD, Roskams T, Van Damme-Lombaerts R, et al. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood. 2005;105:1648–51. doi: 10.1182/blood-2004-08-2997. [DOI] [PubMed] [Google Scholar]
- 38.Krebs P, Crozat K, Popkin D, et al. Disruption of MyD88 signaling suppresses hemophagocytic lymphohistiocytosis in mice. Blood. 2011;117:6582–88. doi: 10.1182/blood-2011-01-329607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pascual V, Allantaz F, Arce E, et al. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fall N, Barnes M, Thornton S, et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007;56:3793–804. doi: 10.1002/art.22981. [DOI] [PubMed] [Google Scholar]
- 41.Behrens EM, Canna SW, Slade K, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 2011;121:2264–77. doi: 10.1172/JCI43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henter JI, Elinder G, Soder O, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78:2918–22. [PubMed] [Google Scholar]
- 43.Henter JI, Andersson B, Elinder G, et al. Elevated circulating levels of interleukin-1 receptor antagonist but not IL-1 agonists in hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1996;27:21–25. doi: 10.1002/(SICI)1096-911X(199607)27:1<21::AID-MPO5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 44.Sumegi J, Barnes MG, Nestheide SV, et al. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood. 2011;117:e151–60. doi: 10.1182/blood-2010-08-300046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gattorno M, Piccini A, Lasiglie D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505–15. doi: 10.1002/art.23437. [DOI] [PubMed] [Google Scholar]
- 46.Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63:545–55. doi: 10.1002/art.30128. [DOI] [PubMed] [Google Scholar]
- 47.Zeft A, Hollister R, LaFleur B, et al. Anakinra for systemic juvenile arthritis: the Rocky Mountain experience. J Clin Rheumatol Pract Rep Rheum Musculoskeletal Dis. 2009;15:161–64. doi: 10.1097/RHU.0b013e3181a4f459. [DOI] [PubMed] [Google Scholar]
- 48.Fitzgerald AA, Leclercq SA, Yan A, et al. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52:1794–803. doi: 10.1002/art.21061. [DOI] [PubMed] [Google Scholar]
- 49.Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial) Ann Rheum Dis. 2011;70:747–54. doi: 10.1136/ard.2010.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2396–406. doi: 10.1056/NEJMoa1205099. [DOI] [PubMed] [Google Scholar]
- 51.Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol. 2008;4:615–20. doi: 10.1038/ncprheum0919. [DOI] [PubMed] [Google Scholar]
- 52.Miettunen PM, Narendran A, Jayanthan A, et al. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology. 2011;50:417–19. doi: 10.1093/rheumatology/keq218. [DOI] [PubMed] [Google Scholar]
- 53.Durand M, Troyanov Y, Laflamme P, Gregoire G. Macrophage activation syndrome treated with anakinra. J Rheumatol. 2010;37:879–80. doi: 10.3899/jrheum.091046. [DOI] [PubMed] [Google Scholar]
- 54.Grom AA, Brunner HI, Ruperto N, et al. Canakinumab in systemic juvenile idiopathic arthritis: impact on the rate and clinical presentation of macrophage activation syndrome. Ann Rheum Dis. 2014;73(Suppl 2) Abstr. FRI 0528. [Google Scholar]
- 55.de Benedetti F, Massa M, Robbioni P, et al. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34:1158–63. doi: 10.1002/art.1780340912. [DOI] [PubMed] [Google Scholar]
- 56.Xu XJ, Tang YM, Song H, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160:984–90. doi: 10.1016/j.jpeds.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 57.Strippoli R, Carvello F, Scianaro R, et al. Amplification of the response to toll-like receptor ligands by prolonged exposure to interleukin-6 in mice: implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheum. 2012;64:1680–88. doi: 10.1002/art.33496. [DOI] [PubMed] [Google Scholar]
- 58.de Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95. doi: 10.1056/NEJMoa1112802. [DOI] [PubMed] [Google Scholar]
