

Abstract
Mycobacterium tuberculosis antigen 85 (Ag85) enzymes catalyze the transfer of mycolic acid (MA) from trehalose monomycolate to produce the mycolyl arabinogalactan (mAG) or trehalose dimycolate (TDM). These lipids define the protective mycomembrane of mycobacteria. The current model of substrate binding within the active sites of Ag85s for the production of TDM is not sterically and geometrically feasible; additionally, this model does not account for the production of mAG. Furthermore, this model does not address how Ag85s limit the hydrolysis of the acyl-enzyme intermediate while catalyzing acyl transfer. To inform an updated model, we obtained an Ag85 acyl-enzyme intermediate structure that resembles the mycolated form. Here, we present a 1.45-Å X-ray crystal structure of M. tuberculosis Ag85C covalently modified by tetrahydrolipstatin (THL), an esterase inhibitor that suppresses M. tuberculosis growth and mimics structural attributes of MAs. The mode of covalent inhibition differs from that observed in the reversible inhibition of the human fatty-acid synthase by THL. Similarities between the Ag85-THL structure and previously determined Ag85C structures suggest that the enzyme undergoes structural changes upon acylation, and positioning of the peptidyl arm of THL limits hydrolysis of the acyl-enzyme adduct. Molecular dynamics simulations of the modeled mycolated-enzyme form corroborate the structural analysis. From these findings, we propose an alternative arrangement of substrates that rectifies issues with the previous model and suggest a direct role for the β-hydroxy of MA in the second half-reaction of Ag85 catalysis. This information affords the visualization of a complete mycolyltransferase catalytic cycle.
Keywords: acyltransferase, hydrolase, serine esterase, structural biology, glycolipid, α/β-hydrolase, antigen 85, lipid esterase, mycolyltransferase, tetrahydrolipstatin
Introduction
Tuberculosis is caused by Mycobacterium tuberculosis, a slowly replicating bacillus that primarily afflicts the respiratory system (1). A signature characteristic of M. tuberculosis is its complex cell envelope, which includes a unique lipid-rich outer membrane (mycomembrane) that provides superior environmental protection and a formidable barrier for antibiotic uptake (2). M. tuberculosis is encapsulated by an inner membrane and a periplasmic region possessing a thick peptidoglycan layer that is covalently linked to the arabinogalactan (AG),3 which is in turn covalently modified on the 5-hydroxy of the terminal arabinose by an ester-linked mycolic acid (MA) (3). The mycolated AG (mAG) accounts for the majority of the inner leaflet of the mycomembrane, whereas the outer leaflet is composed of various glycolipids, phospholipids, sulfolipids, and mycolated forms of trehalose (trehalose mono- and dimycolate (TMM and TDM, respectively)) (3).
MAs are unique to the Mycobacterium genus and consist of two alkyl chains: the shorter α-chain (24–26 carbons) and the longer, variable meromycolate chain capped by a β-hydroxy (48–62 carbons; cis- or trans-methoxy, cis- or trans-keto, and trans-hydroxy structural forms) (4). Biosynthesis of MAs occurs in the cytoplasm with the penultimate form being a mycolic β-keto ester on the 6-hydroxy of a trehalose molecule. Following reduction of the β-ketone to a β-hydroxy by CmrA, production of TMM is complete, and it is transported to the periplasm by MmpL3 (4). TMM serves as the source of MA required for the production of mAG and TDM by the essential antigen 85 (Ag85) complex (Fig. 1A) (4–6).
Figure 1.

M. tuberculosis Ag85 reaction products, mechanism, and trehalose-binding sites. A, Ag85s transfer MA onto the 6′-hydroxy of TMM and the 5-hydroxy of AG to form TDM and mAG. The depicted MA has a cis-keto meromycolate branch (4, 10). B, ping–pong reaction mechanism for Ag85C formation of TDM (R1 and R2 = MA chains) adapted from Ronning et al. (7). C, identified trehalose-binding sites adjacent to the Ag85B active site and a secondary site (Protein Data Bank code 1F0P) (9).
The Ag85 complex is encoded by the fbp genes (fbpA, fbpB, and fbpC) and comprises Ag85A, Ag85B, and Ag85C, three secreted, homologous transesterase enzymes (5). All three enzymes have been shown to be capable of catalyzing the production of TDM and mAG from TMM and respective MA acceptors (5). MA transfer occurs selectively onto the 6′-hydroxy of TMM to yield trehalose 6,6′-dimycolate or the 5-hydroxy of the terminal arabinose to produce mAG (4). Ag85 enzymes are members of the α/β-hydrolase superfamily with the three M. tuberculosis Ag85 homologs possessing a sequence similarity greater than 75% and nearly identical substrate-binding sites (7–9). Indeed, the known trehalose-binding site located within the Ag85 active site is identical in all three M. tuberculosis-encoded homologs. Ag85s utilize a ping–pong reaction mechanism with a catalytic triad composed of a nucleophilic serine (Ag85C: Ser124), histidine base (Ag85C: His260), and glutamic acid (Ag85C: Glu228) (Fig. 1B) (7–9).
Two trehalose-binding sites on Ag85 enzymes have been identified: the secondary site, which is located near the middle of the α9-helix and the base of the α5-helix, and the active site, with the 6-hydroxy pointing toward the nucleophilic serine residue (Fig. 1C) (9). On the basis of these observed binding sites, Anderson et al. (9) proposed an interfacial mechanism model in which TMM initially binds at the secondary site outside the active site, stimulating a conformational change of the side chain of Phe232 in Ag85A and -B (Leu230 in Ag85C) that allows TMM to then enter the active site and undergo nucleophilic attack as the initiating step of the first half-reaction. Following this step, the liberated trehalose molecule transiently resides in the active site until it is released as the product of the first half-reaction. This scheme suggests that, in the acyl-enzyme intermediate form, the α-chain of MA is buried in a hydrophobic hole with the meromycolate chain flipped out away from the enzyme and residing within the mycomembrane (9). The second half-reaction would then proceed upon binding of a second molecule of TMM to the secondary site followed by translocation to the active site (9). TDM is thereby produced following nucleophilic attack on the MA-enzyme intermediate by the second molecule of TMM in the active site (9).
Shortly after the interfacial mechanism model was proposed, an X-ray crystal structure of the octyl thioglucoside in complex with Ag85C was solved, highlighting potential problems with the proposed arrangement of substrates (8). The sugar moiety of octyl thioglucoside sits in the sugar-binding region/pocket of the active site with the non-hydrolyzable thioether linkage located near the nucleophilic serine. The octyl chain resides in a hydrophobic cleft pointing toward the secondary site and the potential hydrophobic hole for the α-chain of MA. The positioning of the octyl chain highlights how sterically hindered the active site would become following acylation of the enzyme, hindering the translocation of the second molecule of TMM from the secondary site to the active site. To decipher potential substrate specificity, a recent study investigated the few non-conserved residues among Ag85 homologs, some of which are near the secondary trehalose site (11). That study suggested that variations in the protein sequence affect the dynamic nature of the α9-helix, resulting in noticeable differences in substrate specificity and negating the relevance of the secondary binding site to catalysis (11).
Mutation and structural studies have shown that a disruption of the hydrogen bond network in the active site results in helical relaxation and loss of enzymatic activity (12). Specifically, when the nucleophilic serine (Ser124) was mutated to an alanine, resulting in the loss of a hydrogen bond to the catalytic histidine (His260), relaxation in the α9-helix was observed (12). A similar structural shift was observed in the Ag85C-diethyl phosphate cocrystal structure, which mimics the tetrahedral transition state (7). These mutagenesis and structural studies, paired with the findings of Backus et al. (11), suggest a direct relationship between the dynamic movement of the α9-helix and the catalytic cycle (7, 12).
