

Abstract
CD16a/Fc γ receptor IIIa is the most abundant antibody Fc receptor expressed on human natural killer (NK) cells and activates a protective cytotoxic response following engagement with antibody clustered on the surface of a pathogen or diseased tissue. Therapeutic monoclonal antibodies (mAbs) with greater Fc-mediated affinity for CD16a show superior therapeutic outcome; however, one significant factor that promotes antibody–CD16a interactions, the asparagine-linked carbohydrates (N-glycans), remains undefined. Here, we purified CD16a from the primary NK cells of three donors and identified a large proportion of hybrid (22%) and oligomannose N-glycans (23%). These proportions indicated restricted N-glycan processing and were unlike those of the recombinant CD16a forms, which have predominantly complex-type N-glycans (82%). Tethering recombinant CD16a to the membrane by including the transmembrane and intracellular domains and via coexpression with the Fc ϵ receptor γ–chain in HEK293F cells was expected to produce N-glycoforms similar to NK cell–derived CD16a but yielded N-glycoforms different from NK cell–derived CD16a and recombinant soluble CD16a. Of note, these differences in CD16a N-glycan composition affected antibody binding: CD16a with oligomannose N-glycans bound IgG1 Fc with 12-fold greater affinity than did CD16a having primarily complex-type and highly branched N-glycans. The changes in binding activity mirrored changes in NMR spectra of the two CD16a glycoforms, indicating that CD16a glycan composition also affects the glycoprotein's structure. These results indicated that CD16a from primary human NK cells is compositionally, and likely also functionally, distinct from commonly used recombinant forms. Furthermore, our study provides critical evidence that cell lineage determines CD16a N-glycan composition and antibody-binding affinity.
Keywords: post-translational modification (PTM), antibody, glycomics, glycosylation, protein secretion, Fc γ receptor, Fc γ receptors IIIa, N-glycan, protein glycosylation
Introduction
Antibodies activate a diverse set of protective responses following the recognition of a previously-encountered epitope. Therapeutic monoclonal antibodies (mAbs) circumvent prior exposure to induce a protective response against diseased tissue by eliciting the same protective mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC)3 through binding Fc γ receptors (FcγRs), and represent a major drug class worth tens of billions of dollars each year in global sales. mAbs are predominantly built from immunoglobulin G (IgG), and a large proportion bind FcγRs expressed on a range of leukocytes to initiate target destruction. However, very little is known about the composition of FcγRs from native leukocytes and how this composition affects IgG binding and thus treatment efficacy.
ADCC is predominantly performed by natural killer (NK) cells (1), and cytolytic lymphocytes present as 1–6% of circulating leukocytes (2, 3) that express FcγRIIIa/CD16a as the predominant FcγR. The activating role of CD16a in ADCC by NK cells is well-established (1). Indeed, investigations of rituximab, trastuzumab, and cetuximab therapeutics demonstrated the importance of CD16a on NK cells for success (4).
Improved therapeutic mAb efficacy can be achieved by enhancing the mAb–CD16a interaction. In vitro binding studies demonstrated the less common CD16a Val-158 allotype binds IgG1 with ∼5-fold greater affinity compared with CD16a Phe-158 (5). Furthermore, increased binding leads to an increased NK cell response and increased ADCC (6–9). Current advances in mAb therapy are targeting optimization of NK cell ADCC activity by Fc glycoengineering to improve CD16a binding (4). These efforts include targeted modifications to the asparagine-linked carbohydrate (N-glycan) attached to Asn-297 of the IgG1 heavy chain (9, 11). A glycoengineered mAb performed better in the clinic when compared with a non-optimized version (4, 12).
N-Glycans contribute to protein function and are targets for new therapies or enhancing existing therapies. N-Glycan processing occurs during protein secretion. The types and extent of modifications are not template-driven; thus, it is not currently possible to predict N-glycan composition from DNA sequence information. Following the en bloc transfer of a 14-residue precursor N-glycan as directed by the presence of an NX(S/T) sequon, where X is any residue except proline, the glycoprotein associates with protein folding-specific chaperones, and if properly folded, it continues through the ER and Golgi-mediated secretory pathway to encounter remodeling glycosyl-hydrolase and glycosyltransferase enzymes (Fig. 1). Although many factors contributing to N-glycan composition on any given protein remain undefined, it is generally believed that modifying enzyme expression, protein structure, and the presence or absence of a membrane anchor affect N-glycan composition in the secreted protein (13). N-Glycans that experience minimal processing emerge from the cell as oligomannose forms (Fig. 1B). More highly processed N-glycans may also include hybrid forms and the predominant complex types found on cell surfaces and circulatory serum proteins. In addition to folding, N-glycans promote secretion, protect against proteolysis, reduce antigenicity, extend serum half-life, and contribute specific epitopes for receptor binding as well as impact protein structure and function (14–16).
