

Abstract
Rap1-interacting protein 1 (Rif1) regulates telomere length in budding yeast. We previously reported that, in metazoans and fission yeast, Rif1 also plays pivotal roles in controlling genome-wide DNA replication timing. We proposed that Rif1 may assemble chromatin compartments that contain specific replication-timing domains by promoting chromatin loop formation. Rif1 also is involved in DNA lesion repair, restart after replication fork collapse, anti-apoptosis activities, replicative senescence, and transcriptional regulation. Although multiple physiological functions of Rif1 have been characterized, biochemical and structural information on mammalian Rif1 is limited, mainly because of difficulties in purifying the full-length protein. Here, we expressed and purified the 2418-amino-acid-long, full-length murine Rif1 as well as its partially truncated variants in human 293T cells. Hydrodynamic analyses indicated that Rif1 forms elongated or extended homo-oligomers in solution, consistent with the presence of a HEAT-type helical repeat segment known to adopt an elongated shape. We also observed that the purified murine Rif1 bound G-quadruplex (G4) DNA with high specificity and affinity, as was previously shown for Rif1 from fission yeast. Both the N-terminal (HEAT-repeat) and C-terminal segments were involved in oligomer formation and specifically bound G4 DNA, and the central intrinsically disordered polypeptide segment increased the affinity for G4. Of note, pulldown assays revealed that Rif1 simultaneously binds multiple G4 molecules. Our findings support a model in which Rif1 modulates chromatin loop structures through binding to multiple G4 assemblies and by holding chromatin fibers together.
Keywords: G-quadruplex, DNA–protein interaction, oligomerization, intrinsically disordered protein, DNA replication, DNA repair, chromatin structure, HEAT repeats
Introduction
Rif1 (Rap1-interacting protein 1) was originally discovered for its role at telomeres in budding yeast, where it controls telomere length via counting the number of telomere-bound Rap1 molecules (1, 2). ScRif1 (budding yeast Rif1) also plays an anti-checkpoint function by covering ssDNA2 generated after DNA lesions near the telomere to prevent RPA and Rad24 from assembling on the ssDNA, shutting down an ATR-signaling pathway (3, 4). In contrast, we found that SpRif1 (fission yeast Rif1) specifies the spatio-temporal program of origin firing during S phase (5) and that it is bound not only at telomeres but also at chromosome arm segments (5, 6). We and others also reported that functions in replication timing regulation are conserved in mammalian Rif1 (7–10). More recently, ScRif1 was also reported to regulate replication timing program in budding yeast (11–13).
Vertebrate Rif1 proteins have been implicated also in a wide variety of cellular processes. Human Rif1 contributes to the ATM-mediated DNA damage response, which is dependent on 53BP1 as well as on ATM (14). Subsequently, it was reported that Rif1 is involved in homologous recombination-mediated repair of double-stranded DNA break (DSB), and Rif1 deficiency results in aberrant aggregates of Rad51 (15). In Xenopus egg extracts, Rif1 was reported to interact with TopBP1, ATM, and an Mre11–Rad50–Nbs1 complex, which are key regulators of checkpoint responses to DSBs (16). Depletion of Rif1 from egg extracts reduces recruitment of these regulatory components and compromises the activation of Chk1 in response to DSBs but not to stalled replication forks. It was more recently reported that mammalian Rif1 interacts with ATM-phosphorylated 53BP1 and that recruitment of Rif1 is essential for 53BP1 to promote non-homologous end-joining (NHEJ)-mediated DSB repair through suppression of 5′-end resection (17–21). It was also reported that Rif1 deficiency impairs DNA repair in the G1 and S phases and 53BP1-dependent class switch recombination (CSR) in B lymphocytes (18, 19). In contrast, 53BP1-dependent toxic NHEJ in BRCA1−/− background is dependent on PTIP, another 53BP1 partner, but not on Rif1 (22).
Human Rif1 was reported to be a component of a complex containing BLM (the helicase defective in Bloom syndrome) and to work with BLM to promote recovery of stalled replication forks (23). Knockout of mouse Rif1 (muRif1) leads to failure in embryonic development, and conditional deletion of muRif1 from mouse embryo fibroblasts affects S phase progression, rendering cells hypersensitive to replication poisons (15). Consistent with a role in S phase progression, muRif1 accumulates at stalled replication forks, preferentially around pericentromeric heterochromatin (15).
Rif1 is highly expressed in totipotent and pluripotent cells during early mouse development, and in male and female germ cells in adult mice, and it has been implicated in maintenance of pluripotency of stem cells (24). Rif1 physically interacts with the telomere-associated protein TRF2 in mouse embryonic stem cells (24). Furthermore, Rif1 is highly expressed in human breast tumors and acts as an anti-apoptotic factor (25).
Despite some discrepancies among different reports, these findings generally point to crucial roles of Rif1 in processing of DSB caused by genotoxins or by stalled replication forks. We have shown that Rif1 is specifically located at the nuclear periphery in insoluble nuclear skeleton-like structures and affects chromatin loop sizes (7). The localization of Rif1 was closely associated with the mid-late replication foci, the appearance of which is suppressed by Rif1. Thus, we hypothesized that Rif1 may facilitate chromatin loop formation, which may generate specific chromatin compartments related to replication timing domains as well as to the chromatin domains responsible for coordinated regulation of other chromosome transactions (9).
Vertebrate Rif1 contains a C-terminal DNA-binding domain that resembles the αCTD domain of bacterial RNA polymerase, and this domain preferentially binds to fork and Holliday junction DNA in vitro and is required for Rif1 to resist replication stress in vivo (23, 26). A recent structural study revealed a tetrameric structure of the ScRif1 C-terminal domain (27), which may correspond to the above αCTD-like domain of vertebrate Rif1. One of the conserved features of Rif1 is the presence of HEAT repeats in the N-terminal segment, although the function of this domain is unknown. Vertebrate Rif1 contains a long intrinsically disordered polypeptide (more than 1000 amino acids long), which is located between the N- and C-terminal segments and encoded by a single exon. Another conserved feature of Rif1 is the presence of a phosphoprotein phosphatase 1 (PP1)-binding motif, which was shown to be important for suppression of late-firing origins through counteracting Cdc7-mediated phosphorylation events (11, 13, 28).
Although increasing evidence points to crucial roles of Rif1 in various chromosome events, links between its biochemical features and its biological functions have been limited. Recently, we reported that SpRif1 specifically binds to G-quadruplex (G4) structures and regulates replication timing over a long distance (6). However, it has not been known whether G4-binding activity is conserved in Rif1 from other species. Thus, we set out to purify and characterize the full-length and various domains of murine Rif1 protein to clarify the mechanisms by which Rif1 regulates chromatin architecture.
Results
Expression and purification of full-length murine Rif1
Isolation of the full-length mammalian Rif1 (2418 amino acids) has not been achieved. To characterize DNA-binding specificity and anticipated oligomer formation of mammalian Rif1, we attempted overexpression of muRif1 in 293T cells. Hexahistidine and 3xFLAG tags were added to muRif1 (2418 amino acids) at its N and C terminus, respectively, for convenient two-step affinity purification (Fig. 1A). Because the addition of a small tag at the N or C terminus did not affect the functions of SpRif1, we assumed that addition of these tags to muRif1 would not affect its functions. Indeed, N-terminally FLAG- or GFP-tagged mammalian Rif1 has been shown to be functional (29, 30). Strong EF1α promoter drives the expression of the recombinant muRif1 (31, 32). After 2 days of transfection, 293T cells were harvested and extracted by Benzonase® and Triton X-100 in CSK buffer (see under “Experimental procedures”). About 30% of muRif1 was solubilized by this treatment. The soluble muRif1 was purified successfully by anti-FLAG and subsequent Ni2+-affinity chromatography, although some extra bands were detected after SDS-PAGE (Fig. 1B). We were not able to remove the extra bands by any additional chromatography such as Mono Q, probably because of continued degradation during chromatography (Fig. 1C, lanes 1 and 2).
Figure 1.

Expression of the full-length and partially truncated murine Rif1 polypeptides in 293T cells and their purification. A, schematic drawing of the primary structures of muRif1 and its domains used in this study. HEAT-repeat, αCTD-, and PP1-binding regions of muRif1 are assigned as reported previously (23, 73). A LID region is assigned according to Sukackaite et al. (26). The number of amino acids in parentheses include those from His tag, 3xFLAG tag, and cloning sites on pCEH_FLAG expression vector. B, silver-stained gel showing Ni2+-NTA affinity column chromatography of muRif1 after anti-FLAG affinity purification. C, muRif1-NC (lanes 3 and 4), muRif1-NTD (lanes 5 and 6), muRif1-LID (lanes 7 and 8), and muRif1-CTD (lanes 9 and 10) were partially purified by anti-FLAG affinity column and subsequent Ni2+-NTA column chromatography (Fig. S1). After dialysis into Buffer F, aliquots (odd lanes, 2 μl; even lanes, 6 μl) were subjected to SDS-PAGE followed by silver staining. A Mono Q peak fraction of the full-length muRif1 was also run on the same gel (lane 1, 5 μl; lane 2, 15 μl). The full-length muRif1 bands are indicated by horizontal arrowheads in B and C.
Expression and purification of partially truncated muRif1
Size-exclusion chromatography of the full-length muRif1 could not lead to precise determination of the Stokes radius (RS), because it eluted in fractions very close to the void volume of a Superose 6 gel-filtration column (data not shown). Thus, we next prepared smaller fragments of muRif1 to analyze its oligomeric nature more precisely. We constructed the four truncated muRif1s (Fig. 1A), i.e. muRif1-NTD (N-terminal domain), muRif1-CTD (C-terminal domain), muRif1-NC (NTD + CTD), and muRif1-LID (long intrinsically disordered region). The four proteins were successfully produced in 293T cells, but muRif1-NC was not efficiently solubilized by the above extraction protocol used for the full length. The increase of NaCl concentration to 0.3 m resulted in solubilization of muRif1-NC to a sufficient extent. The solubilized proteins were purified by the same two-step affinity chromatography, and profiles of Ni2+-affinity chromatography are shown in Fig. S1, A–D. They still contained small amounts of extra bands (Fig. 1C, lanes 3–10) that could not be completely removed by subsequent Mono Q chromatography (data not shown).
Self-interaction of muRif1-NTD and muRif1-CTD
To examine oligomerization potential of muRif1-NTD and muRif1-CTD, HA- and FLAG-tagged polypeptides were coexpressed in 293T cells, and interactions were examined by immunoprecipitation of either tag and detection of coimmunoprecipitation with the other tag. Immunoprecipitation of FLAG-tagged muRif1-NTD by anti-DDDDK antibody from the soluble cell extract pulled down HA-tagged muRif1-NTD (Fig. 2A, lane 1). Conversely, immunoprecipitation of HA-tagged muRif1-NTD by anti-HA antibody pulled down FLAG-tagged muRif1-NTD (Fig. 2B, lane 1). Therefore, muRif1-NTD may form a homodimer or larger homo-oligomers. Similar experiments were conducted with muRif1-CTD (Fig. 2, A and B, lane 6), and the results indicate that muRif1-CTD can interact with each other, showing its potential to form a homodimer or larger homo-oligomer. In contrast, muRif1-LID did not self-associate (Fig. 2, A and B, lane 11), suggesting that muRif1-LID exists as monomer. The interdomain interaction between NTD and CTD was not detected (Fig. 2, A and B, lanes 2 and 5), and muRif1-LID did not interact with NTD or CTD, either (Fig. 2, A and B, lanes 3, 7, 9, and 10).