On the basis of the previous studies described above and a detailed analysis of M. tuberculosis Ag85 structures determined over nearly two decades, we questioned the accuracy of the current model of substrate arrangements and the interfacial mechanism in Ag85 enzymes. Furthermore, we were intrigued as to how the enzyme limits the hydrolysis of the biologically expensive acyl-enzyme intermediate. To address these topics, we pursued a structure of the elusive mycolated form of the acyl-enzyme intermediate. Due to the insoluble nature of MAs, we used tetrahydrolipstatin (THL), which possesses two alkyl chains and yields a β-hydroxy as a result of nucleophilic attack on the β-lactone ring of THL by the enzyme. Both of these chemical characteristics mimic the core attributes of MAs. M. tuberculosis Ag85C was cocrystallized with THL, a covalent lipid esterase inhibitor with a minimum inhibitory concentration of 5 μg/ml against M. tuberculosis (13). The resulting X-ray cocrystal structure of Ag85C-THL was solved to 1.45 Å. This structure and subsequent analyses suggest how the enzyme protects the solvent-exposed acyl-enzyme intermediate form and provides a basis for an updated model of substrate binding that directly influences catalysis.
Results
Covalent inhibition by THL
THL is a well known lipid esterase inhibitor that suppresses M. tuberculosis growth (13). Covalent inhibition results from nucleophilic attack on the carbonyl center in the β-lactone ring of THL by Ag85s (Fig. 2A). To determine the rate of covalent inhibition and thus acylation of the enzyme by THL, kinact/KI was measured to be 7.9 ± 1.0 × 10−3 μm−1 min−1 by monitoring the rate of transesterification by Ag85C in the presence of varying concentrations of THL (Fig. 2B). Inhibition progress curves are provided in Fig. S1.
Figure 2.

Covalent inhibition of Ag85C by THL. A, chemical structure of THL and resulting structure upon covalent attack by Ag85C. B, kinact/KI plot of THL inhibition. Error bars represent S.D. of triplicate reactions.
Ag85C-THL X-ray crystal structure
M. tuberculosis Ag85C was cocrystallized with THL, resulting in a structure of 1.45-Å resolution (crystallographic and refinement statistics are in Table 1). Continuous difference density was present for all atoms of THL. The Fo − Fc omit map for the THL-modified Ser124 and 2Fo − Fc map for surrounding residues is shown in Fig. 3A. Covalent modification of Ser124 by THL results in an ester linkage between the drug and enzyme and yields the β-hydroxy of THL as a result of ring opening. The carbonyl of that ester linkage points directly toward the identified oxyanion hole of the backbone amides of Met125 and Leu40 (Fig. 3B) (7). The alkyl chains of THL lie within a hydrophobic cleft extending back toward the secondary trehalose-binding site, which resides below the terminal methyl carbon of the palmitic core chain. The hexanoyl tail approaches the hydrophobic hole; however, it does not extend down into this channel, which is occupied by seven water molecules (Fig. S2A). The peptidyl side arm of THL is extended toward the α9-helix, displacing the catalytic His260. Additionally, the α9-helix adopts a relaxed conformation relative to the kinked apo structure, displacing the helix away from the protein core (Fig. S2B). Unfortunately, interpretable electron density for residues 216–221, which account for a dynamic loop connecting the α9-helix to the preceding β7-strand, was not present and therefore was not modeled. Two glycerol molecules are present in the trehalose active site location with one hydroxy pointing toward the acyl-enzyme intermediate and the β-hydroxy of THL (Fo − Fc omit map in Fig. 3A). Aside from these noted observations, the overall protein fold is identical to that of apoAg85C with the two structural models exhibiting an r.m.s.d. of 0.23 Å (Fig. S2B).
Table 1.
X-ray data collection and refinement statistics (molecular replacement)
One crystal was used for this structure. Values in parentheses, unless otherwise indicated, represent data in the highest-resolution shell. r.m.s., root mean square.
| Ag85C-THL (Protein Data Bank code 5VNS) | |
|---|---|
| Data collection | |
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 68.38, 122.78, 40.26 |
| α, β, γ (°) | 90.00, 90.00, 90.00 |
| Resolution (Å) | 35.12–1.45 (1.5–1.45) |
| Total reflections (unique) | 773,689 (61,070) |
| Rmerge | 0.06 (0.53) |
| Rmeas | 0.07 (0.57) |
| CC1/2 | 0.97 (0.91) |
| I/σI | 18.4 (3.8) |
| Completeness (%) | 99.91 (99.18) |
| Redundancy | 7.3 (7.3) |
| Refinement | |
| Resolution (Å) | 35.12–1.45 |
| Rwork/Rfree | 0.161/0.173 |
| No. atoms | |
| Protein | 2,190 |
| Ligand/ion | 28 |
| Water | 281 |
| B-factors | |
| Protein | 15.0 |
| Ligand/ion | 28.9 |
| Solvent | 28.70 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.209 |
| Ramachandran | |
| Favored (%) | 97 |
| Outliers (%) | 1 |
Figure 3.

Ag85C-THL structure. A, Fo − Fc likelihood-weighted omit map (blue) contoured to 3.0 σ for Ser124 modified by THL and glycerol molecules in the active site and 2Fo − Fc map (red) contoured to 1.5 σ for surrounding active site residues. B, surface rendering of Ag85C with Ser124 modified with THL and two glycerol molecules occupying the trehalose-binding portion of the active site.
Structural analysis of Ag85s with disrupted catalytic triads
In the catalytically active enzyme, the nucleophilic hydroxy of Ser124 is hydrogen-bonded to the ϵ-nitrogen of the His260 imidazole ring, and the δ-nitrogen of His260 is hydrogen-bonded to Glu228 (Fig. 4A) (7). In this form, the α9-helix is kinked toward the active site with a connecting dynamic loop positioned away from the active site, allowing for substrate binding (Fig. 4A). In structures exhibiting a disrupted catalytic triad, His260 rotates about both χ1 and χ2 to form an interaction with the side chain of Ser148 (Fig. 4A) (7, 12, 14, 15). Disruption of hydrogen bonds with residues of the α9-helix therefore relaxes the helix, allowing the dynamic loop to be positioned over the active site, resulting in the side chain of Leu217 being oriented between His260 and the nucleophilic Ser124 (Fig. 4, A and B) (12). Residue conservation at position 217 is split between leucine and isoleucine in all mycolyltransferases (Fig. S3A). Positioning of the aliphatic side arm of THL is similar to that of Leu217 (Fig. 4B). In both the disrupted catalytic and THL-modified forms, the aliphatic substituents help orient the His260 side chain such that it maintains a hydrogen bond with Ser148 and abrogates potential solvent interactions with His260, decreasing the potential for water activation (Fig. 4B).
Figure 4.