- 59.Kobayashi M, Takahashi Y, Yamashita H, et al. Benefit and a possible risk of tocilizumab therapy for adult-onset Still’s disease accompanied by macrophage-activation syndrome. Mod Rheumatol/Jpn Rheum Assoc. 2011;21:92–96. doi: 10.1007/s10165-010-0348-9. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu M, Nakagishi Y, Kasai K, et al. Tocilizumab masks the clinical symptoms of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome: the diagnostic significance of interleukin-18 and interleukin-6. Cytokine. 2012;58:287–94. doi: 10.1016/j.cyto.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 61.Chiossone L, Audonnet S, Chetaille B, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front Immunol. 2012;3:239–49. doi: 10.3389/fimmu.2012.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Dommelen SL, Sumaria N, Schreiber RD, et al. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity. 2006;25:835–48. doi: 10.1016/j.immuni.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 63.Makay B, Yilmaz S, Turkyilmaz Z, et al. Etanercept for therapy-resistant macrophage activation syndrome. Pediatr Blood Cancer. 2008;50:419–21. doi: 10.1002/pbc.21019. [DOI] [PubMed] [Google Scholar]
- 64.Prahalad S, Bove KE, Dickens D, et al. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol. 2001;28:2120–24. [PubMed] [Google Scholar]
- 65.Aeberli D, Oertle S, Mauron H, et al. Inhibition of the TNF-pathway: use of infliximab and etanercept as remission-inducing agents in cases of therapy-resistant chronic inflammatory disorders. Swiss Med Wkly. 2002;132:414–22. doi: 10.4414/smw.2002.10031. [DOI] [PubMed] [Google Scholar]
- 66.Stern A, Riley R, Buckley L. Worsening of macrophage activation syndrome in a patient with adult onset Still’s disease after initiation of etanercept therapy. J Clin Rheumatol Pract Rep Rheum Musculoskeletal Dis. 2001;7:252–56. doi: 10.1097/00124743-200108000-00013. [DOI] [PubMed] [Google Scholar]
- 67.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1 beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA. 1999;96:2256–61. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dinarello CA. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol. 2007;27:98–114. doi: 10.1016/j.semnephrol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 69.Mazodier K, Marin V, Novick D, et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood. 2005;106:3483–89. doi: 10.1182/blood-2005-05-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maeno N, Takei S, Imanaka H, et al. Increased interleukin-18 expression in bone marrow of a patient with systemic juvenile idiopathic arthritis and unrecognized macrophage-activation syndrome. Arthritis Rheum. 2004;50:1935–38. doi: 10.1002/art.20268. [DOI] [PubMed] [Google Scholar]
- 71.Kawashima M, Yamamura M, Taniai M, et al. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001;44:550–60. doi: 10.1002/1529-0131(200103)44:3<550::AID-ANR103>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 72.Novick D, Schwartsburd B, Pinkus R, et al. A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine. 2001;14:334–42. doi: 10.1006/cyto.2001.0914. [DOI] [PubMed] [Google Scholar]
- 73.Novick D, Elbirt D, Miller G, et al. High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. Cytokine. 2009;48:103–4. doi: 10.1016/j.jaut.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Favilli F, Anzilotti C, Martinelli L, et al. IL-18 activity in systemic lupus erythematosus. Ann N Y Acad Sci. 2009;1173:301–9. doi: 10.1111/j.1749-6632.2009.04742.x. [DOI] [PubMed] [Google Scholar]
- 75.Shimizu M, Yokoyama T, Yamada K, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology. 2010;49:1645–53. doi: 10.1093/rheumatology/keq133. [DOI] [PubMed] [Google Scholar]
- 76.de Jager W, Vastert SJ, Beekman JM, et al. Defective phosphorylation of interleukin-18 receptor beta causes impaired natural killer cell function in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2782–93. doi: 10.1002/art.24750. [DOI] [PubMed] [Google Scholar]