Figure 2.

Self-association of muRif1-NTD and of muRif1-CTD. Various portions of muRif1 were fused to either HA tag or 3xFLAG tag, and combinations of an HA-tagged and a FLAG-tagged polypeptide were transiently expressed in 293T cells as shown. One portion of cell lysates was subjected to immunoprecipitation (IP) with anti-DDDDK antibody (A) and the other portion with anti-HA antibody (B). Precipitated (left) and input (right) polypeptides were detected by Western blotting with anti-HA antibody (upper) or with anti-DDDDK antibody (lower). muRif1-LID showed weak signals, because electrotransfer efficiency of muRif1-LID from polyacrylamide gel to PVDF membrane was low, probably due to its very acidic property (calculated pI = 4.64).
Molecular shape and oligomerization of truncated muRif1
Hydrodynamic behaviors of purified muRif1-NTD and -CTD were analyzed by glycerol-gradient centrifugation (Fig. 3, A and B, and Fig. S2D) and by gel-filtration chromatography (Fig. 3, C and D, and Fig. S2E). muRif1-NTD sedimented at two peak positions after the glycerol-gradient centrifugation (Fig. 3A, p1 and 2) and formed also two peak fractions during the size-exclusion chromatography (Fig. 3C, p3 and 4). When these peak fractions were subjected to non-denaturing (native) electrophoresis on agarose-acrylamide composite gel (33, 34), p1 or p2 migrated in a manner similar to that shown by p4 or p3, respectively (Fig. 3, E and F, lanes 1–4). Thus, p1 and p4 most likely represent one form of native muRif1-NTD, whereas p2 and p3 represent another form. muRif1-CTD also generated two peaks in sedimentation and gel-filtration profiles (Fig. 3, B, p5 and p6, and D, p7 and p8), and non-denaturing electrophoresis indicated that p5 and p6 correspond to p8 and p7, respectively (Fig. 3F, lanes 5–8). Sedimentation coefficient, S value, was determined from the peak positions of glycerol-gradient centrifugation (Fig. S2B), and RS was calculated from the peak positions of gel-filtration chromatography (Fig. S2C). According to Erickson's linear approximation (35), the two hydrodynamic parameters (S and RS) of each protein gave estimation of its native molecular weight (Fig. 3G, column of Native Mw.), and dividing this value by the molecular weight of a unit of polypeptide provides the estimated number of polypeptides in each molecule (Fig. 3G, column of polypeptides). The slow-sedimenting form (p1 and p4) of the muRif1-NTD was estimated to be a tetramer (calculated as 4.46-mer) and the fast-sedimenting form (p2 and p3) to be an octamer (calculated as 7.50-mer). In contrast, the slow-sedimenting form (p5 and p8) of the muRif1-CTD was estimated to be a dimer (calculated as 2.26-mer), and the fast-sedimenting form (p6 and p7) to be a dodecamer (calculated as 11.5-mer). Therefore, muRif1-NTD most likely forms a tetramer and an octamer, whereas muRif1-CTD forms a dimer and a dodecamer, although the dimer seems to be a minor form (Fig. 3, B and D).
Figure 3.

Hydrodynamic analyses of truncated muRif1. A and B, sedimentation profiles of partially purified muRif1-NTD (A) and -CTD (B) through 15–36% glycerol-gradient centrifugation. C and D, size-exclusion chromatography of partially purified muRif1-NTD (C) and -CTD (D) through Superose 6 gel-filtration column. Peak positions are marked with vertical arrowheads and designated as p1–p8 according to Fig. S2, D and E. The size standards used are as follows: a, blue dextran 2000; b, thyroglobulin; c, ferritin; d, catalase; e, aldolase; f, albumin; g, ovalbumin; and h, chymotrypsinogen A. E, non-denaturing electrophoresis of marker proteins (left panel) and muRif1-NTD peak fractions, p1–p4 (right panel) through 0.4% agarose, 2.5% polyacrylamide composite gel. Glycerol-gradient peaks and gel-filtration peaks are marked with G.G. and with SEC, respectively, above the immunoblot images. F, non-denaturing electrophoresis of peak fractions of muRif1-NTD (left panel) and muRif1-CTD (right panel). All peak fractions, p1–p8, were run on the same composite gel. G, calculation of native molecular weights of truncated muRif1 polypeptides and determination of their oligomeric states. R values for muRif1-NTD and -CTD were deduced according to Erickson (35). Similar results were also obtained when the Siegel and Monty method (74) was used to deduce Stokes radii.
Specific interaction of muRif1 with G-quadruplex DNA
We previously reported genome-wide binding sites of SpRif1 on the chromosome arms (6). Furthermore, we found that these binding sites have a potential to form G-quadruplex (G4) structure, and we showed that SpRif1 indeed specifically binds to G4. Therefore, we examined whether the purified muRif1 could recognize G4 DNA in vitro. A 5′-biotinylated oligonucleotide bearing 24 consecutive dG (T6G24 probe) was boiled and then gradually cooled down in the presence of KCl and PEG200 that provides a macromolecular crowding condition (36, 37) to generate a parallel-type G4 structure. The G4-folded oligonucleotide was pulled down by streptavidin-magnetic beads (dynabeads), and the presence of muRif1 was examined (Fig. S3, A and B). When 60 fmol of purified muRif1 was incubated with the G4 DNA beads, a substantial fraction (12–13 fmol) was adsorbed onto the beads (Fig. 4, A and B, lane 4). This binding was lost by addition of 4-fold excess of G4-folded T6G24 (Fig. 4, A and B, lane 5), but it was barely affected by addition of 32-fold excess of T6(GA)12 DNA (Fig. 4, A and B, lanes 8 and 9) or a large excess (2 μg) of calf thymus DNA (Fig. 4, A and B, lanes 1–3). Thus, muRif1 bound specifically to immobilized G4 DNA.
Figure 4.
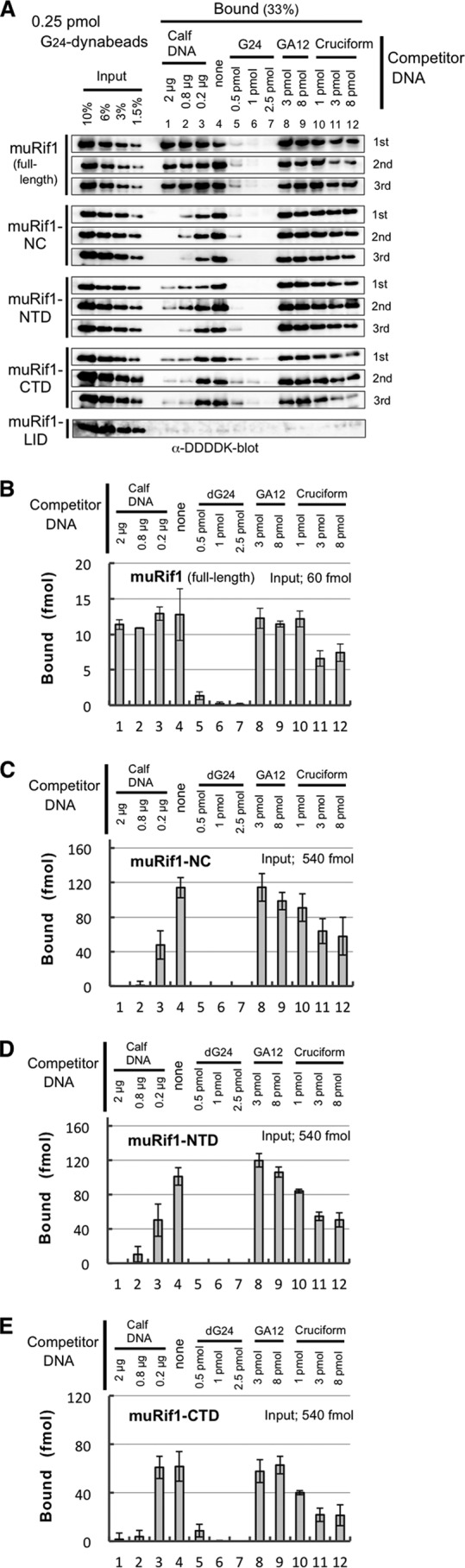
Specific association of muRif1 domains with immobilized G4 DNA. A 5′-biotinylated, G4-folded T6G24 oligonucleotide was conjugated to streptavidin-dynabeads (G24-dynabeads) and used to pull down partially-purified muRif1 or truncated muRif1 polypeptides (Fig. S3). The same experiment was repeated three times for every polypeptide (A) except muRif1-LID. After densitometric scanning of the immunoblots, the amounts of G4 DNA-bound muRif1 (B), muRif1-NC (C), muRif1-NTD (D), and muRif1-CTD (E) are plotted in the bar graphs. A cruciform competitor was made of four oligonucleotides of 40 nucleotides long, identical to those used in Sukackaite et al. (26). The presence of a large excess of T6(GA)12 DNA (3–8 pmol; lanes 8 and 9) or 1 pmol of the cruciform DNA (lane 10) did not significantly affect the bindings of these muRif1 polypeptides to the beads (B–E), whereas addition of >0.5 pmol of G4-folded T6G24 DNA competed out the bindings (lanes 5–7). Addition of >3 pmol of the cruciform DNA reduced the bindings (lane 11 and 12), but less efficiently than the G4 competitor did.
Bipartite G4-binding domains of muRif1
Similar pulldown assay was performed with the four partially truncated muRif1 (Fig. 4, A and C–E). muRif1-NC and -NTD bound efficiently to the G4-folded T6G24-dynabeads (Fig. 4, A, C, and D, lane 4), and muRif1-CTD bound moderately (Fig. 4, A and E, lane 4), but muRif1-LID did not (Fig. 4A, bottom panel, lane 4). Interestingly, both muRif1-NTD and -CTD bound to the G4 beads (Fig. 4, D and E, lane 4), indicating bipartite or multivalent G4-binding activity of muRif1. The purified short C-terminal segments of human and mouse Rif1 were reported to bind to various structured DNA, especially cruciform DNA in vitro (23, 26). However, >38% of muRif1-NTD and >33% of muRif1-CTD still bound to the G4 beads even in the presence of an excess cruciform DNA consisting of four 40 base-oligonucleotides (Fig. 4, D and E, lanes 10–12). Additionally, >50% of muRif1 and muRif1-NC still bound to the G4 beads even in the presence of 32-fold excess of cruciform DNA (Fig. 4, B and C, lanes 10–12). These results suggest that the N- and C-terminal domains of muRif1 cooperate for highly structure-specific recognition of G4 DNA. The binding of three truncated proteins was significantly reduced by the addition of excess amount of calf thymus DNA, presumably due to the G4-like structures in the DNA preparation (Fig. 4, C–E, lanes 1–3), but the binding of the full-length muRif1 was resistant to the same amounts of the competitor DNA (Fig. 4B, lanes 1–3). This suggests a contribution of the internal LID to the highly structure-specific recognition of G4 DNA by muRif1, although no apparent G4 binding was shown by the LID alone.