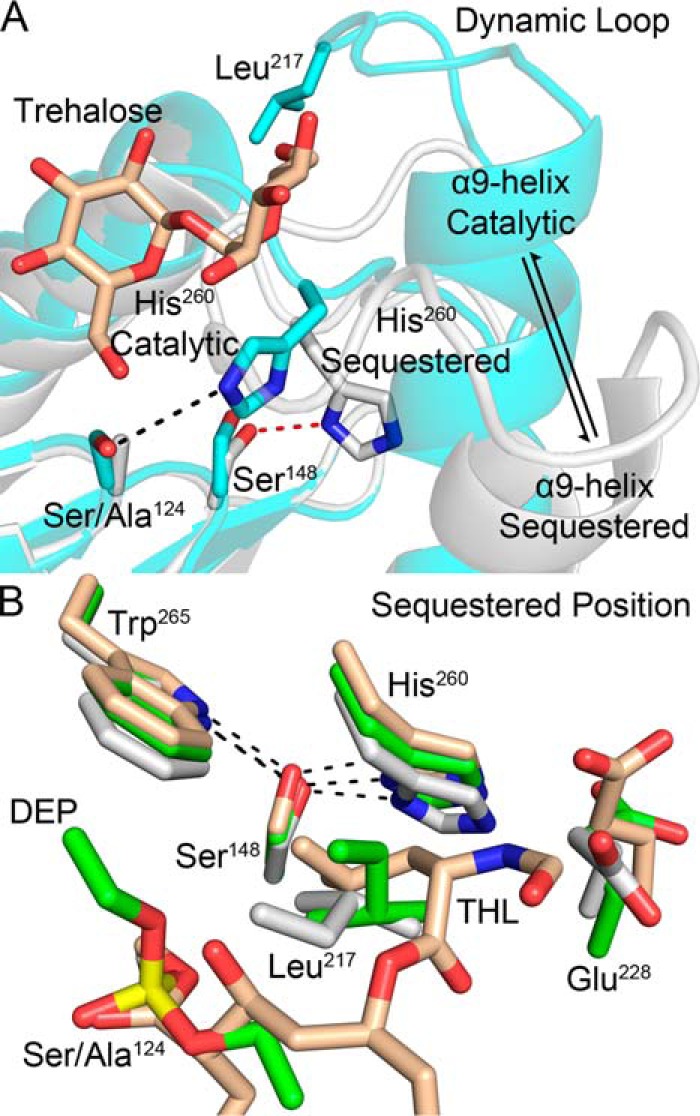
Observed structural changes between the active and acyl-enzyme intermediate forms. A, catalytic triad disruption results in relaxation of the α9-helix with His260 adopting a sequestered position (white, Ag85C-S124A, Protein Data Bank code 4QEK; cyan, trehalose-bound Ag85B, Protein Data Bank code 1F0P) (9, 12). B, consistent displacement of His260 results in hydrogen bond formation with Ser148. The aliphatic side arm of THL mimics the hydrophobic interactions of the side chain of Leu217 in the diethyl phosphate–modified Ag85C and S124A mutant structures (wheat, Ag85C-THL, Protein Data Bank code 5VNS; green, Ag85C-diethyl phosphate, Protein Data Bank code 1DQY; white, Ag85C-S124A, Protein Data Bank code 4QEK) (7, 12).
This repositioning of His260 by disruption of the catalytic triad results in the formation of a hydrogen bond between the δ-nitrogen of the imidazole ring and the neighboring hydroxy of Ser148. Ser148 and the neighboring Trp265, which forms a hydrogen bond with Ser148 through its indole nitrogen, are both highly conserved among all known mycolyltransferases (Fig. S3, B and C). In every Ag85C structure with disrupted catalytic triads, a similar displacement of His260 that mimics this acylated form is present (Fig. 4B) (7, 12, 14, 15).
WT, S148A, and S148T enzymatic activity
Based on the structural alignments and high sequence conservation, we sought to determine whether Ser148 plays a direct role in sequestering the catalytic His260 to limit hydrolysis of the acyl-enzyme intermediate. To investigate potential effects of Ser148 on enzymatic activity, we mutated Ser148 to alanine and threonine. Both variants lacked observable transesterase activity but exhibited hydrolase activity at a lower level than that of WT (43.46 ± 3.1 and 25.06 ± 1.4% of observed WT hydrolysis for S148A and S148T, respectively) (Fig. S4).
Molecular dynamics
To investigate the conformational dynamics of the acyl-enzyme intermediate and THL-inhibited forms of Ag85C, we performed conventional molecular dynamics (MD) simulations of four models. Specifically, we simulated Ag85C in complex with THL (Ag85C-THL), MA (Ag85C-MA-His260seq and Ag85C-MA-His260cat), and trehalose with MA (Ag85C-MA-trehalose). The Ag85C-MA models contained two different conformations of His260, i.e. the sequestered position and the catalytic position. In the sequestered position, His260 is hydrogen-bonded to Ser148, whereas in the catalytic position, His260 is hydrogen-bonded to the proximal β-hydroxy of MA (Ag85C-MA-His260cat). The fourth model (Ag85C-MA-trehalose) is with His260 in the catalytic position and an acceptor molecule of trehalose in the active site sugar-binding site. The Ag85C-MA-trehalose simulation was conducted to study our proposed substrate-binding model and to gain insights into nucleophilic activation for the second half-reaction. Details on how each of the four MD starting models were generated are given under “Experimental procedures.”
Each Ag85C model maintained important active site interactions over the time scale of the simulations (see results below). In addition, each model exhibited moderate backbone r.m.s.d. fluctuations (<2.5 Å) of the entire protein throughout the simulations (Fig. S5). Specifically, for Ag85C-THL, His260 remained sequestered to Ser148, and the overall complex was found to be stable throughout the simulation. When THL was modeled to mimic the natural substrate, i.e. in the Ag85C-MA-His260seq model, His260 remained in the sequestered conformation. His260 remained within hydrogen-bonding distance of Ser148 for the entire simulation despite the removal of the peptidyl side arm (Fig. 5, A and C). When His260 was positioned in the catalytic orientation, a hydrogen bond between His260 and the β-hydroxy of MA was present in fewer than 25% of the simulation trajectory, which is substantially more transient than in the Ag85C-MA-His260seq model. Specifically, the Ag85C-MA-His260seq model maintained a hydrogen bond between His260 and Ser148 for 98% of the simulation trajectory (Fig. 5, B and C). When an acceptor molecule of trehalose was present and His260 was in the catalytic position (Ag85C-MA-trehalose), His260 was within a weak-to-moderate hydrogen-bonding distance (N···H distance within 2.5 Å) with either the β-hydroxy of MA or the 6′-hydroxy of trehalose (Fig. 6A). Interestingly, in the presence of trehalose, His260 did not interact with the solvent through hydrogen bonding but rather alternated between interacting with the β-hydroxy of MA and the 6′-hydroxy of trehalose (Fig. 6B). Therefore, we propose that the binding of the incoming acceptor molecule drives the protein to form an active conformation. Based on the two hydrogen-bonding configurations of His260 that were observed in the Ag85C-MA-trehalose MD simulation, the second half-reaction could therefore proceed through a direct or indirect activation pathway upon acceptor molecule binding (Fig. 7).
Figure 5.

MD models of the mycolated Ag85C intermediate. A, Ag85C-MA-His260seq. B, Ag85C-MA-His260cat. C, hydrogen bonding of His260 with Ser148 in the sequestered position was stable during MD simulations. When His260 was placed in the catalytic position without an acceptor molecule present the side chain freely samples conformations, making limited hydrogen-bonding interactions with the β-hydroxy of MA.
Figure 6.

MD model of the mycolated Ag85C intermediate with acceptor molecule present. A, Ag85C-MA-trehalose MD model. B, in the catalytic position when an acceptor molecule is present, His260 exclusively sampled both the 6′-hydroxy of trehalose and the β-hydroxy of MA.
Figure 7.