- 77.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukocyte Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 78.Mosser DM. The many faces of macrophage activation. J Leukocyte Biol. 2003;73:209–12. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 79.Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 80.Zoller EE, Lykens JE, Terrell CE, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med. 2011;208:1203–14. doi: 10.1084/jem.20102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 82.Sikora KA, Fall N, Thornton S, Grom AA. The limited role of interferon-gamma in systemic juvenile idiopathic arthritis cannot be explained by cellular hyporesponsiveness. Arthritis Rheum. 2012;64:3799–808. doi: 10.1002/art.34604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogilvie EM, Khan A, Hubank M, et al. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:1954–65. doi: 10.1002/art.22644. [DOI] [PubMed] [Google Scholar]
- 84.Bracaglia C, Caiello I, de Graaf K, et al. Interferon-gamma in macrophage activation syndrome associated with systemic juvenile idiopathic arthritis: high levels in patients and a role in a murine MAS model. Presented at Eur Paediatr Rheumatol Congr. 2014 21st, Belgrade (Abstr) [Google Scholar]
- 85.Osugi Y, Hara J, Tagawa S, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–3. [PubMed] [Google Scholar]
- 86.Takada H, Takahata Y, Nomura A, et al. Increased serum levels of interferon-gamma-inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol. 2003;133:448–53. doi: 10.1046/j.1365-2249.2003.02237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Saint Basile G, Menasche G, Latour S. Inherited defects causing hemophagocytic lymphohistiocytic syndrome. Ann N Y Acad Sci. 2011;1246:64–76. doi: 10.1111/j.1749-6632.2011.06307.x. [DOI] [PubMed] [Google Scholar]
- 88.Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immunity. 2012;13:289–98. doi: 10.1038/gene.2012.3. [DOI] [PubMed] [Google Scholar]
- 89.Ibarra MF, Klein-Gitelman M, Morgan E, et al. Serum neopterin levels as a diagnostic marker of hemophagocytic lymphohistiocytosis syndrome. Clin Vaccine Immunol. 2011;18:609–14. doi: 10.1128/CVI.00306-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Quesnel B, Catteau B, Aznar V, et al. Successful treatment of juvenile rheumatoid arthritis associated haemophagocytic syndrome by cyclosporin A with transient exacerbation by conventional-dose G-CSF. Br J Haematol. 1997;97:508–10. [PubMed] [Google Scholar]
- 91.Gupta AA, Tyrrell P, Valani R, et al. Experience with hemophagocytic lymphohistiocytosis/macrophage activation syndrome at a single institution. J Pediatr Hematol/Oncol. 2009;13:81–84. doi: 10.1097/MPH.0b013e3181923cb4. [DOI] [PubMed] [Google Scholar]
- 92.Sung L, King SM, Carcao M, et al. Adverse outcomes in primary hemophagocytic lymphohistiocytosis. J Pediatr Hematol/Oncol. 2002;24:550–54. doi: 10.1097/00043426-200210000-00011. [DOI] [PubMed] [Google Scholar]
- 93.Coca A, Bundy KW, Marston B, et al. Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol. 2009;132:10–18. doi: 10.1016/j.clim.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 94.Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120:e622–28. doi: 10.1542/peds.2006-3164. [DOI] [PubMed] [Google Scholar]
- 95.Su IJ, Wang CH, Cheng AL, Chen RL. Hemophagocytic syndrome in Epstein-Barr virus-associated T-lymphoproliferative disorders: disease spectrum, pathogenesis, and management. Leukemia Lymphoma. 1995;19:401–6. doi: 10.3109/10428199509112197. [DOI] [PubMed] [Google Scholar]
- 96.Larroche C, Mouthon L, Casadevall N, et al. Successful treatment of thymoma-associated pure red cell aplasia with intravenous immunoglobulins. Eur J Haematol. 2000;65:74–76. doi: 10.1034/j.1600-0609.2000.9c212.x. [DOI] [PubMed] [Google Scholar]
- 97.Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol/Oncol. 2007;29:569–73. doi: 10.1097/MPH.0b013e3180f61be3. [DOI] [PubMed] [Google Scholar]
- 98.Bosman G, Langemeijer SM, Hebeda KM, et al. The role of rituximab in a case of EBV-related lymphoproliferative disease presenting with haemophagocytosis. Netherlands J Med. 2009;67:364–65. [PubMed] [Google Scholar]
- 99.Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60:101–9. doi: 10.1002/pbc.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Keith MP, Pitchford C, Bernstein WB. Treatment of hemophagocytic lymphohistiocytosis with alemtuzumab in systemic lupus erythematosus. J Clin Rheumatol Pract Rep Rheum Musculoskeletal Dis. 2012;18:134–37. doi: 10.1097/RHU.0b013e31824e8d9b. [DOI] [PubMed] [Google Scholar]