High-affinity interaction of muRif1 with G4 DNA
The G4 DNA binding was evaluated more quantitatively by EMSA. A 5′-labeled T6G24 probe was folded into G4 structure as described above. As a negative control, a T6(GA)12 probe was similarly treated, and both probes were subjected to EMSA with the full-length or truncated forms of muRif1 (Fig. 5, A–C). The full-length muRif1 efficiently shifted the mobility of the T6G24 probe (Fig. 5A, lanes 2–5). The shifted bands were detected at the gel top and at the position slightly below it. Neither a lower non-G4 band of the same probe nor a T6(GA)12 probe was shifted (Fig. 5A, lanes 2–5 and lanes 7–10). The shift was detected even at the lowest concentration of the protein (0.33 nm). In contrast, muRif1-NTD and muRif1-CTD did not induce significant shift of the same G4 probe at <5 nm of the proteins (Fig. 5A, lanes 11 and 12 and lanes 17 and 18), whereas the full-length muRif1 exhibited efficient shift at 0.67–1.33 nm. When assuming a 1:1 interaction, the dissociation constant (Kd) of the G4-folded probe was calculated as 6.49 nm for muRif1-NTD and as 33.0 nm for muRif1-CTD (Fig. 5A, right graph). If we assume that a protomer of the full-length muRif1 binds to a single G4 molecule, i.e. if n = 1, the Kd value could not be calculated with a reasonable curve fit. This would be due to the presence of multiple G4-binding sites on a muRif1 protomer, and the above pulldown assays show that indeed is the case (Fig. 4). Because both NTD and CTD of muRif1 bind to G4 DNA (Fig. 4, D and E), we assume that a muRif1 protomer has two independent G4-binding sites. We then re-calculated the Kd value under the assumption that both sites have equivalent affinity for the G4 probe. (The calculation would involve resolution of a complicated cubic equation if two sites have a different affinity for G4. We assume here an equal Kd value for both sites for simplicity.) Kd is estimated to be 0.197 nm for each binding site with a better curve fit than those for NTD and CTD (n = 2; Fig. 5A, right graph). Thus, it appears that both G4-binding sites exhibit >32-fold higher affinity to the G4 probe when present in the full-length muRif1 than when present alone as separate polypeptides. Next, EMSA was conducted using higher amounts of muRif1-NC, -NTD, and -CTD proteins (Fig. 5B). Addition of >15 nm muRif1-NC or muRif1-NTD induced substantial shift of the G4 probe (Fig. 5B, lanes 4 and 5 and lanes 11 and 12), whereas >45 nm muRif1-CTD was required to detect a comparable shift (Fig. 5B, lane 17). The Kd for the G4-folded probe was calculated to be 8.68 nm (when n = 1) and 20.7 nm (when n = 2) with muRif1-NC, roughly comparable with that with muRif1-NTD. Because the affinity of muRif1-NC to G4 is lower than that of the full-length protein by more than 1 order of magnitude, the LID also plays an unexpected role in increasing the affinity and specificity of the G4 binding. It may coordinate the N- and C-terminal domains through its versatile morphology so that Rif1 can adjust its shape for efficient and multivalent binding to various forms of G4 structures (see below). The above EMSA used the T6G24 probe at 8.33 nm, with the G4 form at 5.1–5.5 nm, and it would be too low to determine convincing Kd values for truncated muRif1 proteins. Thus, we repeated the same EMSA with 16.7 nm (11–12 nm of the G4 form) and 38.9 nm (26.4–28 nm of the G4 form) of the probe (Fig. S4), and the calculated Kd values are listed in Table 1. Kd of 1.51–2.35 nm would be more reliable for muRif1-NTD because the G4 probe concentrations (>11 nm) are higher than its Kd value. Similarly, Kd of 18.1 nm would be more reliable for muRif1-CTD because the G4 probe concentrations (>26 nm) are higher than its Kd. However, it may be possible that either the isolated NTD or CTD binds cooperatively to the G4 probe, because the more probe input seems to result in less Kd value, i.e. more increase in affinity (Table 1). The above EMSA used PEG200-containing 12% polyacrylamide gels to separate the G4-folded probe from non-G4 (predominantly single-stranded) forms. However, most of the muRif1 recombinant proteins accumulated at the gel top in this gel system, raising the possibility that they just aggregated on G4 rather than forming biologically relevant protein–DNA complexes. We exploited a low percentage agarose-acrylamide composite gel system to examine these possibilities (Fig. 5C and Fig. S5). For all muRif1 recombinants, most of the G4-specific protein–DNA complexes entered the gels and generated discrete bands (Fig. 5C, a–d), instead of being trapped at the gel top. Thus, G4 complexes containing full-length and truncated muRif1 proteins are most likely not non-specific aggregates but represent specific complexes.
Figure 5.

High-affinity binding of muRif1 to G4 DNA. A, EMSA was conducted on radiolabeled T6G24 and T6(GA)12 probes using increasing amounts of purified full-length muRif1 (lanes 1–5), muRif1-NTD (lanes 11–13), or muRif1-CTD (lanes 17–19). Prior to the assays, both probes were boiled for 5 min and then cooled down gradually to 25 °C in 50 mm KCl, 40% PEG200, 1 mm EDTA, and 10 mm Tris-HCl (pH 7.5). Apparent dissociation constants (Kd values) were calculated from the right graph after plotting the amount of unbound G4 probe (bands indicated by the vertical line to left of panel) versus concentrations of added proteins. For the full-length muRif1, a Kd value was calculated under the assumption that a Rif1 protomer has two equal-affinity binding sites to the G4 DNA (n = 2). B, EMSA was conducted on T6G24 probe, as in A, with increasing amounts (different ranges) of muRif1-NC (lanes 1–5), muRif1-NTD (lanes 10–12), or muRif1-CTD (lanes 15–17). Shifts were not observed when T6(GA)12 probe was used (lanes 6–9, 13 and 14, and 18 and 19). Kd values were calculated from the right graph as in A. For muRif1-NC, two Kd values were calculated under the assumptions that a Rif1-NC protomer has a single (n = 1) or two (n = 2) equal-affinity binding sites to the G4 DNA. These analyses were performed with increasing concentrations of the G4 probe in Fig. S4. C, EMSA was conducted on the T6G24 probe using a 0.4% agarose, 2% polyacrylamide composite gel. muRif1-NTD (a), -CTD (b), -NC (d), and full-length muRif1 (c) generated discrete shifted bands of specific protein–DNA complexes that entered the gel (lanes 2–4, 7–9, 12–14, and 17–19). The higher mobility of the G4-bound full-length muRif1 would be due to its acidic property originating from extremely lower isoelectric point of LID (calculated pI = 4.64). No shifted band was detected when T6(GA)12 probe was used (lanes 6, 10, 16, and 20).
Table 1.
Dissociation constant (Kd) for interaction between T6G24 G4-probe and muRif1 proteins
Differential recognition of G4 and other structured DNA by muRif1
We conducted EMSAs in the presence of various DNA competitors such as cruciform or forked DNA (23, 26) Although all these structured DNAs were previously shown to be bound by Rif1-CTD (26), none of them, when present at 30-fold molar excess over the labeled G4 probe, canceled muRif1-induced shift of the G4 probe (Fig. 6, lanes 5–12). Under the same condition, the G4 competitor itself efficiently reduced it, whereas T6(GA)12 competitor did not affect the shift (Fig. 6, lanes 15–18). This result suggests that muRif1 has much higher affinity to G4 than to cruciform DNA or other structures. It is also possible that Rif1 has more than two DNA-binding domains that bind to different sets of structured DNA, although this possibility would be rather unlikely in light of weak but significant competition by a cruciform DNA in the above pulldown assays (Fig. 4, lanes 10–12).
Figure 6.

muRif1 prefers G4 DNA much more than other structured DNA. EMSA was conducted on the T6G24 probe (5.6 nm) with the full-length muRif1 (2.7 nm) in the presence of various structured DNAs as competitors. Competitors used were dsDNA (lanes 3 and 4), Y-fork (lanes 5 and 6), flap (lanes 7 and 8), fork (lanes 9 and 10), cruciform (4WJ, lanes 11 and 12), telomere repeat (Tel-ds, lanes 13 and 14), and T6(GA)12 (lanes 15 and 16), all of which failed to compete out the muRif1 binding to T6G24 probe at 30-fold excess (×30). Lanes 17 and 18 contain cold T6G24 DNA as a competitor, and substantial competition was obvious even at 3-fold excess (×3). The relative amounts of unbound G4 probe (bands indicated by the vertical line to the left of panel) are shown in a bar graph at the bottom, with the level in the absence of muRif1 being taken as 100.
Interaction of muRif1 with characteristic subsets of G4 DNA
There are several well-known G4 oligonucleotides that have been structurally characterized (38–46). We used 10 of them for EMSA with purified muRif1 (Fig. S6). Four parallel-stranded G4 probes were shifted significantly with 1.65 nm of muRif1 (Fig. S6, T2G15T, VEGF, c-kit2, and c-myc). A bcl-2 gene-derived 23-mer, a hybrid-type G4 probe, was also shifted with muRif1 (Fig. S6, bcl-2). In contrast, muRif1 did not induce significant band shift of G4 probes with anti-parallel conformation or those with telomere-derived sequences (Fig. S6, TBA, Telo22, Telo24, Telo23, and Telo25). Thus, muRif1 shows some preference for G4 structures with particular topology (parallel, hybrid) over other topology (anti-parallel, telomere-derived). Then, we focused on parallel-type G4 oligonucleotides consisting of different nucleotide sequences (47), and we evaluated their binding to the full-length muRif1 in EMSA (Fig. 7A). A Telo23 probe, a hybrid-type G4 DNA, was used for comparison. muRif1 showed higher affinity to the three G4 probes (CEB1, 93del, and G15) but not to T95-2T, TERC18, or Telo23 probe (Fig. 7, A and B). Intriguingly, the same preference was observed for muRif1-NTD and -CTD, as well as for muRif1-NC (Fig. 7, C–E). More detailed characterization is needed to clarify the structure/topology specificity of Rif1 G4 binding.
Figure 7.