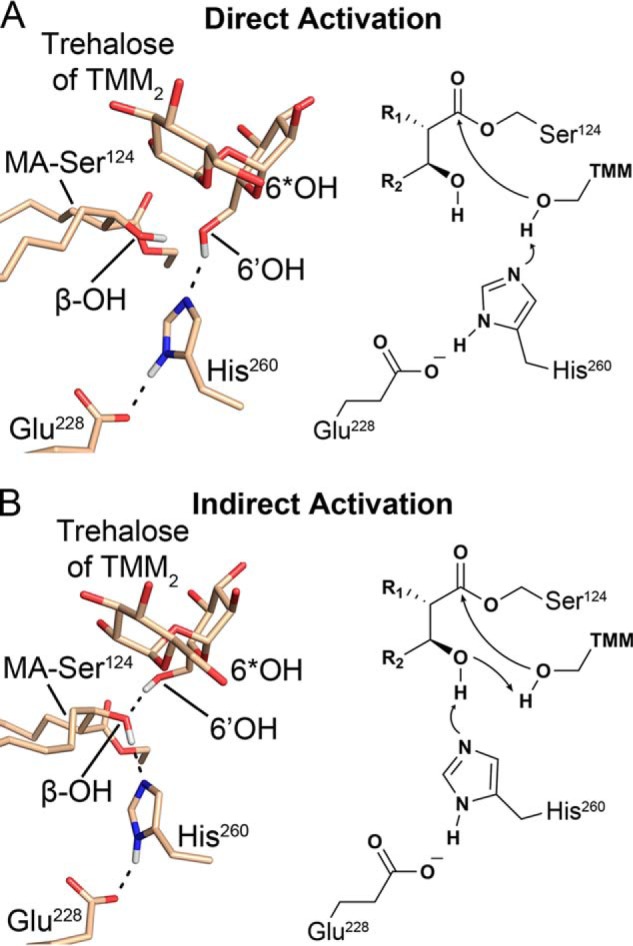
Nucleophilic activation pathways for the second half-reaction (6*OH denotes the mycolated hydroxy of TMM). A, direct activation of the 6′-hydroxy of TMM for the second half-reaction by His260, stabilized by the β-hydroxy of MA. B, indirect activation scheme proceeding through a proton transfer from the β-hydroxy of MA to His260 followed by the proton transfer from the 6′-hydroxy to deprotonated β-hydroxy of MA.
To achieve more thorough phase space sampling and obtain an unbiased representation of thermodynamically accessible conformations in the acyl-enzyme intermediate, we also performed temperature replica-exchange molecular dynamics (T-REMD) simulations on the Ag85C-MA-His260cat model. T-REMD is an enhanced sampling method that affords a more extensive and efficient exploration of phase space than conventional MD simulations (16–18). For the T-REMD simulations of Ag85C-MA-His260cat, the potential energy of each temperature replica had sufficient overlap with the neighboring replicas, resulting in an exchange rate of ∼33% between each replica (Fig. S6A). Sufficient overlap of the potential energy between the individual replicas ensured that each replica could randomly “walk” through the temperature range, thereby enhancing phase space sampling. The backbone r.m.s.d. distribution of the replicas remained constant after 8 ns (320 ns total = 8 ns × 40 replicas) of simulation, indicating system convergence (Fig. S6B). The distance between His260 and the β-hydroxy of MA and the distance between the nucleophilic Ser124 and the α9-helix are plotted as a 2D heat map (Fig. 8). When both of these distances are small, the enzyme is in the catalytic state. Alternatively, when the distance between His260 and β-hydroxy of MA was large (>5 Å), two states were observed. In state 1, the α9-helix was kinked toward the active site, similar to what was observed in the catalytic state, but the distance between His260 and β-hydroxy of MA was between 5 and 7 Å. In the T-REMD simulations, the enzyme transitioned toward the relaxed form of the enzyme (state 2). In state 2, the α9-helix was farther away from the active site, whereas His260 remained dynamic and sampled many conformations. Importantly, in many of these states, His260 moved closer to Ser148 but never formed a hydrogen bond with the hydroxy of Ser148. The conformations of His260 observed in the initial conventional MD simulation of Ag85C-MA-His260cat are consistent with the corresponding REMD simulations, suggesting that the shorter simulations were sufficient to obtain meaningful conformational sampling of the active site.
Figure 8.
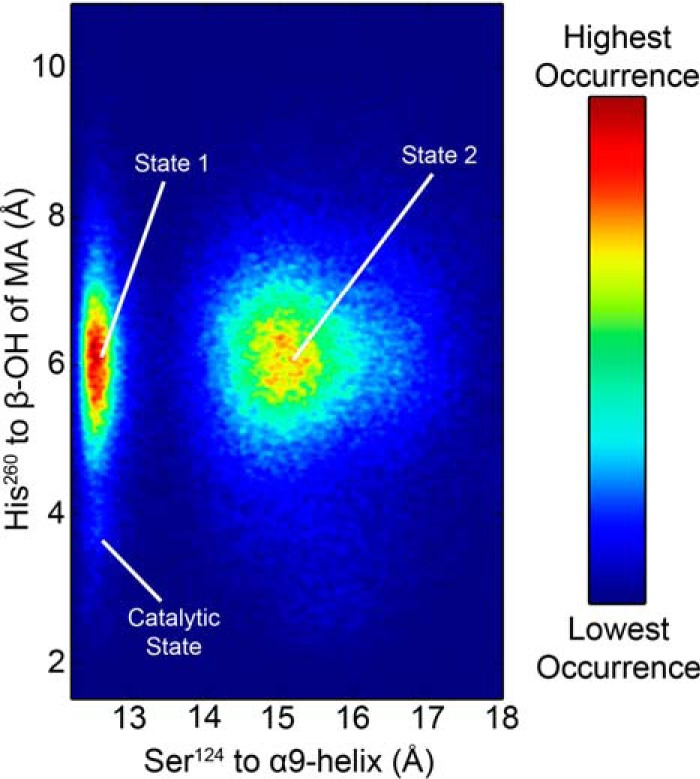
Heat map of Ag85C-MA-His260cat conformations sampled during the REMD simulation. The starting catalytic position was not frequently sampled when the α9-helix remained kinked toward the active site in the catalytic position; instead His260 randomly sampled phase space, similar to what was observed in the initial MD simulation (state 1). In state 2, the α9-helix relaxes, similar to what is observed in the sequestered form. Although His260 is positioned toward Ser148, a stable hydrogen bond to Ser148 is never observed.
Discussion
THL, known commercially as Orlistat, is a synthetically stable derivative of lipstatin, a natural product synthesized by Streptomyces toxytricini that inhibits the human pancreatic lipase (19, 20). THL was shown to inhibit the thioesterase domain of the human fatty-acid synthase (FAS) (21). Functioning as a versatile lipid esterase inhibitor, THL exhibits growth inhibition on M. tuberculosis via covalent modification of various endogenous lipid esterases; Ag85C is reported as one of 14 validated targets (13). Here, we have shown that covalent inhibition occurs within minutes, and initial binding affinities are in the low μm range (8.7 ± 0.7 μm), resulting in a kinact/KI of 7.9 ± 1.0 × 10−3 μm−1 min−1. This value falls within the range of previously reported values for ebselen-derived covalent inhibitors of Ag85C that are known to efficiently inhibit M. tuberculosis growth (15).
A significant difference in THL inhibition of Ag85C compared with human FAS is the lack of observed hydrolysis of the covalent enzyme adduct. The structure of FAS in complex with THL contains two molecules in the asymmetric unit: one with an intact ester linkage and the other in a hydrolyzed form (22). A later study found that movement of the hexanoyl tail resulted in the repositioning of the β-hydroxy of THL (23). Repositioning of the β-hydroxy disrupts hydrogen bonding to the stationary catalytic histidine, allowing for water activation and subsequent hydrolysis of the acyl-enzyme intermediate (23). However, in the Ag85C-THL structure, we found that covalent modification results in structural changes, specifically the catalytic histidine, His260, is physically displaced by the peptidyl side arm and is instead within hydrogen-bonding distance to neighboring Ser148 (Fig. 4A). The sequestered positioning of His260 to Ser148 was shown to be stable via MD simulations, thereby limiting water activation and subsequent hydrolysis of Ag85C-THL. This stable interaction is experimentally supported as Ag85C was successfully crystallized with THL in only a slight molar excess of THL to enzyme, 1.2:1, respectively.