Figure 1.
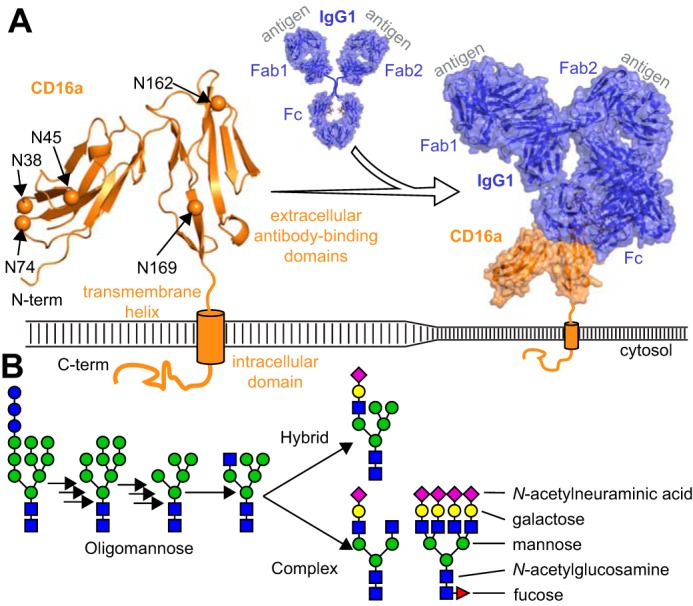
Membrane-anchored CD16a binds IgG antibodies. A, CD16a (orange ribbon) contains five potential N-glycosylation sites in the extracellular domains (orange spheres). B, N-glycan processing occurs in the ER and Golgi following transfer of a large glycan en bloc to the protein and produces oligomannose, hybrid, and complex-type N-glycans. Individual carbohydrate residues are colored according to the SNFG convention (14).
The CD16a sequence encodes five N-glycosylation sites, although little is known about either the N-glycan composition or how CD16a N-glycans impact function. Edberg and Kimberly (17) report that NK cell CD16a contained a high degree of oligomannose-type N-glycans using immobilized lectin chromatography and found little evidence for complex or hybrid types. This characterization was in stark contrast to later mass spectrometry-based reports on the glycosylation of recombinant CD16A, which identified predominantly complex types with low percentages (2–5%) of oligomannose N-glycans (18–21). However, these recombinant expressions did not include the membrane anchor and intracellular domains nor the coexpressed Fc ϵ receptor γ- or CD3 ζ-chains that may influence N-glycan processing. It is well-known that N-glycan composition can influence protein function (15, 22–24), with some evidence indicating composition might affect the antibody-binding function of CD16a (21). Thus, the in vitro analyses of monoclonal antibody binding by CD16a should include proper N-glycoforms for representative results. At this point, there has not been a report on the high-resolution N-glycan composition of CD16a from primary human leukocytes.
Access to a sufficient quantity of primary human tissue represents a significant barrier to high-resolution composition studies because CD16a is expressed by a limited subset of leukocytes, including natural killer cells and CD16a+ monocytes that constitute 1–6 and 0.4–2.4% of circulating leukocytes, respectively (3, 25). We predict that it would require as much as 1.25 liters of human blood to recover a minimum of 100 × 106 natural killer cells and 1 μg of CD16a from a single donor to provide sufficient material for characterization. It is untenable to harvest one-quarter or more of a donor's blood volume for obvious reasons. The adoption of apheresis systems by blood donation centers to collect plasma, platelets, and other blood components provides a valuable source of viable primary human lymphocytes from peripheral blood through leukocyte reduction system (LRS) filters that are normally discarded following donation (26). In this study, we describe a method to purify CD16a from primary human NK cells from a single donor to analyze N-glycan composition.