High-affinity binding of muRif1 to subsets of parallel-stranded G4 DNA. A, EMSA was conducted on six parallel-type G4 DNA (CEB1, T95-T2, 93del, T6G24, G15, TERC18), a hybrid-type G4 DNA (Telo23), and an RNA probe corresponding to TERC18 DNA. As a negative control, a T6(GA)12 probe was also assayed. 4.17 nm radiolabeled and heated oligonucleotide was run on each lane with none (lane 1), 0.17 nm (lane 2), or 0.5 nm (lane 3) purified muRif1. The radioactivity (cpm/pmol) of every probe was slightly different, which is indicated in Fig. S7. B, fraction of the Rif1-bound probe was normalized and presented as a bar graph. C–E, EMSA was performed as above with partially-purified muRif1-NC (C), muRif1-NTD (D), or muRif1-CTD (E). The amount of the bound probe was calculated as in B and presented as bar graphs. See Fig. S7 for raw gel images of EMSA with muRif1-NC, -NTD, and -CTD.
Simultaneous interaction of muRif1 with multiple G4 DNA
Our previous studies suggested the possibility that Rif1 participates in physical interactions of distant chromatin segments, contributing to the formation of chromatin loops (6, 7, 9). As shown in the previous section of this report, purified muRif1 behaves as oligomers and bears at least two G4-binding domains (NTD and CTD). These findings prompted us to examine whether muRif1 could mediate physical interaction between different G4 DNAs. As muRif1 was pulled down with G4-folded, T6G24-conjugated magnetic beads (Fig. 4, A and B), similar pulldown assays were conducted three times in the presence of a radiolabeled G4 probe in the presence or absence of muRif1, and the pulled down G4 probe was quantified (Fig. 8, A and B). A significant amount of the radiolabeled G4 probe was pulled down by the G4 magnetic beads when muRif1 was present (Fig. 8B, columns 2 and 3). The binding of G4 probe was lost by addition of 5-fold excess of cold T6G24 (Fig. 8B, column 4) but unaffected by addition of even 10-fold excess of T6(GA)12 DNA (Fig. 8B, column 5). When T6(GA)12 DNA was used as a radiolabeled probe, it was not pulled down even in the presence of muRif1 (Fig. 8B, columns 7 and 8). These results indicate that muRif1 simultaneously bound to both immobilized G4 and radiolabeled G4 DNA, supporting the idea that muRif1 could hold together multiple G4 DNA molecules.
Figure 8.

Simultaneous association of muRif1 with multiple molecules of G4 DNA. A, G24-dynabeads were mixed with radiolabeled G4-folded T6G24 (lanes 1–5) or T6(GA)12 probe (lanes 6–8) in the presence (lanes 2–5 and 7 and 8) or the absence (lanes 1 and 6) of purified muRif1 and were pulled down. The bound materials were eluted by brief boiling in 80% formamide and run on 10% polyacrylamide gel containing 8 m urea. The same assay was repeated three times. The association of the radioactive G4 probe was lost by addition of 1 pmol of unlabeled T6G24 (lane 4) but was unaffected by addition of 2 pmol of T6(GA)12 DNA (lane 5). B, amount of the associated radioactive DNA is shown in a bar graph according to quantitation of relevant radioactive bands on denaturing polyacrylamide gel.
Discussion
Accumulating genetic and cell biological evidence points to wide varieties of cellular functions of Rif1. These include programming genome-wide DNA replication timing, repair of DNA lesion, restart from fork collapse, anti-apoptosis, replicative senescence, and transcriptional regulation in addition to its classical role in telomere length regulation (1–30). Whereas multiple physiological functions of Rif1 have become unraveled, biochemical and structural information on Rif1 is still limited, mainly due to difficulty in purifying Rif1 protein in a full-length form. We have previously developed convenient mammalian expression system for protein production using 293T cells and low-cost transfection reagent, PEI (31, 32). By exploiting this efficient expression system, we succeeded in purification of the 2418-amino-acid-long full-length murine Rif1 in a soluble form.
Because a C-terminal peptide derived from budding yeast Rif1 (ScRif1) was shown to form a tetramer (27), we first observed hydrodynamic behaviors of the purified muRif1 to analyze its oligomer formation. However, its native form was found to be too large to determine its RS and native molecular weight accurately, although our preliminary analysis suggested that muRif1 adopts an elongated or extended shape (data not shown). Therefore, we have decided to produce and purify smaller segments of muRif1 to define its oligomeric state more precisely and to localize the segment(s) responsible for oligomerization. Interestingly, both muRif1-CTD and muRif1-NTD formed oligomers in the cell extracts (Fig. 2) and after purification (Fig. 3). The LID region does not self-associate. The hydrodynamic analyses of muRif1-NTD demonstrated the presence of tetramers and octamers (Fig. 3G). Because the HEAT repeats of TOR kinase were shown to form a dimer (48, 49), oligomerization of muRif1-NTD could also depend on its HEAT repeats. After our initial submission of this paper, Mattarocci et al. (50) reported that the isolated HEAT repeat domain of ScRif1 forms a dimer, but the dimer formation depends on dsDNA bound by the two HEAT repeat domains (50). In contrast, muRif1-NTD forms a tetramer or an octamer on its own in the absence of DNA. Similar analyses of muRif1-CTD demonstrated the presence of dimers and dodecamers, in contrast to the tetramer formation of the C-terminal 60-amino acid segment of ScRif1 (27). Thus, the nature of oligomer formation seems quite different among Rif1 proteins from different species.
The results presented in this report suggest that there may be a unique muRif1 oligomer composed of assemblies of a CTD dodecamer and three NTD tetramers (or an NTD octamer plus an NTD tetramer) as illustrated in Fig. S8. This also suggests that much larger muRif1 oligomers could be generated through association between NTD tetramers, and such a large complex might have prevented the determination of the precise oligomer size for the full-length muRif1 in our hydrodynamic analysis.
The results of these analyses also provide information on the molecular shape of each identified homo-oligomer. The calculated S/Smax (=R/Rmin) values indicate that both muRif1-NTD and -CTD adopts an elongated or extended shape (Fig. 3G, right-most column). muRif1-NTD contains 21 HEAT repeats, consistent with its elongated shape. Intriguingly, the values we obtained suggest that muRif1-CTD is more elongated than muRif1-NTD, whereas the X-ray crystallography of the tetrameric C-terminal peptide of yeast Rif1 indicated a rather globular shape (27). The difference may be due to the use of a longer polypeptide (299 amino acids) for our analyses of muRif1-CTD compared with the 60-amino-acid polypeptide used for the atomic structure analyses of yeast Rif1 CTD. In conclusion, our hydrodynamic analyses demonstrated oligomer formation and very elongated shapes of muRif1-NTD and -CTD, thus leading us to speculate similar characteristics for the full-length muRif1.
In budding yeast, association of Rif1 with chromosomal DNA is thought to be mediated by its binding to Rap1 protein (27). In contrast, the C-terminal domains of human and mouse Rif1 were shown to interact directly with structured DNA, most notably with cruciform DNA in vitro (23, 26). We recently reported that SpRif1 specifically binds to a G4 structure, which is the functional binding target of SpRif1 (6). Thus, we examined interaction of muRif1 with G4 DNA. Our EMSA and pull-down analyses demonstrated that purified muRif1 indeed specifically binds to G4 DNA (Figs. 4–8). Competition assays indicate that its affinity to G4 DNA seemed much higher than that to cruciform DNA. muRif1 bound to parallel-type G4 more efficiently than to telomere repeat G4 or anti-parallel-type G4. Interestingly, high-affinity G4 binding by muRif1 was not attributed to a single domain, and apparently it required all the three domains (NTD, LID, and CTD). Our results indicate that isolated muRif1-NTD and -CTD interact with G4 DNA, although they bind to it much less efficiently than the full-length muRif1 does. Additionally, the pattern of their preferences for different G4 probes is similar to that of the full-length muRif1, although the selectivity for G4-fold seemed less stringent than the full-length muRif1. muRif1-LID lacks G4-binding activity but is likely to contribute to the high-affinity binding of the full-length muRif1 to G4, because muRif1-NC could not recapitulate the G4-binding affinity of the full-length muRif1. Naturally unstructured and intrinsically disordered segments have been found in many chromatin-associated proteins (51–54), but their physiological functions and biochemical activities are still largely unknown. Through its ability to modulate the structure, LID may coordinate the NTD and CTD for proper complex formation with the target G4 structures.
We previously suggested the possibility that Rif1 mediates interaction of distant chromosomal sites in the nucleus, thereby organizing chromosomal domains or compartments (6, 7, 9). Thus, we next examined the possibility of chromatin loop formation by direct simultaneous interaction of Rif1 with multiple DNA strands. Pull-down assays demonstrated that one muRif1 molecule bound to at least two G4 DNAs simultaneously (Fig. 8). Therefore, muRif1 appears to have an intrinsic ability to hold together distant DNA strands via the association with multiple G4 DNAs. This ability is likely to depend on oligomer formation of muRif1 as well as on its bipartite G4-binding sites present on its NTD and CTD (Fig. 9A). Because muRif1 could form a tetramer or an octamer or even a dodecamer, multiple chromatin fibers could be assembled by muRif1 (Fig. 9B), as was proposed before (6). Our novel findings of oligomer formation of muRif1 and of its bipartite G4 binding would provide the mechanical basis for inter-chromatin interactions mediated by Rif1.
Figure 9.
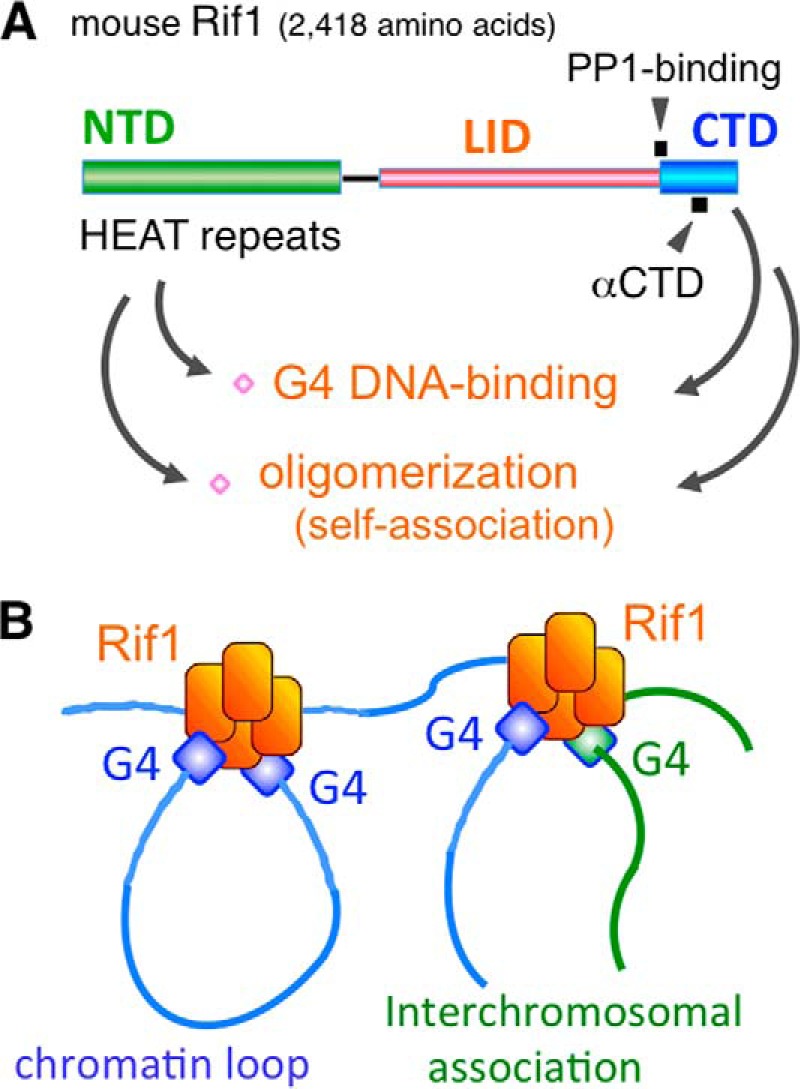
Multivalent G4 binding by muRif1 oligomer could mediate association between distant chromatin loci. A, schematic representation of at least two G4-binding and oligomerization domains present in the N- and C-terminal segments of muRif1. B, model on G4-based assembly of multiple chromatin fibers mediated by Rif1 that binds to multiple G4-containing DNA due to its oligomeric structure.