A similar displacement of His260 toward Ser148 is observed in both the tetrahedral intermediate (represented by the Ag85C-diethyl phosphate complex) and the nucleophilic S124A mutant structures (Fig. 4A) (7, 12). Although both of these structures lack a bulky substituent to force the dislocation of His260, the side chain of His260 occupies the sequestered conformation due to structural rearrangement resulting from hydrogen bond disruption between His260 and the nucleophilic serine (12). Therefore, we hypothesized that the acyl-enzyme intermediate form of Ag85C increases the number of conformations that are energetically accessible for His260, which results in limiting the hydrolysis of the acyl-enzyme intermediate. The sequestered conformation is one of the many conformations that His260 can adopt in the acyl-enzyme intermediate that may prevent water binding at the site necessary for nucleophilic attack on the acyl-enzyme intermediate. Indeed, many conformations of His260 were observed in the MD simulation of the Ag85C-MA-His260cat model. In general, His260 was observed to be >5 Å from the β-hydroxy of MA, positioning the imidazole ring away from the acyl-enzyme ester moiety, prohibiting His260 to act as a general base. However, His260 did sample conformations near the acyl-enzyme ester, allowing for potential water activation. This observation would therefore be consistent with the low levels of hydrolysis observed in solution (Fig. S4). To enhance the sampling of thermodynamically accessible conformations, we conducted T-REMD simulations of the acyl-enzyme intermediate. In addition, during the T-REMD simulations, the system transitioned toward the sequestered form, but a hydrogen bond did not form between His260 and Ser148. In the mutant activity studies, low levels of hydrolytic activity persisted when the highly conserved Ser148 was mutated to alanine or threonine to investigate a potential role for Ser148 in sequestering His260. Similar to the wildtype enzyme, we attributed this residual hydrolytic activity to His260 sampling active conformations. Interestingly, the Ser148 mutants did lose all detectable acyltransferase activity, indicating that Ser148 plays a role in the molecular positioning of the acceptor molecule with the active site of Ag85C.
In the relaxed structural form, the α9-helix and the connecting loop to the β7-strand accompany the displacement of His260, which has been observed in numerous Ag85C structures with either allosteric covalent modification or perturbation of the catalytic triad (7, 12, 14, 15). In each case, the dynamic loop falls into the active site due to α9-helix relaxation (Fig. 4, A and B). Restructuring of the dynamic loop results in the side chain of Leu217 being positioned in the same location as the peptidyl side arm of THL in the Ag85C-THL structure, further hindering water activation by His260 (Fig. 4B). Unfortunately, the loop configuration associated with the fully sequestered form of the enzyme, which is observed crystallographically, was not sampled in any of the performed MD or REMD simulations. Whether the enzyme fully adapts the hypothesized sequestered form or not, there is sufficient structural, computational, and enzymatic evidence to suggest that M. tuberculosis Ag85s evolved to limit hydrolysis of the acyl-enzyme intermediate through structural rearrangements stimulated by enzyme acylation.
The previous understanding of the mycolated form of Ag85s was that the α-chain of MA was buried in the hydrophobic hole while the meromycolate chain was flipped outward into the mycomembrane (9). However, the positioning of the resulting bound mycolic acid in relation to the secondary trehalose-binding site is problematic for catalysis of TDM for two reasons. First, this mycolated form would be too sterically hindered to allow an incoming acceptor molecule from the secondary site to enter the active site (9). Second, this positioning would preclude nucleophilic attack by TMM on the carbonyl intermediate because geometric requirements for nucleophilic attack could not be met (24). The incorrect alignment of the carbonyl relative to the oxyanion hole would not allow for stabilization of the oxyanion intermediate (7). Additionally, this scheme does not account for mAG synthesis. Together, these factors further negate the interfacial mechanism model and the proposed coordination of substrate as a function of enzymatic reaction progression. Therefore, an alternative arrangement of MA and the incoming acceptor molecule within the Ag85 active site is required.
We propose that both the α-alkyl chain of the MA and the portion of the meromycolate chain proximal to the ester linkage most likely lie within the active site hydrophobic cleft. The region of the meromycolate chain distal to the ester linkage may remain embedded in the mycomembrane. This positioning properly orients the carbonyl of the acyl-enzyme intermediate toward the oxyanion hole. Furthermore, it allows for nucleophilic attack from the respective 6′- or 5-hydroxy of either trehalose of TMM or arabinose of the AG upon binding to the identified sugar-binding site of the active site. Two potential pathways for the activation of the incoming nucleophile are therefore chemically viable based on this binding mode and were exclusively sampled in the Ag85C-MA-trehalose MD simulation (Figs. 6 and 7). Central to both pathways is the β-hydroxy of MA as it is positioned to allow for either direct or indirect activation of the incoming nucleophile based on the stable catalytic conformation of His260 in the presence of trehalose (Fig. 7).
Indirect activation would proceed through a concerted proton transfer from the β-hydroxy of MA to His260 while the proton is being transferred from the incoming nucleophilic hydroxy of the acceptor molecule to the β-hydroxy of MA (Fig. 7B). The now deprotonated hydroxy is free to undergo nucleophilic attack on the acyl-enzyme ester. The indirect pathway is chemically feasible and would be similar to a proton shuttle mechanism (25–27). The more conventional, direct activation pathway would consist of a simple proton transfer from the incoming hydroxy nucleophile by His260 (Fig. 7A). Again, the deprotonated hydroxyl is now free to undergo nucleophilic attack on the acyl-enzyme ester. Regardless of the pathway chosen, the β-hydroxy of MA stabilizes the deprotonated nucleophile prior to nucleophilic attack. Therefore, the final reduction of the β-ketone of MA to β-hydroxy during biosynthesis of MA is required for Ag85 catalysis (4).
Our proposed organization of substrates and intermediates as the reaction proceeds is depicted in Fig. 9 and outlined in the corresponding figure legend. The proposed alternative model satisfies the issues raised with the previous model and highlights the importance of the dynamic nature of the α9-helix in the enzymatic activities of Ag85A, -B, and -C (11). Specifically, this arrangement of substrates in the enzyme explains how both TMM and AG can act as acceptor molecules and why both are selectively mycolated on the 6′- or 5-hydroxy, respectively. Therefore, the secondary trehalose-binding site is not necessary for this catalytic reaction scheme, but the affinity for trehalose and sugar-based detergents cannot be ignored (8, 9). As a consequence of the conformational change between native and acyl-enzyme forms, the secondary binding site changes shape and thereby likely changes the affinity of this site for carbohydrates. Therefore, it seems most likely that the secondary binding site of Ag85 has affinity to trehalose to maintain Ag85 at the surface of the mycomembrane. This association is most important in the native form of the enzyme where it interacts with the mycomembrane surface through non-covalent interactions but is less important when Ag85 is in a covalent complex with an MA that is embedded in the mycomembrane.
Figure 9.

Proposed structure-based catalytic cycle of Ag85s. The light blue surface corresponds to the α9-helix and dynamic loop, yellow corresponds to the MA-binding site, and red corresponds to the sugar-binding site. A, apoenzyme exhibits a large TMM1-binding site (Protein Data Bank code 1DQZ) (7). B, Ag85C octyl thioglucoside structure mimics the initial TMM1 binding event (Protein Data Bank code 1VA5) (8). C, tetrahedral transition state (TS) of the first half-reaction (RXN) represented by the Ag85C-diethyl phosphate structure with His260 sequestered and the α9-helix relaxed (Protein Data Bank code 1DQY) (7). A free trehalose molecule leaves, completing the first half-reaction. D, model of the Ag85C-MA intermediate based on the Ag85C-THL structure. E, the second half-reaction proceeds through the binding of TMM2 or AG to the sugar-binding site. The α9-helix and His260 are restored to the catalytic position as a result of acceptor binding. The model of Ag85C-MA-trehalose is shown. F, transition state for the second half-reaction, again modeled by Ag85C-diethyl phosphate, leading to the formation of TDM or mAG and subsequent product release (Protein Data Bank code 1DQY) (7).