Results
NK cell isolation and CD16 purification
Light scattering of isolated NK cells combined with CD56 and CD16 staining assessed by flow cytometry revealed >90% cell purity, >87% viability, and a high degree of CD16 expression (Fig. 2 and Fig. S1). A blot of NK cell lysate probed with an anti-CD16 antibody showed a band from 53 to 59 kDa (Fig. 2C). Despite differences in CD16 staining intensity measured from more than a dozen donors, NK cell lysate repeatedly showed indistinguishable CD16 migration characteristics (data not shown). The mobility of the NK cell CD16 band in an SDS-PAGE experiment increased following digestion with PNGase F, an enzyme that specifically removes N-glycans. However, the resulting two bands at ∼33 kDa are still larger than expected for the 27-kDa CD16a polypeptide (Fig. 2C). This treatment indicates that N-glycans retard CD16 mobility in a polyacrylamide gel. The CD16 identified here is consistent with CD16 from multiple previous reports based on migration in SDS-PAGE (27–30).
Figure 2.
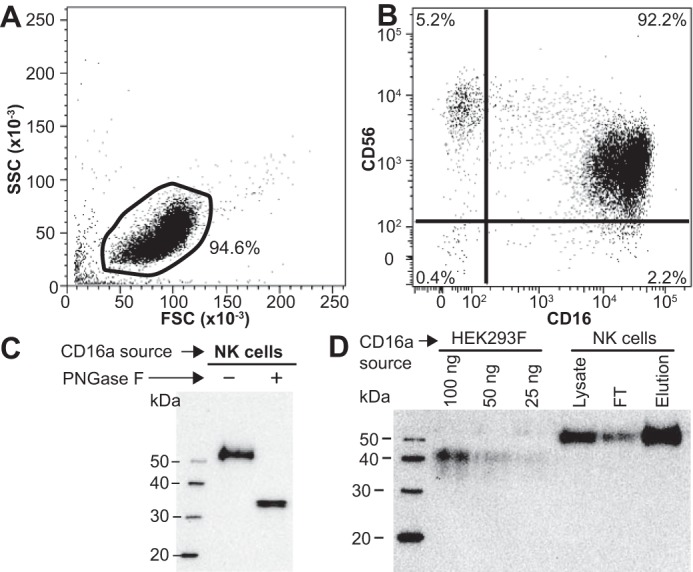
Representative NK cell isolation and CD16a purification. A, light scattering; B, double-antibody staining of negatively-selected NK cells. Isotype and negative-staining controls are shown in Fig. S1. C, anti-CD16 Western blot of PNGase F-digested CD16a shows an increase in mobility following N-glycan removal. D, anti-CD16 Western blot of CD16a purification from an NK cell lysate compared with recombinant CD16a truncated at the transmembrane domain (HEK293F).
A monoclonal anti-CD16 mouse IgG1 antibody, 3G8 (31), is widely used for flow cytometry but not for Western blotting applications because CD16 denaturation destroys the epitope (data not shown). Thus, 3G8 is a good candidate to precipitate folded and processed CD16 from cell lysates at a preparative scale. Our purification scheme first lyses NK cells in detergent, followed by incubation with protein G resin to remove IgG that may obscure the CD16 epitope recognized by the 3G8 antibody, adsorption to a 3G8-agarose resin, extensive washing to remove material that weakly interacts with the resin, and finally elution with 45:55:0.1 water/acetonitrile/TFA (Fig. S2). This procedure showed clear depletion of CD16 from the NK cell lysate and enrichment in the elution fraction with recovery of ∼1 μg of CD16/donor (Fig. 2D).
CD16a validation with ESI-MS/MS
The observed mobility of CD16 from primary human NK cells in SDS-polyacrylamide gels is similar to previous reports and likely represents similar material, although peptide sequencing or other comparable techniques were not previously applied for validation. Thus, we applied high-resolution peptide sequencing to verify the immunoprecipitated material. Purified and trypsin-digested CD16a from a single donor's NK cells generated multiple polypeptide fragments in an HPLC(C18)-ESI-MS/MS experiment that fit with high confidence to 47% of the CD16a sequence (Fig. 3). One peptide revealed this donor expressed the predominant CD16a Phe-158 allele (Fig. 3D). The donor allotype was also confirmed by cDNA sequencing (data not shown). This analysis did not detect peptides from other proteins nor did it detect peptides containing predicted CD16a N-glycosylation sites, transmembrane regions, or intracellular regions; however, the observed peptides were enough to confirm CD16a identity. It is possible that the immunoprecipitated material includes low levels of contaminating proteins. To address this potential shortcoming, we analyzed PNGase F-treated peptides from a single donor using HPLC(C18)-ESI-MS/MS. This sample contained the Fc ϵ receptor γ-chain and the ubiquitous contaminant keratin from human hair and skin cells. Again, CD16a accounted for a majority of the identified peptides. Surprisingly, human actin and myosin coeluted with CD16a, as well as trace amounts of trypsin and the IgG1 heavy chain. With the exception of the IgG1 heavy chain that leached from the immunoprecipitation column, none of the identified proteins were N-glycoproteins. These results indicate that the PNGase F treatment of the elution fraction will produce only CD16a N-glycans from NK cells if the immunoprecipitation antibody is treated to remove N-glycans prior to immobilization.