Notably, the G4 EMSA experiments indicated that muRif1 bound efficiently to the parallel-type G4 probes, CEB1 (18-mer) and 93del (16-mer), although less efficiently than to G15, also known to adopt a parallel-fold (Fig. 7). The two oligonucleotides were reported to fold into self-stacked G4 dimers (Fig. S9A) (55 and 56). The oligomerization of G4 through stacking would more readily occur with parallel-type G4s than anti-parallel or hybrid-type G4s, because loops are positioned along the side of G4. In contrast, the loops in anti-parallel and hybrid-type G4s are either diagonal or edgewise (crossing or protruding, respectively, over outward G-quartet planes (57)), interfering with G4 stacking (Fig. S9B). Our data suggest that Rif1 prefers G4s that have intrinsic tendency to form oligomers (Fig. 7). Thus, Rif1's binding preference to parallel-type G4s may be due to their propensity to form stacked G4 oligomers. We cannot exclude the possibility that Rif1 also prefers interstrand G4s in addition to stacked G4s. There appear to be bands representing dimer or larger oligomers of T6G24 and G15 probes that run more slowly on EMSA gels and are preferentially bound by muRif1 (Figs. 5–7), consistent with the notion that muRif1 has higher affinity to oligomeric G4s than to the monomer. These observations suggest that muRif1 preferentially and with a high affinity binds to sequences capable of forming G4 oligomer(s) in cells as well. Further cellular experiments need to be conducted to validate our model that G4-to-G4 interactions mediated by Rif1 are the basis for establishing chromatin domain structures.
Hänsel-Hertsch et al. (58) recently reported 10,560 high-confidence cellular G4 sites in a human epidermal keratinocyte HaCaT cell line by ChIP-seq analyses using BG4 polypeptide antibody that is known to specifically bind to G4 (59). This number is quite low, given the initial bioinformatics estimation of ∼375,000 potential G4-forming sequences (60, 61) and more recent experimental evidence for 716,310 sites capable of generating G4 deduced by the G4-seq method (based on a polymerase stop assay) (62). However, we speculate that this number under-represents the actual numbers of G4 on the human genome for the following reasons. First, in Hänsel-Hertsch et al. (58), chromatin is fixed by formaldehyde prior to the incubation with BG4. This would result in failure of the detection of a large fraction of intrinsic cellular G4, because those G4 that are bound by cellular proteins or those that are present in a non-accessible chromatin environment may escape detection. Second, BG4, a single polypeptide monoclonal antibody, may not detect all classes of cellular G4 that form on duplex DNA and may adopt structures distinct from the known canonical G4 structures generated on single-stranded DNA. Third, detection of cellular G4 by 1H6, another G4-specific monoclonal antibody, revealed numerous fine nuclei foci, sometimes homogeneously, which suggested the existence of cellular G4 in numbers much larger than that estimated by ChIP-seq (63). Finally, formation and elimination of G4, with varying stability, may be dynamically regulated in cells. Fixation and staining only capture a snapshot of a given moment and would lead to the underestimation of the actual numbers of cellular G4.
These considerations led us to estimate that the numbers of cellular G4 may be quite large, although some of them may be only transiently formed or dynamically undergo formation/elimination cycles due to its instability. We propose that Rif1 may generate chromatin architecture by facilitating the dynamic interactions between G4 structures present throughout the genome. We envision these interactions may be very dynamic, not static, and not stably associating with each other. We point out that these Rif1-mediated G4–G4 interactions may be somehow confined in the late-replicating domain, in accordance with the observation made by the Hi-C analyses (64). This could be due to enrichment of G4 structures in the late replicating domain, a prediction that needs to be tested in the future.
One common feature of fission yeast and mammalian Rif1 in their role in replication timing regulation is their close association with late firing origins or late-replicating domains (6, 64). Thus, Rif1 needs to be somehow recruited to specific chromatin segments that are associated with late replication. Although it is an attractive model that Rif1 is recruited to chromatin by directly recognizing G4 (Fig. 9B), it could be recruited through other proteins as well. Indeed, in budding and fission yeast, other telomere factors, Rap1 and Taz1, respectively, appear to recruit Rif1 not only to the telomere but also to a subset of late-firing origins (65, 66). There are many other Rif1-interacting proteins, including BLM (23), 53BP1 (17–21), histones, lamin B, and PP1 (67, 68). Among them, direct molecular interaction with Rif1 was shown for BLM, 53BP1, and PP1. Lamin B may play a role in localization of Rif1 near the nuclear periphery but not in recruiting Rif1 to chromatin (64). 53BP1 recruits Rif1 to chromatin, but it does so only upon DNA damage (17–21). Xu et al. (23) reported that Rif1 (not BLM) provides a DNA-binding interface for the BLM–Rif1 complex. Other chromatin proteins, including histones, may play a role for recruiting Rif1 to chromatin. Although Rif1 co-localizes partially with heterochromatic marks, it is unlikely that heterochromatin recruits Rif1, because its formation occurs downstream of Rif1 (69, 70). Potential roles of these proteins in recruiting Rif1 to chromatin and inducing chromatin architecture that regulates replication, transcription, or recombination are intriguing and deserve future studies.
Rif1 plays critical roles in the choice of DSB repair pathways downstream of 53BP1 (17–22). It suppresses HRR and thereby promotes repair by NHEJ at DSB. G4 structures may be generated during the course of replication and transcription, increasing the risk of DNA breaks by causing their temporal arrest. As HRR could cause deletion or expansion between the G-tract sequences, NHEJ would be favored at DSB near G4 sites. Thus, the G4-binding property of Rif1 might also contribute to quick choice of repair pathways in a manner independent of 53BP1. Rif1 and other NHEJ components participate in CSR at immunoglobulin class switch in mammalian B cells (18, 19). The genomic regions involved in class switch are enriched for G-tracts, and copy numbers of G-tracts affect efficiency of CSR (71). Thus, Rif1 may regulate CSR through direct binding to the class switch target DNAs that are likely to adopt G4-like structures.
In summary, we show here that mammalian Rif1 specifically and simultaneously binds to multiple molecules of G4 DNA probably due to its highly oligomeric structures and bipartite G4-binding sites. These biochemical features of Rif1 may contribute to its ability to assemble multiple chromatin fibers at nuclear periphery to generate specific functional chromatin domains that may affect the efficiency of various chromosome transactions, including replication, recombination, repair, and transcription. Further studies aimed to clarify amino acid residues of Rif1 essential for oligomerization and for G4 recognition and their structural basis will lead to a better understanding of how G4 and Rif1 regulates chromatin architecture and other aspects of chromosome dynamics.
Experimental procedures
Reagents, antibodies, and cells
Anti-DDDDK (mouse FLA-1) and anti-HA (rabbit 561) antibodies were obtained from MBL Co. Monoclonal anti-HA antibodies (mouse 16B12 and rat 3F10) were from Roche Applied Science. Anti-FLAG M2-agarose beads, 3xFLAG peptides, and polyglutamic acid were purchased from Sigma. Polyethylene glycol 200 (PEG200) was from Nakalai Tesque. Dynabeads-streptavidin M280 was from Invitrogen. Human IgM was purchased from Wako Chemical, and other molecular size standards for hydrodynamic analysis were from GE Healthcare. Benzonase was purchased from Novagen. Other nucleases and molecular biology enzymes were from Takara-Bio, TOYOBO, Roche Applied Science, Sigma, or New England Biolabs. Oligonucleotides were synthesized commercially by Hokkaido System Science or by Eurofins Genomics. 293T cells were cultured in Dulbecco's modified Eagle's medium (Nissui Co.) plus 10% fetal bovine serum (Nichirei Co.). PEI was from Polysciences Inc. Protease inhibitors (Complete EDTA-free, Roche Applied Science), 10 μm MG132 (Peptide Institute, Inc.), and 1 mm phenylmethylsulfonyl fluoride (Sigma) were included in the reagents for cell extraction and for protein purification unless otherwise indicated.
Recombinant plasmids
BamHI sites were added to N and C termini of muRif1 cDNA (GenBankTM JX255737) by conventional PCR. pCSII-EF-His-MCS_FLAG (version 3-4) and pCSII-EF-His-MCS_HA (version 3-5) were described previously (31). A viral 5′-sequence (SnaBI 588 nt to PpuMI 2311 nt) was removed from both vectors, and a viral 3′-sequence (KpnI 4546 nt to ApaI 5118 nt) was also deleted in the version 3-4 (3xFLAG version). The resulting expression vectors, pCEH_FLAG and pCEH_HA, were used for insertion of the full-length muRif1 cDNA into a unique BamHI site or of parts of the coding frame between the BamHI and XhoI sites. The strong EF1α promoter drives expression of muRif1 recombinants bearing His6 tag at the N terminus and another tag (3xFLAG or HA) at the C terminus.
Preparation of 293T cell extract and purification of recombinant proteins
According to a PEI-transfection protocol (31, 32), 16–18 150-mm inner diameter dishes of 293T cells were transfected with pCEH-muRif1_FLAG for 2 days. The cells producing His6-muRif1-FLAG3 were solubilized for 30 min in 1 ml/dish of CSK buffer (10 mm Pipes-KOH (pH 6.8), 100 mm potassium glutamate, 300 mm sucrose, 1 mm MgCl2, 1 mm EGTA, and 1 mm DTT) containing 0.1% Triton X-100 and 10 units/ml Benzonase (CSK-T-Bz). The cleared lysate was mixed with 1.3–1.5 ml of anti-FLAG M2 beads for 90 min. After washing the column with 21–24 ml of FLAG-B buffer (20 mm Tris-HCl (pH 8.0), 0.3 m NaCl, 0.02% Triton X-100, and 10% glycerol), the bound polypeptides were eluted six times with 1.3–1.5 ml of 0.2 mg/ml 3xFLAG peptide in FLAG-B buffer. Finally, the residual material was eluted with 4× 1.3–1.5 ml of 0.1 m glycine-HCl (pH 3.5). The fractions 2–6 (total 6.5–7.5 ml) of the above anti-FLAG column were mixed with 2-ml bed volume of Ni2+-NTA resin (equilibrated with ½× FLAG-B buffer). After mild rocking for ∼60 min at 4 °C, the resin was poured into a disposable column and washed with ∼30 ml of FLAG-B buffer. The bound proteins were eluted with a step gradient of imidazole (20–240 mm, 1.8 ml/fraction). His6-muRif1-NTD-FLAG3, His6-muRif1-CTD-FLAG3, and His6-muRif1-LID-FLAG3 were expressed and purified almost in the same manner. His6-muRif1-NC-FLAG3 was difficult to solubilize in CSK-T-Bz. Therefore, it was solubilized by adding NaCl at 0.3 m to the CSK-T-Bz lysate and purified as described for His6-muRif1-FLAG3.