The Ag85C-THL structure and findings presented in this study should facilitate the design of new THL-derived drugs with higher specificity for M. tuberculosis Ag85s while avoiding other, non-essential M. tuberculosis and human lipid esterases. Conversely, the findings of this study can influence modifications to THL that potentially enhance inhibition of other lipid esterases outside of M. tuberculosis. In particular, this structure highlights the important role of the peptidyl side arm in extending the lifetime of the covalent enzyme-inhibitor complex and should inform the design of covalent α/β-hydrolase inhibitors to prevent activation of water. With regard to M. tuberculosis and general acyltransferase chemistry, the mycolated enzyme intermediate model affords visualization of every step of the catalytic cycle and a better understanding of mycolyltransferase catalysis while rationalizing the chemical need for the β-hydroxy of MA. Ultimately, these catalytic insights can be applied to the entire α/β-hydrolase superfamily, providing a better understanding of how these enzymes conduct unique catalytic functions despite a common fold and catalytic triad.
Experimental procedures
WT and mutant molecular cloning
The M. tuberculosis fbpC gene was previously cloned into a pET-29a plasmid as described (14). The recombinant Ag85C expression construct included a non-cleavable C-terminal polyhistidine tag. S148A and S148T mutants were generated using the Agilent QuikChange® Lightning kit. The plasmid harboring the WT fbpC gene was used as the template for both mutagenesis reactions with primers 5′-ggttgaggaagcccgccaacgacgcggcgta-3′, 5′-ggttgaggaagcccgtcaacgacgcggcgta-3′, and corresponding complements used for the S148A and S148T mutants, respectively.
Protein expression and purification
Recombinant M. tuberculosis WT Ag85C was expressed and purified as previously published with S148A and S148T following identical experimental procedures (14). In short, chemically competent T7 Express Escherichia coli cells were transformed with the desired pET-29a C-terminal polyhistidine construct. Inoculated cultures were grown at 37 °C in Luria-Bertani broth to a density of 0.6 A600 nm. Incubation temperature of the cultures were then dropped to 16 °C, and protein expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside. After 24–36 h, induced cultures were harvested and resuspended in 20 mm Tris pH 8.0 buffer containing 5 mm β-mercaptoethanol and placed at −80 °C for storage.
Induced cells were thawed, lysozyme and DNase I were added and incubated on ice, and cell lysis was complete following sonification. The crude cellular lysate was clarified by centrifugation and loaded onto a metal (cobalt) affinity chromatography column equilibrated with lysis buffer. Following washing with lysis buffer, protein was eluted with a gradient of imidazole (0–150 mm). Eluted protein was pooled and loaded onto a 5-ml anion exchange column equilibrated with washing buffer (20 mm Tris pH 8.0 buffer containing 1 mm EDTA and 0.3 mm tris(2-carboxyethyl)phosphine). Following washing, protein was eluted with a 0–1 m NaCl gradient. Eluted protein was pooled and subjected to ammonium sulfate precipitation (2.8 m). Precipitated protein was pelleted via centrifugation and resuspended in 10 mm Tris, pH 7.5, 2 mm EDTA for crystallization or 50 mm sodium phosphate, pH 7.5, for enzymatic assays. Resuspended protein was dialyzed overnight against the respective buffer to remove residual ammonium sulfate.
Crystallization and data collection
Recombinant Ag85C was concentrated to 150 μm (∼5 mg/ml). Ag85C-THL crystals were obtained through cocrystallization via incubation of THL (20 mm stock in DMSO) at a 1:1.2 molar ratio of protein to compound for 90 min on ice prior to drop setup. Using the hanging drop method, crystals formed after a week at 16 °C in a 1:1 protein to well solution (0.1 m calcium chloride dehydrate, 0.05 m Bis-Tris, pH 6.5, 22.5% (v/v) (±)-2-methyl-2,4-pentanediol, Hampton Research Index HR2-144). Crystals were cryoprotected through the addition of 0.2 μl of glycerol (10% (v/v) final drop concentration) immediately prior to looping and flash cooling in liquid nitrogen. X-ray diffraction data were collected at 100 K using synchrotron radiation (λ = 0.97872 Å) on beam line F of Life Sciences Collaborative Access Team, Advanced Photon Source at Argonne National Laboratory.
Structure determination and refinement
Diffraction data were indexed, integrated, and scaled using HKL2000 (28). The resulting diffraction data set was indexed and scaled as P21212. The phase solution came from molecular replacement using the native Ag85C structure (Protein Data Bank code 1DQZ) with one molecule determined to be in the asymmetric unit. Residues not fitting 2Fo − Fc electron density were deleted, and the resulting model was subjected to a rigid body refinement followed by simulated annealing (phenix.refine) (29). Deleted residues and the THL-modified Ser124 were built manually using Coot (30). Restraints for the THL-modified Ser124 were generated with eLBOW (31). Glycerol and (±)-2-methyl-2,4-pentanediol molecules were added using Ligand Fit (32). The progressing model was subjected to rounds of XYZ coordinate, real-space, occupancy, and B-factor refinements in between manual builds and ligand additions (phenix.refine) (29). Model building and refinement ended when Rwork and Rfree values of 0.163 and 0.167, respectively, were reached with 97% of residues being Ramachandran favored and 1% being outliers. Model statistics were validated with MolProbity (33). The two outlier residues, Gly29 and Ser86, have been flagged as outliers in prior Ag85C structures (12). These residues are found within loop regions and are positioned based on the difference density maps giving correlation coefficients of 0.97 and 0.86 for those two residues, respectively.
Enzymatic and inhibition assays
A previously described fluorescence-based assay was used to monitor transesterase activity (14). For inhibition studies, THL was serially diluted from a 30 mm stock in DMSO, resulting in a range of final reaction concentrations of 300–12.5 μm. Kinetic reads were initiated following the titration of resorufin butyrate immediately after the titration of THL or an equal volume of DMSO. Kinetic reads were conducted at 37 °C in a 50 mm sodium phosphate buffer, pH 7.5, using λex = 500 nm and λemit =590 nm. Relative fluorescent units were converted to product concentration using a previously established standard curve methodology (15). Background water hydrolysis of resorufin butyrate was subtracted from the triplicate data, and the rates were determined using Prism 7 with a one-phase association equation: Y = Y0 + (Plateau − Y0)(1 − exp(−kx)) where x = time, Y = [Product], (Y0 + (Plateau − Y0) = Vi/kobs, and k = kobs. kinact/KI was determined by plotting the kobs versus inhibitor concentration and fitting the data with the equation kobs = kinact/(1 + (KI/[I])) (34).
Enzymatic activity for mutants was determined with an identical assay as described above. Conditions for transesterase activity were as follows: 500 nm respective enzyme, 4 mm trehalose (500 mm buffer stock), and 100 μm resorufin butyrate (10 mm DMSO stock). Kinetic reads were initiated immediately following the titration of resorufin butyrate. For hydrolase activity, reactions were identical sans trehalose. Data were converted to product concentration, background water hydrolysis of resorufin butyrate was subtracted from all triplicate data, and rates were determined using Prism 7 with a linear fit. Reported transesterase activity has the enzymatic rate of resorufin butyrate hydrolysis subtracted.
Sequence alignments
The sequences of 464 known mycolyltransferases were aligned using Clustal Omega (35). The resulting alignment was used to generate the probability of a given amino acid at a determined position, indicating sequence conservation. Figures were generated using WebLogo 3 (36).