Figure 3.

ESI-MS/MS analysis identifies only CD16a following immunoprecipitation from NK cells. A, peptides identified are shown with a blue bar below the CD16a sequence. Expected trypsin cleavage sites are indicated with a vertical line, and N-glycosylation sites are denoted with a red “N.” B–D, MS/MS spectra of select peptides.
N-Glycan analysis of CD16a from primary human NK cells
We isolated peripheral NK cells from three male donors ranging in age from 66 to 78 to characterize N-glycan composition; blood remaining in the LRS filter after apheresis provided genetic material to determine the donor's CD16a allotype (Table 1 and Figs. S3 and S4). Following cell lysis, CD16a immunoprecipitation utilized EndoS-treated 3G8 antibody to eliminate potential contamination with antibody N-glycans (Fig. S5). Analysis of procainamide-modified CD16a N-glycans by hydrophilic interaction liquid chromatography (HILIC)-ESI-MS showed a strong correlation between N-glycan elution time and N-glycan mass (Fig. 4). We identified 33, 27, and 19 CD16a N-glycans from donors 1–3, respectively, with an average mass error of 0.0010 Da for the three datasets (supporting Spreadsheet). The CD16a glycans contained minimally processed forms, including a high percentage of oligomannose (23%) and hybrid types (22%; Fig. 5). Of the remaining complex-type N-glycans (55%), biantennary forms comprised more than half of the complex-type species (51%). CD16a N-glycan fucosylation proved pervasive with 63% of the total and 89% of the complex-type N-glycans containing this modification (Fig. 5B). CD16a N-glycans also revealed a high degree of sialylation with 100% of the hybrid and 78% of the complex-type forms displaying at least one N-acetylneuraminic acid residue (Fig. 5C).
Table 1.
NK cell donor information
| Donor 1 | Donor 2 | Donor 3 | |
|---|---|---|---|
| Year of birth | 1947 | 1951 | 1939 |
| Sex | M | M | M |
| ABO group | A | A | A |
| Rh(D) | + | − | + |
| CD16a allotype | V/F | V/V | V/F |
| Collection date | 5/31/17 | 5/18/17 | 5/18/17 |
| NK cell count (106) | 206 | 50 | 113 |
| NK cell viability | 93% | 83% | 86% |
| CD56+ in NK cell preparation | 83.8% | 94.1% | 95.6% |
Figure 4.

N-Glycans identified from donor 1 NK cell CD16a. The retention time from HILIC for the procainamide-derivatized N-glycans is plotted versus the exact mass. Insets for three N-glycans show MS identification. Carbohydrate diagrams show one possible configuration of the N-glycan species; isobaric species were not distinguished.
Figure 5.

Comparison of CD16a N-glycan characteristics from different sources. A, pie charts indicate what percentage of identified N-glycans fall into each of the three categories; extracted bar graphs show the breakdown of branching types for the complex-type N-glycans only. B, percentage of each N-glycan species that contained at least one fucose residue. C, percentage of each N-glycan species that contained at least one N-acetylneuraminic acid residue.
Analysis of the individual glycoforms corresponding to the 10 most intense N-glycan species identified by MS showed complex-type, core-fucosylated, and sialylated biantennary N-glycans produced the two strongest signals in spectra of CD16a N-glycans from all three donors (Fig. 6). However, the next eight most intense ion species from each donor were predominantly hybrid forms (13/24) with comparable levels of complex-type (6) and oligomannose (5) forms, including Man9 (2), Man6 (2), and Man5 (1). This prevalence of N-glycan species with restricted processing in the three datasets is surprising given the reports of N-glycans from recombinant CD16a that are primarily of complex type with a high degree of branching (21).
Figure 6.

Possible N-glycan configurations corresponding to the most intense ions are shown from each CD16a source. Carbohydrate diagrams show one possible organization of the N-glycans; isobaric species were not distinguished. Values in the lower right corner of each cell indicate the relative ion intensities of these species, compared with the most abundant species from the same source.