Co-immunoprecipitation assay
On 6-well dishes, 293T cells were co-transfected with pCEH-Domain1_FLAG and pCEH-Domain2_HA (domain 1 or 2 representing muRif1-NTD, -CTD, -NC, or -LID) for co-expression of 3xFLAG-tagged domain 1 and HA-tagged domain 2. The transfected cells were harvested after 48 h and solubilized for 30 min in 0.24 ml/well of CSK-T-Bz. 100 μl of the cleared lysate was mixed with 2.5 μl of protein G-dynabeads pre-conjugated with 0.5 μg of mouse anti-DDDDK antibody (FLA-1) or those with 0.25 μg of rat anti-HA antibody (3F10) for 60 min on ice. The protein G-dynabeads were then separated by a magnet and washed with 300 μl of Wash solution K100 (0.1 m KCl, 0.05% Triton X-100, 0.1 mm DTT in Buffer GD (72)). The protein-bound beads were resuspended with 300 μl of the same buffer and transferred to fresh tubes. The beads were recollected and suspended finally in 50 μl of 1.2× SDS-sample buffer, and the bound polypeptides were eluted at 95 °C. The eluates were subjected to SDS-PAGE followed by Western blotting with anti-DDDDK antibody (FLA-1) to detect 3xFLAG-tagged proteins or by anti-HA antibody (rabbit 561) to detect HA-tagged ones.
Glycerol-gradient centrifugation
In 2.2-ml centrifuge tubes, 15–36% glycerol gradient was made in 20 mm Tris-HCl (pH 7.8), 150 mm KCl, 1 mm EDTA, 0.5 mm DTT and appropriate protease inhibitors. 0.2 ml of a Ni2+-NTA fraction 11 of truncated muRif1 was layered on top of the gradient and centrifuged at 40,000 rpm for 16 h at 4 °C in Beckman TLS55 rotor. 0.1-ml fractions were removed from top to bottom and subjected to SDS-PAGE followed by anti-DDDDK blotting.
Analytical gel filtration
50 μl of Ni2+-NTA fraction 11 (0.22-μm filtrated) of truncated muRif1 was applied to pre-equilibrated Superose 6 PC3.2/30 column (GE Healthcare), and eluent (20 mm Tris-HCl (pH 7.8), 150 mm KCl, 1 mm EDTA, 0.5 mm DTT, 10% glycerol, and protease inhibitors (×1/5, Roche Applied Science)) was flowed at 40 μl/min at 4 °C. 80-μl fractions were collected and subjected to SDS-PAGE followed by anti-DDDDK blotting.
Non-denaturing electrophoresis on agarose-polyacrylamide composite gel
0.4% agarose, 2.5% acrylamide (37.5:1) composite gels were prepared as 2-mm-thick mini-slab gels according to the literature (33, 34). Peak fractions of muRif1-NTD and -CTD as well as molecular size standards (IgM, catalase, and ovalbumin) were loaded on the gel and run for 110 min at 50 V in 25 mm Tris, 192 mm glycine buffer. The lane containing size standard proteins was stained with Coomassie Brilliant Blue. The other lanes were immersed in the electrode buffer containing 0.1% SDS and 14 mm 2-mercaptoethanol for 16 h at 4 °C and then electroblotted to Immobilon-P membrane followed by anti-DDDDK blotting. When the composite gel was used for EMSA, 0.4% agarose, 2% acrylamide (37.5:1) composite gel in 2-mm-thick large slab gel was made with 1× TBE plus 50 mm KCl. EMSA was conducted basically as described below except that electrophoresis was for 4.5 h at 40 V.
Preparation of structured oligonucleotides
To induce G4 folding in oligonucleotides, synthetic ssDNA (18–30-mer) was boiled for 5 min and then annealed in 50 mm KCl, 40% PEG200, 1 mm EDTA, 10 mm Tris-HCl (pH 7.5) by gradual cooling to room temperature (36, 37). Various competitor DNAs were prepared by annealing synthetic oligonucleotides of 20–40-mer, most of which were described in Sukackaite et al. (26). 40-bp dsDNA, Y-fork, flap, fork, 4WJ (cruciform), and telomere repeat (6 times) DNAs (Tel-ds) were made by annealing 1 nmol of the oligonucleotides (shown in Table 2) in 0.1 ml of 10 mm Tris-HCl (pH 8), 1 mm EDTA, 50 mm KCl, and 40% PEG200. The annealed DNA bands were excised after PAGE. The excised gel pieces were homogenized in 2-ml tubes and extracted with 0.6 ml of 0.5 m ammonium acetate, 10 mm Mg(OAc)2, 1 mm EDTA, and 0.1% SDS by shaking at 37 °C for 16 h. After a brief centrifugation, supernatant was transferred into 0.22-μm microfiltration tubes (COSTAR 8160). The each tube was spun for 10 min at 12,000 rpm, and the filtrate was subjected to ethanol precipitation. Precipitated DNA was dissolved in 50 μl of pure water.
Table 2.
DNA oligonucleotides used in this study
DNA substrates for EMSA (Fig. 7C) were prepared by annealing: (H1 + H5), dsDNA; (H1 + H4), Y-fork; (H1 + H3.5 + H4), flap; (H1 + H2.5 + H3.5 + H4), fork; (H1 + H2 + H3 + H4), 4WJ; (TelG-Fw,+ TelC-Rv),Tel-ds.
| Oligonucleotide name | Length | Sequence (5′-to-3′) | Refs. |
|---|---|---|---|
| G-quadruplex-forming oligonucleotides | |||
| T6G24 | 30-mer | TTTTTTGGGGGGGGGGGGGGGGGGGGGGGG | This study |
| T2G15T | 18-mer | TTGGGGGGGGGGGGGGGT | 38 |
| VEGF | 22-mer | CGGGGCGGGCCTTGGGCGGGGT | 39 |
| c-kit2 | 21-mer | CGGGCGGGCTCGAGGGAGGGT | 40 |
| c-myc | 22-mer | TGAGGGTGGGTAGGGTGGGTAA | 41 |
| Telo24 | 24-mer | TTAGGGTTAGGGTTAGGGTTAGGG | 42 |
| Thrombin aptamer | 15-mer | GGTTGGTGTGGTTGG | 43 |
| Telo22 | 22-mer | GGGTTAGGGTTAGGGTTAGGGT | 44 |
| bcl-2 | 23-mer | GGGCGCGGGAGGAATTGGGCGGG | 45 |
| Telo23 | 23-mer | TAGGGTTAGGGTTAGGGTTAGGG | 46 |
| Telo25 | 25-mer | TAGGGTTAGGGTTAGGGTTAGGGTT | 46 |
| CEB1 | 18-mer | AGGGGGGAGGGAGGGTGG | 55 |
| T95–2T | 18-mer | TTGGGTGGGTGGGTGGGT | 47 |
| 93del | 16-mer | GGGGTGGGAGGAGGGT | 56 |
| G15 | 15-mer | GGGGGGGGGGGGGGG | This study |
| TERC18 | 18-mer | GGGTTGCGGAGGGTGGGC | 47 |
| Other DNA oligonucleotides | |||
| T6(GA)12 | TTTTTTGAGAGAGAGAGAGAGAGAGAGAGA | This study | |
| TelG-Fw | TTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGG | This study | |
| TelC-Rv | CCCTAACCCTAACCCTAACCCTAACCCTAACCCTAA | This study | |
| sub01(H1) | GTGACCGTCTCCGGGAGCTGGAAACGCGCGAGACGAAAGG | 26 | |
| sub05(H5) | CCTTTCGTCTCGCGCGTTTCCAGCTCCCGGAGACGGTCAC | 26 | |
| sub04(H4) | CGAGTTGCTCTTGCCCGGCGCAGCTCCCGGAGACGGTCAC | 26 | |
| sub07(H3.5) | CGCCGGGCAAGAGCAACTCG | 26 | |
| sub06(H2.5) | CCTTTCGTCTCGCGCGTTTC | 26 | |
| sub02(H2) | CCTTTCGTCTCGCGCGTTTCGCCAGCCCCGACACCCGCCA | 26 | |
| sub03(H3) | TGGCGGGTGTCGGGGCTGGCCGCCGGGCAAGAGCAACTCG | 26 | |
Pull-down assay with G4 DNA-magnetic beads
100 pmol of 5′-biotinylated T6G24 (synthetic 30-mer) was boiled for 5 min and then folded to form a G4 structure as described above. It was then conjugated to 1 mg of streptavidin-dynabeads for 3.5 h at room temperature according to the manufacturer's protocol using a kilobase BINDER kit (Invitrogen). The DNA-dynabeads (equivalent to ∼0.25 pmol of biotin-T6G24) were mixed wit the indicated amount of muRif1 proteins in the presence of 1 μg of BSA in a 40-μl reaction for 30 min at room temperature. For competition experiments, the indicated amount of calf thymus DNA, T6(GA)12, G4-formed T6G24 DNA, or cruciform DNA was preincubated with muRif1 proteins before addition to T6G24-dynabeads. The DNA-dynabeads were then separated by magnet, and washed twice with Wash solution-K100 (Buffer GD containing 0.1 m KCl, 0.05% Triton X-100, 0.1 mm DTT, and protease inhibitors). The bound polypeptides were eluted in 45 μl of 1.2× SDS-sample buffer at 95 °C, and subjected to SDS-PAGE followed by anti-DDDDK blotting.
EMSA
To end-label oligonucleotides, 10 pmol of PAGE-purified oligonucleotides were dried, labeled at the 5′-end with [γ-32P]ATP by T4 polynucleotide kinase, and purified using microspin G-25. Then, 40 μl (∼73%) of the labeled ssDNA were heated at 96 °C for 5 min and then cooled down gradually (in 80 μl; ∼5 h) to induce G4 folding in 50 mm KCl, 40% PEG200, 1 mm EDTA, and 10 mm Tris-HCl (pH 7.5). In an 18-μl reaction, the indicated amount of pre-heated 32P-labeled substrate DNA (T6G24 or T6(GA)12) was mixed with muRif1 or with its truncated polypeptides in 20 mm Hepes-KOH (pH 7.6), 50 mm KCl, 1 mm EDTA, 0.01% Triton X-100, and 10% PEG200. After incubation for 30 min at 30 °C, the mixture was electrophoresed in 1× TBE plus 50 mm KCl on 12% polyacrylamide (29:1) gel containing 50 mm KCl and 40% PEG200 at 150 V for 16–17 h. In competition assays, 0.1 pmol of heated 32P-probe (T6G24) was mixed with 0.3 pmol (3 times) or 3.0 pmol (30 times) of various competitor DNAs (see Table 2) before addition of proteins.