Molecular modeling and MD simulations
The X-ray crystal structure presented (Protein Data Bank code 5VNS) was used as the model for the Ag85C-THL simulations. For the Ag85C-MA sequestered/catalytic and Ag85C-MA-trehalose classical MD simulations, the models generated were adapted from the Ag85C-THL and Ag85-trehalose X-ray structures. Because of the structural similarities of THL and MA, the Ag85C-THL crystal structure permitted a straightforward transformation to the mycolated acyl-enzyme intermediate form of the enzyme. For the starting Ag85C-MA model, we kept the alkyl chain lengths the same as those observed in the THL structure while inverting the observed stereochemistry of THL to that of MA; additionally, the peptidyl side arm of THL was removed. THL was modified to mimic MA and then used to generate the ligand using eLBOW (31). The resulting MA molecule was manually positioned in the active site with PyMOL (37), and the final coordinates were then inserted into the Ag85C model in place of THL. The MA alkyl chain atoms were modeled based on the 2Fo − Fc density for THL. The final MA model was subjected to bond angle refinement in Coot (30). Upon alignment of the Ag85C-THL structure or the resulting Ag85C-MA model with the previously published Ag85B-trehalose structure (Protein Data Bank code 1F0P), it became apparent that the 6′-hydroxy is positioned for nucleophilic attack on the carbonyl intermediate (9). We therefore modeled trehalose as an incoming acceptor molecule. In the crystal structure of Ag85C-THL, His260 is displaced away from the catalytic position by the peptidyl moiety of THL. Thus, His260 was shifted back to the catalytic position by aligning Ag85C-THL to the Ag85B-trehalose structure. The dynamic loop in each model was rebuilt using the program Coot (30). The protonation states of Ag85C at pH 7.5 were assigned using PROPKA with the PDB2PQR sever (38). The net charge of Ag85C was −6. Each substrate used in the MD simulations had an overall neutral charge.
Molecular mechanics force field parameters were generated for MA, THL, and trehalose as follows. Each substrate model was subjected to geometry optimization with Gaussian 09 (39) at the HF/6–31G* level of theory. Specifically, for the covalently linked substrates (THL-Ser124 and MA-Ser124) the serine backbone was capped with a (-CO-CH3) C-terminal cap and an (-NH-CH3) N-terminal cap. Following geometry optimization, the electrostatic potential of each substrate was computed at the same level of theory. Atomic partial charges were calculated by restrained electrostatic potential charge fitting using the program R.E.D. (40). The antechamber module of AMBER16 was then used to assign atom types from the general AMBER force field for both THL and MA, and the GLYCAM_06j-1 force field was used for trehalose (41–43).
Each model was generated with the leap module of AMBER16 using the ff14SB force field to describe the protein (43). Crystallographic water molecules were retained, and each system was further solvated in a cubic box of TIP3P water with at least 20 Å between the protein and the nearest face of the solvent box (44). Six Na+ counterions were added to neutralize the charge of the system. The system was then subjected to 1000 steps of steepest descent minimization followed by 250 steps of conjugate gradient minimization. For the MD simulations, periodic boundary conditions were applied, and the particle mesh Ewald method was used to evaluate long-range electrostatic interactions (45). A cutoff of 8 Å was used for real-space non-bonded interactions. The SHAKE algorithm was used to constrain all bonds to hydrogen, allowing the use of a 2-fs time step (46). Prior to production MD simulation, the system was equilibrated in three steps. First, the system was heated from 0 to 300 K over 500 ps with Langevin dynamics and a collision frequency of 1 ps−1. During the first equilibration step, all heavy atoms of the protein and ligand(s) were subjected to a harmonic restraint potential of 5 kcal mol−1 Å−2. The second equilibration step consisted of a 500-ps simulation at constant pressure (1 atm) with isotropic scaling, again with heavy atoms harmonically restrained. In the third 500-ps equilibration step, only the Cα atoms were restrained. Each system was then subjected to 100-ns unrestrained production MD.
For the T-REMD simulations, the systems were equilibrated using the same approach as for the standard MD equilibration except that the final temperature of each replica was selected based on the temperature distribution, which was calculated as follows. Four systems were equilibrated at 280, 306, 332, and 358 K. The average energy was then computed from these four systems. The average energy as a function of temperature was fitted to a polynomial, allowing iterative solution of the Monte Carlo criterion with an exchange rate of 0.3 (18). The resulting temperature distribution was 299.2, 300.4, 301.6, 302.8, 304.1, 305.3, 306.5, 307.7, 308.9, 310.2, 311.4, 312.7, 313.9, 315.2, 316.4, 317.7, 319.0, 320.3, 321.5, 322.8, 324.1, 325.4, 326.7, 328.0, 329.3, 330.6, 331.9, 333.2, 334.5, 335.9, 337.2, 338.5, 339.9, 341.2, 342.6, 343.9, 345.3, 346.6, 348.0, and 349.4 K. The production T-REMD simulations were performed in the NVT ensemble. The time between exchanges was 0.5 ps, and each system was evolved for 12 ns, yielding a total of 12 ns × 40 replicas = 480-ns cumulative simulation time. During the simulation, the frames were recorded every ps, and all frames were used during the data analysis, which was performed with the pytraj module (47, 48).
Author contributions
C. M. G., S. D., M. D. S., J. M. P., and D. R. R. formal analysis; C. M. G., S. D., M. D. S., J. M. P., and D. R. R. validation; C. M. G., S. D., M. D. S., and J. M. P. investigation; C. M. G. and S. D. methodology; C. M. G. and S. D. writing-original draft; C. M. G., S. D., M. D. S., J. M. P., and D. R. R. writing-review and editing; J. M. P. and D. R. R. data curation; J. M. P. and D. R. R. supervision; J. M. P. and D. R. R. visualization; D. R. R. conceptualization; D. R. R. funding acquisition; D. R. R. project administration.
Supplementary Material
Acknowledgments
This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. This work used resources of the Compute and Data Environment for Science (CADES) at Oak Ridge National Laboratory, which is managed by UT–Battelle, LLC for the United States Department of Energy under Contract DE-AC05-00OR22725.
This work was supported in part by National Institutes of Health Grant AI105084. This work has been co-authored by UT–Battelle, LLC under Contract DE-AC05-00OR22725 with the United States Department of Energy. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and the United States Department of Energy.
This article contains Figs. S1–S6.
The atomic coordinates and structure factors (code 5VNS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AG
- arabinogalactan
- Ag85
- antigen 85
- MA
- mycolic acid
- TMM
- trehalose monomycolate
- TDM
- trehalose dimycolate
- mAG
- mycolyl arabinogalactan
- THL
- tetrahydrolipstatin
- r.m.s.d.