N-Glycan analysis of recombinant CD16a from HEK293 cells
It is possible that CD16a N-glycans isolated from NK cells differ from recombinant CD16a N-glycans because the soluble recombinant fragment lacks the membrane anchor and experiences a different environment during secretion, or that NK cells produce a different mixture of glycan-modifying enzymes, or a combination of both possibilities. We expressed CD16a in HEK293F cells to assess the impact of the transmembrane and intracellular domains on N-glycan composition using two forms: soluble recombinant CD16a that contains the two extracellular antibody-binding domains (srCD16a) and full-length recombinant CD16a containing the complete CD16a ORF, including the signal peptide, coexpressed with the Fc ϵ receptor γ-chain (frCD16a). It is important to note that recombinant expression of frCD16a requires coexpression with the Fc ϵ receptor γ-chain or CD3 ζ-chain to achieve cell-surface localization (data not shown) (32).
Migration in SDS-PAGE of srCD16a (28–46 kDa) and frCD16a (38–43 and 47–59 kDa) showed much broader migration distributions than CD16a from NK cells (53–59 kDa), indicating a greater degree of heterogeneity for the recombinant forms (Fig. S6). We identified 122 and 86 individual procainamide-derivatized srCD16a and frCD16a N-glycans, respectively, with an average mass error of 0.0016 Da (Fig. S7). Predominant N-glycans species from srCD16a and frCD16a were of a complex-type (82 and 73%, respectively) with low levels of oligomannose forms (5 and 9%, respectively), which stands in marked contrast to NK cell CD16a (Fig. 5 and supporting Spreadsheet). Furthermore, the complex types showed much greater representation of tri- and tetra-antennary forms than NK cell CD16a. Fucosylation of all srCD16a and frCD16a species was comparable (71%), and both were greater than NK cell CD16a (63%). Furthermore, fewer srCD16a and frCD16a N-glycans contained N-acetylneuraminic acid when compared with NK cell CD16a. This characterization of srCD16a is highly comparable with expressions in NS0 cells (19) with 6% oligomannose (Man4–6) or BHK cells with no oligomannose (33).
Closer inspection of N-glycan species revealed stark differences between the recombinant forms. The most intense N-glycan signals from srCD16a included a majority of complex-type forms (9/10) in contrast to frCD16a (6/10) that included a significant contribution from oligomannose forms (4/10; including Man9, Man8, Man7, and Man5; Fig. 6). Furthermore, complex-type glycans from frCD16a showed a low level of hexose incorporation (2/6; likely galactose) in contrast to srCD16a with a very high degree of branching (7/9 w/3+ branches) and hexose incorporation (8/9). By these measures, the CD16a membrane anchor and Fc ϵ receptor γ-chain coexpression resulted in the synthesis of a higher degree of hybrid and oligomannose N-glycans, but neither the soluble nor membrane-anchored recombinant CD16a forms recapitulated the N-glycan species found on NK cell CD16a. It is important to note that by using a detergent to solubilize the cell membrane, folded but incompletely-processed CD16a residing in the ER and the Golgi at the moment of cell lysis will be purified in addition to CD16a from the cell surface. However, frCD16a was extracted using the same technique as NK cell CD16a, and thus a direct comparison between these two sources is justifiable. Furthermore, the impact of incompletely-processed CD16a from the ER and Golgi cannot alone account for the increased prevalence of hybrid forms when compared with frCD16a or srCD16a because 100% of the NK cell CD16a hybrid glycoforms are sialylated, a hallmark of late-stage N-glycan processing.
Oligomannose N-glycans increase receptor affinity
The difference between CD16a N-glycan species from primary NK cells and recombinant expression systems appears compelling because both cell lineage and protein sequence impacted N-glycan remodeling. N-Glycan composition can likewise impact protein function, as is the case for IgG1, but it is unknown how composition affects the antibody-binding function of CD16a. To test whether N-glycan composition contributes to CD16a function, we expressed srCD16a in a HEK293S cell line that makes only oligomannose N-glycans with Man5 as the predominant species to compare binding to HEK293F-expressed srCD16a, characterized above, with predominantly complex-type and highly-branched N-glycans (Figs. 5 and 6) (34).