Calculation of Kd between muRif1 and G4-folded T6G24
We use the equation, Kd = Pf·Gf/Gb = {n(Pt) − Gb}·Gf/Gb, where the concentration of each molecule is as follows: Gf, unbound G4 oligonucleotide; Gb, bound G4 oligonucleotide; Pf, free G4-binding sites; Pt, total protein, and n. number of G4-binding sites on a protein (protomer). If the concentration of total G4 oligonucleotide was designated as Gt, Kd = {n(Pt) + Gf − Gt}·Gf/(Gt − Gf), thus leading to a quadratic equation, Gf2 + {Kd + n(Pt) − Gt}·Gf − Gt·Kd = 0. This equation can be converted to Gf = {−β + √(−β2 + 4·Gt·Kd)}/2, where β = Kd + n(Pt) − Gt. Then, curve fitting to the real plots was performed in the assumption that n = 1 with “solver” function of Microsoft Excel. Kd was also calculated in the assumption that n = 2 for full-length muRif1 and for muRif1-NC.
Author contributions
K. M. and H. M. conceptualization; K. M. formal analysis; K. M., N. Y.-S., and H. M. investigation; K. M. methodology; K. M. writing-original draft; K. M., N. Y.-S., and H. M. writing-review and editing; H. M. funding acquisition.
Supplementary Material
Acknowledgments
We are very grateful to Naoko Kakusho, Rino Fukatsu, Satoshi Yamazaki, and Yutaka Kanoh for technical instructions and assistance. We thank Akiko Minagawa for secretarial assistance.
This work was supported by JSPS KAKENHI Grants-in-aid for Scientific Research (A) 23247031 and 26251004, Grant-in-aid for Scientific Research on Priority Areas (“non-coding RNA”) 24114520 (to H. M.), and Grant-in-aid for Scientific Research (C) 15K07164 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to N. Y.), and the Uehara Memorial Foundation Research Support (to H. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S9.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) JX255737.
- ssDNA
- single-stranded DNA
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- LID
- long intrinsically disordered domain
- NC
- N-terminal plus C-terminal domain
- G4
- guanine-quadruplex
- EMSA
- electrophoretic mobility shift assay
- PP1
- phosphoprotein phosphatase 1
- ATM
- ataxia telangiectasia mutated
- NHEJ
- non-homologous end-joining
- CSR
- class switch recombination
- DSB
- double-stranded DNA break
- PEI
- polyethyleneimine
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- dsDNA
- double-stranded DNA
- nt
- nucleotide
- mu
- murine
- -seq
- -sequence
- ATR
- ataxia telangiectasia and Rad3-related
- RPA
- replication protein A
- TOR
- target of rapamycin
- HRR
- homologous recombination repair.
References
- 1. Hardy C. F., Sussel L., and Shore D. (1992) A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 6, 801–814 10.1101/gad.6.5.801 [DOI] [PubMed] [Google Scholar]
- 2. Kyrion G., Liu K., Liu C., and Lustig A. J. (1993) RAP1 and telomere structure regulate telomere position effects in Saccharomyces cerevisiae. Genes Dev. 7, 1146–1159 10.1101/gad.7.7a.1146 [DOI] [PubMed] [Google Scholar]
- 3. Xue Y., Rushton M. D., and Maringele L. (2011) A novel checkpoint and RPA inhibitory pathway regulated by Rif1. PLoS Genet. 7, e1002417 10.1371/journal.pgen.1002417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ribeyre C., and Shore D. (2012) Anticheckpoint pathways at telomeres in yeast. Nat. Struct. Mol. Biol. 19, 307–313 10.1038/nsmb.2225 [DOI] [PubMed] [Google Scholar]
- 5. Hayano M., Kanoh Y., Matsumoto S., Renard-Guillet C., Shirahige K., and Masai H. (2012) Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 26, 137–150 10.1101/gad.178491.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kanoh Y., Matsumoto S., Fukatsu R., Kakusho N., Kono N., Renard-Guillet C., Masuda K., Iida K., Nagasawa K., Shirahige K., and Masai H. (2015) Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 22, 889–897 10.1038/nsmb.3102 [DOI] [PubMed] [Google Scholar]
- 7. Yamazaki S., Ishii A., Kanoh Y., Oda M., Nishito Y., and Masai H. (2012) Rif1 regulates the replication timing domains on the human genome. EMBO J. 31, 3667–3677 10.1038/emboj.2012.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cornacchia D., Dileep V., Quivy J. P., Foti R., Tili F., Santarella-Mellwig R., Antony C., Almouzni G., Gilbert D. M., and Buonomo S. B. (2012) Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 31, 3678–3690 10.1038/emboj.2012.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamazaki S., Hayano M., and Masai H. (2013) Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing. Trends Genet. 29, 449–460 10.1016/j.tig.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 10. Renard-Guillet C., Kanoh Y., Shirahige K., and Masai H. (2014) Temporal and spatial regulation of eukaryotic DNA replication: from regulated initiation to genome-scale timing program. Semin. Cell Dev. Biol. 30, 110–120 10.1016/j.semcdb.2014.04.014 [DOI] [PubMed] [Google Scholar]
- 11. Hiraga S., Alvino G. M., Chang F., Lian H. Y., Sridhar A., Kubota T., Brewer B. J., Weinreich M., Raghuraman M. K., and Donaldson A. D. (2014) Rif1 controls DNA replication by directing protein phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 28, 372–383 10.1101/gad.231258.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peace J. M., Ter-Zakarian A., and Aparicio O. M. (2014) Rif1 regulates initiation timing of late replication origins throughout the S. cerevisiae genome. PLoS ONE 9, e98501 10.1371/journal.pone.0098501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mattarocci S., Shyian M., Lemmens L., Damay P., Altintas D. M., Shi T., Bartholomew C. R., Thomä N. H., Hardy C. F., and Shore D. (2014) Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. 7, 62–69 10.1016/j.celrep.2014.03.010 [DOI] [PubMed] [Google Scholar]
- 14. Silverman J., Takai H., Buonomo S. B., Eisenhaber F., and de Lange T. (2004) Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S phase checkpoint. Genes Dev. 18, 2108–2119 10.1101/gad.1216004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buonomo S. B., Wu Y., Ferguson D., and de Lange T. (2009) Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 187, 385–398 10.1083/jcb.200902039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar S., Yoo H. Y., Kumagai A., Shevchenko A., Shevchenko A., and Dunphy W. G. (2012) Role for Rif1 in the checkpoint response to damaged DNA in Xenopus egg extracts. Cell Cycle 11, 1183–1194 10.4161/cc.11.6.19636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zimmermann M., Lottersberger F., Buonomo S. B., Sfeir A., and de Lange T. (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339, 700–704 10.1126/science.1231573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A. D., Feldhahn N., Resch W., Oliveira T. Y., Chait B. T., Nussenzweig A., Casellas R., Robbiani D. F., and Nussenzweig M. C. (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715 10.1126/science.1230624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chapman J. R., Barral P., Vannier J. B., Borel V., Steger M., Tomas-Loba A., Sartori A. A., Adams I. R., Batista F. D., and Boulton S. J. (2013) RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 10.1016/j.molcel.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J. T., Tkáč J., Cook M. A., Rosebrock A. P., Munro M., Canny M. D., Xu D., and Durocher D. (2013) A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 10.1016/j.molcel.2013.01.001 [DOI] [PubMed] [Google Scholar]
- 21. Feng L., Fong K. W., Wang J., Wang W., and Chen J. (2013) RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 288, 11135–11143 10.1074/jbc.M113.457440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Callen E., Di Virgilio M., Kruhlak M. J., Nieto-Soler M., Wong N., Chen H. T., Faryabi R. B., Polato F., Santos M., Starnes L. M., Wesemann D. R., Lee J. E., Tubbs A., Sleckman B. P., Daniel J. A., et al. (2013) 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153, 1266–1280 10.1016/j.cell.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu D., Muniandy P., Leo E., Yin J., Thangavel S., Shen X., Ii M., and Agama K., Guo R., Fox D. 3rd., Meetei A. R., Wilson L., Nguyen H., Weng N. P., Brill S. J., et al. (2010) Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 29, 3140–3155 10.1038/emboj.2010.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams I. R., and McLaren A. (2004) Identification and characterisation of mRif1: a mouse telomere-associated protein highly expressed in germ cells and embryo-derived pluripotent stem cells. Dev. Dyn. 229, 733–744 10.1002/dvdy.10471 [DOI] [PubMed] [Google Scholar]
- 25. Wang H., Zhao A., Chen L., Zhong X., Liao J., Gao M., Cai M., Lee D. H., Li J., Chowdhury D., Yang Y. G., Pfeifer G. P., Yen Y., and Xu X. (2009) Human RIF1 encodes an anti-apoptotic factor required for DNA repair. Carcinogenesis 30, 1314–1319 10.1093/carcin/bgp136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sukackaite R., Jensen M. R., Mas P. J., Blackledge M., Buonomo S. B., and Hart D. J. (2014) Structural and biophysical characterization of murine rif1 C terminus reveals high specificity for DNA cruciform structures. J. Biol. Chem. 289, 13903–13911 10.1074/jbc.M114.557843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi T., Bunker R. D., Mattarocci S., Ribeyre C., Faty M., Gut H., Scrima A., Rass U., Rubin S. M., Shore D., and Thomä N. H. (2013) Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 153, 1340–1353 10.1016/j.cell.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 28. Davé A., Cooley C., Garg M., and Bianchi A. (2014) Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep. 7, 53–61 10.1016/j.celrep.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang H., Liu H., Chen Y., Yang X., Wang P., Liu T., Deng M., Qin B., Correia C., Lee S., Kim J., Sparks M., Nair A. A., Evans D. L., Kalari K. R., et al. (2016) A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun. 7, 10201 10.1038/ncomms10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boersma V., Moatti N., Segura-Bayona S., Peuscher M. H., van der Torre J., Wevers B. A., Orthwein A., Durocher D., and Jacobs J. J. (2015) MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 521, 537–540 10.1038/nature14216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uno S., You Z., and Masai H. (2012) Purification of replication factors using insect and mammalian cell expression systems. Methods 57, 214–221 10.1016/j.ymeth.2012.06.016 [DOI] [PubMed] [Google Scholar]
- 32. Uno S., and Masai H. (2011) Efficient expression and purification of human replication fork-stabilizing factor, Claspin, from mammalian cells: DNA-binding activity and novel protein interactions. Genes Cells 16, 842–856 10.1111/j.1365-2443.2011.01535.x [DOI] [PubMed] [Google Scholar]