- root mean square deviation
- MD
- molecular dynamics
- T-REMD
- temperature replica-exchange molecular dynamics
- FAS
- fatty-acid synthase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Pai M., Behr M. A., Dowdy D., Dheda K., Divangahi M., Boehme C. C., Ginsberg A., Swaminathan S., Spigelman M., Getahun H., Menzies D., and Raviglione M. (2016) Tuberculosis. Nat. Rev. Dis. Primers 2, 16076 10.1038/nrdp.2016.76 [DOI] [PubMed] [Google Scholar]
- 2. Jackson M., McNeil M. R., and Brennan P. J. (2013) Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol. 8, 855–875 10.2217/fmb.13.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., and Jackson M. (2009) Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78 10.1016/S0065-2164(09)69002-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marrakchi H., Lanéelle M. A., and Daffé M. (2014) Mycolic acids: structures, biosynthesis, and beyond. Chem. Biol. 21, 67–85 10.1016/j.chembiol.2013.11.011 [DOI] [PubMed] [Google Scholar]
- 5. Belisle J. T., Vissa V. D., Sievert T., Takayama K., Brennan P. J., and Besra G. S. (1997) Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276, 1420–1422 10.1126/science.276.5317.1420 [DOI] [PubMed] [Google Scholar]
- 6. Armitige L. Y., Jagannath C., Wanger A. R., and Norris S. J. (2000) Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect. Immun. 68, 767–778 10.1128/IAI.68.2.767-778.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ronning D. R., Klabunde T., Besra G. S., Vissa V. D., Belisle J. T., and Sacchettini J. C. (2000) Crystal structure of the secreted form of antigen 85C reveals potential targets for mycobacterial drugs and vaccines. Nat. Struct. Biol. 7, 141–146 10.1038/72413 [DOI] [PubMed] [Google Scholar]
- 8. Ronning D. R., Vissa V., Besra G. S., Belisle J. T., and Sacchettini J. C. (2004) Mycobacterium tuberculosis antigen 85A and 85C structures confirm binding orientation and conserved substrate specificity. J. Biol. Chem. 279, 36771–36777 10.1074/jbc.M400811200 [DOI] [PubMed] [Google Scholar]
- 9. Anderson D. H., Harth G., Horwitz M. A., and Eisenberg D. (2001) An interfacial mechanism and a class of inhibitors inferred from two crystal structures of the Mycobacterium tuberculosis 30 kDa major secretory protein (antigen 85B), a mycolyl transferase. J. Mol. Biol. 307, 671–681 10.1006/jmbi.2001.4461 [DOI] [PubMed] [Google Scholar]
- 10. Vander Beken S., Al Dulayymi J. R., Naessens T., Koza G., Maza-Iglesias M., Rowles R., Theunissen C., De Medts J., Lanckacker E., Baird M. S., and Grooten J. (2011) Molecular structure of the Mycobacterium tuberculosis virulence factor, mycolic acid, determines the elicited inflammatory pattern. Eur. J. Immunol. 41, 450–460 10.1002/eji.201040719 [DOI] [PubMed] [Google Scholar]
- 11. Backus K. M., Dolan M. A., Barry C. S., Joe M., McPhie P., Boshoff H. I., Lowary T. L., Davis B. G., and Barry C. E. 3rd (2014) The three Mycobacterium tuberculosis antigen 85 isoforms have unique substrates and activities determined by non-active site regions. J. Biol. Chem. 289, 25041–25053 10.1074/jbc.M114.581579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Favrot L., Lajiness D. H., and Ronning D. R. (2014) Inactivation of the Mycobacterium tuberculosis antigen 85 complex by covalent, allosteric inhibitors. J. Biol. Chem. 289, 25031–25040 10.1074/jbc.M114.582445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ravindran M. S., Rao S. P., Cheng X., Shukla A., Cazenave-Gassiot A., Yao S. Q., and Wenk M. R. (2014) Targeting lipid esterases in mycobacteria grown under different physiological conditions using activity-based profiling with tetrahydrolipstatin (THL). Mol. Cell. Proteomics 13, 435–448 10.1074/mcp.M113.029942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Favrot L., Grzegorzewicz A. E., Lajiness D. H., Marvin R. K., Boucau J., Isailovic D., Jackson M., and Ronning D. R. (2013) Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat. Commun. 4, 2748 10.1038/ncomms3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goins C. M., Dajnowicz S., Thanna S., Sucheck S. J., Parks J. M., and Ronning D. R. (2017) Exploring covalent allosteric inhibition of antigen 85C from Mycobacterium tuberculosis by ebselen derivatives. ACS Infect. Dis. 3, 378–387 10.1021/acsinfecdis.7b00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanbonmatsu K. Y., and García A. E. (2002) Structure of Met-enkephalin in explicit aqueous solution using replica exchange molecular dynamics. Proteins 46, 225–234 10.1002/prot.1167 [DOI] [PubMed] [Google Scholar]
- 17. Zang W., Wu C., and Duan Y. (2005) Convergence of replica exchange molecular dynamics. J. Chem. Phys. 123, 154105 10.1063/1.2056540 [DOI] [PubMed] [Google Scholar]
- 18. Periole X., and Mark A. E. (2007) Convergence and sampling efficiency in replica exchange simulations of peptide folding in explicit solvent. J. Chem. Phys. 126, 014903 10.1063/1.2404954 [DOI] [PubMed] [Google Scholar]
- 19. Weibel E. K., Hadvary P., Hochuli E., Kupfer E., and Lengsfeld H. (1987) Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. J. Antibiot. 40, 1081–1085 10.7164/antibiotics.40.1081 [DOI] [PubMed] [Google Scholar]
- 20. Hadváry P., Lengsfeld H., and Wolfer H. (1988) Inhibition of pancreatic lipase in vitro by the covalent inhibitor tetrahydrolipstatin. Biochem. J. 256, 357–361 10.1042/bj2560357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kridel S. J., Axelrod F., Rozenkrantz N., and Smith J. W. (2004) Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 64, 2070–2075 10.1158/0008-5472.CAN-03-3645 [DOI] [PubMed] [Google Scholar]
- 22. Pemble C. W. 4th, Johnson L. C., Kridel S. J., and Lowther W. T. (2007) Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 14, 704–709 10.1038/nsmb1265 [DOI] [PubMed] [Google Scholar]
- 23. Fako V. E., Zhang J. T., and Liu J. Y. (2014) Mechanism of orlistat hydrolysis by the thioesterase of human fatty acid synthase. ACS Catal. 4, 3444–3453 10.1021/cs500956m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgi H. B., Dunitz J. D., Lehn J. M., and Wipff G. (1974) Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 30, 1563–1572 10.1016/S0040-4020(01)90678-7 [DOI] [Google Scholar]
- 25. Fisher S. Z., Kovalevsky A. Y., Domsic J. F., Mustyakimov M., McKenna R., Silverman D. N., and Langan P. A. (2010) Neutron structure of human carbonic anhydrase II: implications for proton transfer. Biochemistry 49, 415–421 10.1021/bi901995n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen L., Kong X., Liang Z., Ye F., Yu K., Dai W., Wu D., Luo C., and Jiang H. (2011) Theoretical study of the mechanism of proton transfer in the esterase Estb from Burkholderia gladioli. J. Phys. Chem. B 115, 13019–13025 10.1021/jp206297d [DOI] [PubMed] [Google Scholar]
- 27. Tomanicek S. J., Standaert R. F., Weiss K. L., Ostermann A., Schrader T. E., Ng J. D., and Coates L. (2013) Neutron and X-ray crystal structures of perdeuterated enzyme inhibitor complex reveal the catalytic proton network of the Toho-1 β-lactamase for the acylation reaction. J. Biol. Chem. 288, 4715–4722 10.1074/jbc.M112.436238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moriarty N. W., Grosse-Kunstleve R. W., and Adams P. D. (2009) Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 10.1107/S0907444909029436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Terwilliger T. C., Klei H., Adams P. D., Moriarty N. W., and Cohn J. D. (2006) Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr. 62, 915–922 10.1107/S0907444906017161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Copelan R. A. (2005) Evaluation of Enzyme Inhibitors in Drug Discovery, pp. 214–248, John Wiley and Sons, Hoboken, NJ [Google Scholar]
- 35. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeLano W. L. (2013) The PyMOL Molecular Graphics System, version 1.6, Schrödinger, LLC, New York [Google Scholar]
- 38. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., and Baker N. A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525 10.1093/nar/gkm276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frisch M. J., Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., et al. (2009) Gaussian 09, revision E.01, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 40. Dupradeau F. Y., Pigache A., Zaffran T., Savineau C., Lelong R., Grivel N., Lelong D., Rosanski W., and Cieplak P. (2010) The R.E.D. tools: advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 12, 7821–7839 10.1039/c0cp00111b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 42. Kirschner K. N., Yongye A. B., Tschampel S. M., González-Outeiriño J., Daniels C. R., Foley B. L., and Woods R. J. (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates, J Comput. Chem. 29, 622–655 10.1002/jcc.20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Case D. A., Betz R. M., Cerutti D. S., Cheatham T. E., Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T. S., et al. (2016) AMBER 2016, University of California, San Francisco, CA [Google Scholar]
- 44. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 45. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 46. Ryckaert J. P., Ciccotti G., and Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 47. Gowers R. J., Linke M., Barnoud J., Reddy T. J. E., Melo M. N., Seyler S. L., Domanski J., Dotson D. L., Buchous S., Kenney I. M., and Beckstein O. (2016) MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations, in Proceedings of the 15th Python in Science Conference, Austin, July 11–17, 2016 (Benthall S., and Rostrup S., eds) pp. 98–105, Enthought, Inc., Austin, TX [Google Scholar]
- 48. Roe D. R., and Cheatham T. E. 3rd (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.