CD16a with complex-type N-glycans bound IgG1 Fc in the G0F glycoform with a KD of 310 ± 100 nm and 12-fold less affinity than CD16a with oligomannose N-glycans (25 ± 17 nm; Fig. 7). Furthermore, two of the five CD16a N-glycans previously identified as positive contributors to antibody binding, Asn-45 and Asn-162 (35–38), but not the remaining three N-glycans at positions 38, 74, and 169, were required for the enhanced antibody binding by CD16a displaying oligomannose N-glycans. Removing CD16a N-glycans at positions 45 and 162 by Asn to Gln mutations abolished enhancement from the oligomannose N-glycans. These data indicate that composition of the Asn-45 and Asn-162 CD16a N-glycans impact function.
Figure 7.

CD16a N-glycan composition affects IgG1 Fc binding. A and B, representative SPR sensograms for IgG1 Fc with an agalactosyl core-fucosylated complex-type biantennary N-glycan (G0F) binding to recombinant CD16a with complex-type N-glycans (A) or oligomannose N-glycans (B). C, summary of dissociation constants for CD16a variants. Material was expressed either with HEK293F cells to synthesize primarily complex-type N-glycans (red bars) or with HEK293S cells to synthesize only oligomannose N-glycans (green bars). Values represent the average of at least two independent experiments, and error bars are ± the maximum error of the fitting.
NMR of different srCD16a N-glycoforms
Our laboratory previously observed that different IgG1 Fc N-glycoforms led to different CD16a binding. These changes correlated with differences in cross-peak positions observed with solution NMR spectroscopy, and these differences reflected changes in secondary structure for one IgG1 Fc loop (39). We applied a similar approach and incorporated selective backbone 15N labels into Tyr, Lys, and Phe residues to probe whether N-glycan composition affected 1H-15N heteronuclear single quantum coherence (HSQC) spectra of srCD16a. The 26 Tyr, Phe, and Lys residues are evenly distributed throughout both CD16a extracellular domains. We expect changes in peak positions as these amino acid types will reflect CD16a structural changes in a manner analogous to characterizing ligand binding through NMR cross-peaks that change position during a titration. Indeed, a spectrum of srCD16a expressed in HEK293S cells with oligomannose N-glycans showed different cross-peak positions than srCD16a with complex-type N-glycans (Fig. 8). The largest deviations appeared in the center of the spectrum. We observed 24 peaks, which is close to the 26 expected, and of these, eight showed different cross-peak positions in the two spectra with a combined 1H/15N chemical shift perturbation for each peak of >0.04 ppm as calculated with Equation 2. Furthermore, one peak present in the spectrum of CD16a with complex-type N-glycans was absent in the spectrum of CD16a with oligomannose N-glycans, and two peaks appeared in the spectrum of the oligomannose form. It is important to note that only two of the 26 labeled residues are within five amino acid residues, by primary sequence, from N-glycosylation sites, indicating extensive regions of the protein are affected by N-glycan composition. This result is substantially different from hIgG1 Fc, where changes are limited to a single loop and not observed throughout the protein (39).
Figure 8.
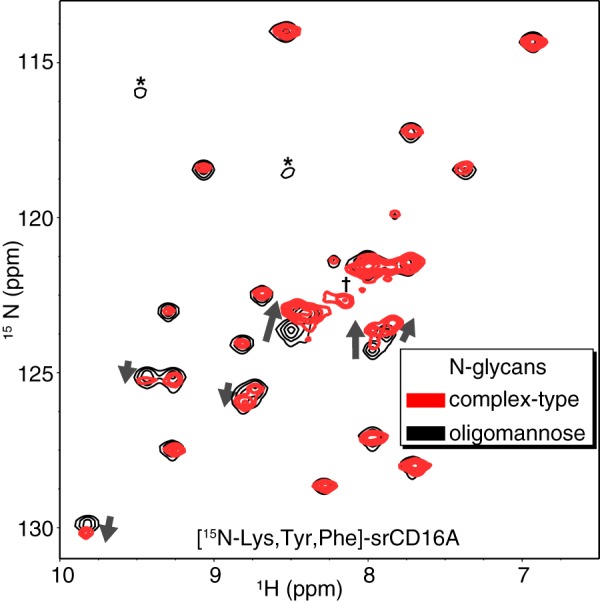
CD16a N-glycan composition impacts polypeptide structure. Select backbone amide cross-peaks observed in an HSQC experiment using CD16a expressed with either complex-type (red contours) or oligomannose (black contours) N-glycans. One peak present in the spectrum of CD16a with complex-type N-glycans but absent in the oligomannose form is indicated (†). Two peaks appearing in the spectrum with oligomannose N-glycans are indicated (*).