- 33. Suh M. H., Ye P., Datta A. B., Zhang M., and Fu J. (2005) An agarose-acrylamide composite native gel system suitable for separating ultra-large protein complexes. Anal. Biochem. 343, 166–175 10.1016/j.ab.2005.05.016 [DOI] [PubMed] [Google Scholar]
- 34. Choi K. H., Farrell A. S., Lakamp A. S., and Ouellette M. M. (2011) Characterization of the DNA binding specificity of Shelterin complexes. Nucleic Acids Res. 39, 9206–9223 10.1093/nar/gkr665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Erickson H. P. (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 10.1007/s12575-009-9008-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kan Z. Y., Lin Y., Wang F., Zhuang X. Y., Zhao Y., Pang D. W., Hao Y. H., and Tan Z. (2007) G-quadruplex formation in human telomeric (TTAGGG)4 sequence with complementary strand in close vicinity under molecularly crowded condition. Nucleic Acids Res. 35, 3646–3653 10.1093/nar/gkm203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng K. W., Chen Z., Hao Y. H., and Tan Z. (2010) Molecular crowding creates an essential environment for the formation of stable G-quadruplexes in long double-stranded DNA. Nucleic Acids Res. 38, 327–338 10.1093/nar/gkp898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sengar A., Heddi B., and Phan A. T. (2014) Formation of G-quadruplexes in poly-G sequences: structure of a propeller-type parallel-stranded G-quadruplex formed by a G15 stretch. Biochemistry 53, 7718–7723 10.1021/bi500990v [DOI] [PubMed] [Google Scholar]
- 39. Agrawal P., Hatzakis E., Guo K., Carver M., and Yang D. (2013) Solution structure of the major G-quadruplex formed in the human VEGF promoter in K+: insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 41, 10584–10592 10.1093/nar/gkt784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu S. T., Varnai P., Bugaut A., Reszka A. P., Neidle S., and Balasubramanian S. (2009) A G-rich sequence within the c-kit oncogene promoter forms a parallel G-quadruplex having asymmetric G-tetrad dynamics. J. Am. Chem. Soc. 131, 13399–13409 10.1021/ja904007p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ambrus A., Chen D., Dai J., Jones R. A., and Yang D. (2005) Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry 44, 2048–2058 10.1021/bi048242p [DOI] [PubMed] [Google Scholar]
- 42. Heddi B., and Phan A. T. (2011) Structure of human telomeric DNA in crowded solution. J. Am. Chem. Soc. 133, 9824–9833 10.1021/ja200786q [DOI] [PubMed] [Google Scholar]
- 43. Schultze P., Macaya R. F., and Feigon J. (1994) Three-dimensional solution structure of the thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol. 235, 1532–1547 10.1006/jmbi.1994.1105 [DOI] [PubMed] [Google Scholar]
- 44. Lim K. W., Amrane S., Bouaziz S., Xu W., Mu Y., Patel D. J., Luu K. N., and Phan A. T. (2009) Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two G-tetrad layers. J. Am. Chem. Soc. 131, 4301–4309 10.1021/ja807503g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dai J., Chen D., Jones R. A., Hurley L. H., and Yang D. (2006) NMR solution structure of the major G-quadruplex structure formed in the human BCL2 promoter region. Nucleic Acids Res. 34, 5133–5144 10.1093/nar/gkl610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Phan A. T., Kuryavyi V., Luu K. N., and Patel D. J. (2007) Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucleic Acids Res. 35, 6517–6525 10.1093/nar/gkm706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heddi B., Cheong V. V., Martadinata H., and Phan A. T. (2015) Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: solution structure of a peptide-quadruplex complex. Proc. Natl. Acad. Sci. U.S.A. 112, 9608–9613 10.1073/pnas.1422605112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takahara T., Hara K., Yonezawa K., Sorimachi H., and Maeda T. (2006) Nutrient-dependent multimerization of the mammalian target of rapamycin through the N-terminal HEAT repeat region. J. Biol. Chem. 281, 28605–28614 10.1074/jbc.M606087200 [DOI] [PubMed] [Google Scholar]
- 49. Baretić D., Berndt A., Ohashi Y., Johnson C. M., and Williams R. L. (2016) Tor forms a dimer through an N-terminal helical solenoid with a complex topology. Nat. Commun. 7, 11016 10.1038/ncomms11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mattarocci S., Reinert J. K., Bunker R. D., Fontana G. A., Shi T., Klein D., Cavadini S., Faty M., Shyian M., Hafner L., Shore D., Thomä N. H., and Rass U. (2017) Rif1 maintains telomeres and mediates DNA repair by encasing DNA ends. Nat. Struct. Mol. Biol. 24, 588–595 10.1038/nsmb.3420 [DOI] [PubMed] [Google Scholar]
- 51. Radivojac P., Iakoucheva L. M., Oldfield C. J., Obradovic Z., Uversky V. N., and Dunker A. K. (2007) Intrinsic disorder and functional proteomics. Biophys. J. 92, 1439–1456 10.1529/biophysj.106.094045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fuxreiter M., Simon I., and Bondos S. (2011) Dynamic protein-DNA recognition: beyond what can be seen. Trends Biochem. Sci. 36, 415–423 10.1016/j.tibs.2011.04.006 [DOI] [PubMed] [Google Scholar]
- 53. Vuzman D., and Levy Y. (2012) Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol. Biosyst. 8, 47–57 10.1039/C1MB05273J [DOI] [PubMed] [Google Scholar]
- 54. Oldfield C. J., and Dunker A. K. (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 83, 553–584 10.1146/annurev-biochem-072711-164947 [DOI] [PubMed] [Google Scholar]
- 55. Adrian M., Ang D. J., Lech C. J., Heddi B., Nicolas A., and Phan A. T. (2014) Structure and conformational dynamics of a stacked dimeric G-quadruplex formed by the human CEB1 minisatellite. J. Am. Chem. Soc. 136, 6297–6305 10.1021/ja4125274 [DOI] [PubMed] [Google Scholar]
- 56. Phan A. T., Kuryavyi V., Ma J. B., Faure A., Andréola M. L., and Patel D. J. (2005) An interlocked dimeric parallel-stranded DNA quadruplex: a potent inhibitor of HIV-1 integrase. Proc. Natl. Acad. Sci. U.S.A. 102, 634–639 10.1073/pnas.0406278102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Burge S., Parkinson G. N., Hazel P., Todd A. K., and Neidle S. (2006) Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 34, 5402–5415 10.1093/nar/gkl655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hänsel-Hertsch R., Beraldi D., Lensing S. V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M., Tannahill D., and Balasubramanian S. (2016) G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 48, 1267–1272 10.1038/ng.3662 [DOI] [PubMed] [Google Scholar]
- 59. Biffi G., Tannahill D., McCafferty J., and Balasubramanian S. (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 5, 182–186 10.1038/nchem.1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huppert J. L., and Balasubramanian S. (2005) Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 33, 2908–2916 10.1093/nar/gki609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Todd A. K., Johnston M., and Neidle S. (2005) Highly prevalent putative quadruplex sequence motif in human DNA. Nucleic Acids Res. 33, 2901–2907 10.1093/nar/gki553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chambers V. S., Marsico G., Boutell J. M., Di Antonio M., Smith G. P., and Balasubramanian S. (2015) High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 33, 877–881 10.1038/nbt.3295 [DOI] [PubMed] [Google Scholar]
- 63. Henderson A., Wu Y., Huang Y. C., Chavez E. A., Platt J., Johnson F. B., Brosh R. M. Jr, Sen D., and Lansdorp P. M. (2014) Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 42, 860–869 10.1093/nar/gkt957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Foti R., Gnan S., Cornacchia D., Dileep V., Bulut-Karslioglu A., Diehl S., Buness A., Klein F. A., Huber W., Johnstone E., Loos R., Bertone P., Gilbert D. M., Manke T., Jenuwein T., and Buonomo S. C. (2016) Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 61, 260–273 10.1016/j.molcel.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kanoh J., and Ishikawa F. (2001) spRap1 and spRif1, recruited to telomeres by Taz1, are essential for telomere function in fission yeast. Curr. Biol. 11, 1624–1630 10.1016/S0960-9822(01)00503-6 [DOI] [PubMed] [Google Scholar]
- 66. Tazumi A., Fukuura M., Nakato R., Kishimoto A., Takenaka T., Ogawa S., Song J. H., Takahashi T. S., Nakagawa T., Shirahige K., and Masukata H. (2012) Telomere-binding protein Taz1 controls global replication timing through its localization near late replication origins in fission yeast. Genes Dev. 26, 2050–2062 10.1101/gad.194282.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hiraga S. I., Ly T., Garzón J., Hořejší Z., Ohkubo Y. N., Endo A., Obuse C., Boulton S. J., Lamond A. I., and Donaldson A. D. (2017) Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 18, 403–419 10.15252/embr.201641983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sukackaite R., Cornacchia D., Jensen M. R., Mas P. J., Blackledge M., Enervald E., Duan G., Auchynnikava T., Köhn M., Hart D. J., and Buonomo S. B. C. (2017) Mouse Rif1 is a regulatory subunit of protein phosphatase 1 (PP1). Sci. Rep. 7, 2119 10.1038/s41598-017-01910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dan J., Liu Y., Liu N., Chiourea M., Okuka M., Wu T., Ye X., Mou C., Wang L., Wang L., Yin Y., Yuan J., Zuo B., Wang F., Li Z., et al. (2014) Rif1 maintains telomere length homeostasis of ESCs by mediating heterochromatin silencing. Dev. Cell 29, 7–19 10.1016/j.devcel.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li P., Wang L., Bennett B. D., Wang J., Li J., Qin Y., Takaku M., Wade P. A., Wong J., and Hu G. (2017) Rif1 promotes a repressive chromatin state to safeguard against endogenous retrovirus activation. Nucleic Acids Res. 45, 12723–12738 10.1093/nar/gkx884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang Z. Z., Pannunzio N. R., Hsieh C. L., Yu K., and Lieber M. R. (2014) The role of G-density in switch region repeats for immunoglobulin class switch recombination. Nucleic Acids Res. 42, 13186–13193 10.1093/nar/gku1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moriyama K., Yoshizawa-Sugata N., Obuse C., Tsurimoto T., and Masai H. (2012) Epstein-Barr nuclear antigen 1 (EBNA1)-dependent recruitment of origin recognition complex (Orc) on oriP of Epstein-Barr virus with purified proteins: stimulation by Cdc6 through its direct interaction with EBNA1. J. Biol. Chem. 287, 23977–23994 10.1074/jbc.M112.368456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sreesankar E., Senthilkumar R., Bharathi V., Mishra R. K., and Mishra K. (2012) Functional diversification of yeast telomere associated protein, Rif1, in higher eukaryotes. BMC Genomics 13, 255 10.1186/1471-2164-13-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Siegel L. M., and Monty K. J. (1966) Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta 112, 346–362 10.1016/0926-6585(66)90333-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.