Discussion
A high percentage of oligomannose and hybrid structures, as well as biantennary forms constituting the largest subgroup of the complex-type species, characterize the CD16a N-glycans from primary human NK cells and indicate restricted processing compared with many serum proteins. This limited processing of NK cell CD16a N-glycans stands in stark contrast to N-glycans from recombinant CD16a with highly-modified and highly-branched carbohydrate structures. Furthermore, we provide clear evidence that N-glycan composition impacts antibody binding: CD16a modified with minimally processed oligomannose N-glycans bound IgG1 Fc with 12-fold greater affinity than CD16a with a mixture of highly-processed N-glycans.
Recombinant CD16a is often used to characterize antibody-binding affinity in many reports, including some of our own, due to practical considerations that include isolating sufficient material for in vitro studies. The differences in N-glycan composition from these different sources highlight the need to describe and develop the tools to mimic native source-specific composition in studies of receptor function. N-Glycan composition is a neglected component of eukaryotic biology, particularly in the context of cell-surface proteins, because access to primary materials is limited, and the absolute mass of each specific receptor on a cell surface is very low (we estimate ∼20–40 fg CD16a/NK cell). Furthermore, detailed studies linking N-glycan composition with function are challenging and likewise represent a barrier to a complete characterization of N-glycan roles in protein function. The characterization presented here provides a detailed description of these variables for CD16a from NK cells. N-Glycosylation of CD16a was targeted due to its relevance in mAb therapy, but the other Fc γ receptors are heavily glycosylated. This approach can be used to characterize N-glycosylation profiles of other Fc γ receptors from primary human tissues.
The relatively limited distribution of migration rates for NK cell CD16a in polyacrylamide gels and the bias toward N-glycoforms with restricted processing were striking in comparison with frCD16a from HEK293F cells and indicate cell type-specific differences in N-glycan processing. Both frCD16a and NK cell CD16a were coexpressed with a coreceptor that is required for localization to the cell surface. Differences in primary structure do not account for the observed differences in N-glycan processing: the CD16a polypeptide sequence and the presence of the coexpressed Fc ϵ receptor γ-chain were the same in both NK cells and that used in HEK293F cells to express frCD16a. Cell type-specific differences in the glycosylation of a specific protein are known (40–44) and are justifiable given the complexity of gene expression and activity for glycan-processing enzymes in different tissues (45–50).
Changes in receptor function resulting from limited N-glycan processing were mirrored by changes in a 2D spectrum of [15N]Lys/Tyr/Phe-labeled CD16a (Figs. 7 and 8). The magnitude of peak shifts and the extent of peaks experiencing shifts in CD16a are comparable with analyses of proteins that experience a conformational rearrangement upon ligand binding. In the latter example, ligand docking stabilizes a specific protein conformation. Surprisingly, CD16a N-glycan composition imposed a similar conformational adjustment that was experienced by a large number of backbone amides, indicating that specific features of the N-glycans interact with residues on the polypeptide surface. Our group recently described the formation of intramolecular interactions between the polypeptide and N-glycans at Asn-45 and Asn-162 (38). N-Glycans at these positions likely differentially impact protein conformation, based on N-glycan composition at each site. Thus, the CD16a N-glycans do not behave as largely inert modifications, but rather they interact with polypeptide residues to modulate protein function. The IgG1 Fc N-glycan composition also impacts receptor binding through intramolecular interactions (15, 39, 51, 52).
Numerous prior studies focused on the modulation of CD16a affinity by IgG N-glycan composition. This wealth of information on IgG N-glycosylation is due primarily to the availability of IgG; here, we focused on the effect of CD16a N-glycosylation on antibody binding. The observation that CD16a N-glycosylation affects binding affinity, potentially to a greater extent than IgG, makes CD16a N-glycosylation an important aspect of immune system function with the potential for clinical implications. There are two important limitations to this study. 1) The restricted donor demographic variability could mask potential N-glycan composition variability present in the larger population. 2) It is currently unclear whether changes to CD16a N-glycan composition that increase antibody binding in SPR experiments enhance NK cell response. Moreover, CD16a-expressing cells, including NK cells, are involved with multiple pathologies (53–56). Therefore, this information regarding specific NK cell N-glycans on CD16a can be applied to probe the connection between patient-specific changes in autoimmune conditions, malignancy, or infection.
Experimental procedures
All materials were purchased from Sigma unless noted otherwise.
Cell isolation
NK cells were isolated from leukocyte reduction filters (LRS filters), obtained from LifeServe (Ames, IA) and DeGowin Blood Centers (Iowa City, IA), after the plateletpheresis procedure. Donors signed consent forms permitting the use of donated blood products for research purposes. We do not directly enroll donors because we obtain materials from the LifeServe and DeGowin Blood Centers. Cell isolation procedures were performed within 3 h of the time the donor completed the apheresis procedure. NK cells were isolated by negative selection using the RosetteSep Enrichment Mixture following the recommended protocol (Stem Cell Tech) with the following minor changes: LRS filters were drained into a sterile 50-ml tube, and the contents were diluted to 30 ml using PBS with 2% IgG-depleted FBS. The cell suspension was then split into 2 × 15-ml aliquots, and each was mixed with 750 μl of the negative selection mixture. After a 20-min incubation, the steps mentioned in the protocol were followed. Contaminating erythrocytes were then removed by incubating the cells with erythrocyte lysis buffer (155 mm ammonium chloride, 12 mm sodium bicarbonate, and 0.1 mm EDTA, pH 7.4) for 10 min at room temperature. Finally, cells were counted, and viability was analyzed with trypan blue and then frozen at −80 °C. The CD16a allotype was determined by sequencing cDNA corresponding to CD16a (cDNA isolation is described below under the “Recombinant expression”; Iowa State University DNA Facility).
Flow cytometry
NK cells were first blocked with 5 μg of hIgG1 (Athens Research and Technology) before incubation with primary antibodies. For each flow cytometry experiment, 5 × 105 NK cells were stained with primary antibody for 40 min in the dark on ice, including 2 μg/ml mIgG2a anti-hCD56 (MEM-188, BioLegend) and 2 μg/ml mIgG1 anti-hCD16 (3G8) or isotype controls, mIgG1 (Research and Diagnostics Systems) and mIgG2a (BioLegend). The cells were washed with 2× 2 ml of phosphate-buffered saline, 0.05% sodium azide by centrifuging at 600 × g for 8 min, and removing the supernatant after each centrifugation. Fluorophore-conjugated secondary antibodies were added, including anti-mIgG1-APC (RMG1–1, BioLegend) and anti-mIgG2a-PE (RMG2a-62, BioLegend), and incubated on ice for 40 min. Cells were fixed in 1% paraformaldehyde before loading them onto a BD FACSCanto (BD Biosciences). For cell purity assessment, NK cells were gated in the side and forward scatter plot to exclude cell debris and cells (primarily erythrocytes) smaller than lymphocytes. Gating of double-stained cells was determined by comparing with the fluorescence intensity of the negative (no primary) control. Isotype controls consistently showed no positive staining for either primary antibody for the first 15 NK cell isolations performed from donors of both gender and a wide age range.
Anti-hCD16 expression and purification
Open reading frames encoding the anti-hCD16 mouse IgG1 (3G8) heavy and light chains were synthesized (IDT). The heavy chain sequence was cloned into pGEef1Puro vector (provided by Dr. Kelley Moremen, University of Georgia) using the Gateway cloning system (Life Technologies, Inc.). The flanking attB sites for gateway cloning of heavy chain were included in the synthesized heavy chain gene. The transfer of gene to final pGEef1Puro vector was performed in a two-step gateway reaction following the manufacturer's protocol with pDONR221 (Life Technologies, Inc.) as the intermediate vector. The light chain sequence was cloned into the pGEN2 vector using the NotI and HindIII restriction sites (57).
The 3G8 monoclonal antibody was generated by cotransfecting HEK293F cells with 3 μg/ml pGEef1Puro/(heavy chain) and 1.5 μg/ml pGEN2/(light chain) as described (57). Antibody was secreted into the medium, adsorbed to a protein A-Sepharose fast flow resin (GE Healthcare), and eluted with 100 mm glycine, pH 3.0, followed by immediate neutralization with 33% 1 m Tris, pH 8.0. Purified antibody was washed to replace the buffer with a buffer containing 25 mm MOPS, 100 mm sodium chloride, pH 7.2, and then coupled to AminoLink aldehyde-functionalized agarose beads at 1 mg/ml of resin according to the product protocol (Thermo Fisher Scientific).
Antibody used to immunoprecipitate CD16a for N-glycan analysis differed in two important ways. First, the mouse IgG1 Fc region was replaced with human IgG1 Fc. Second, this chimeric antibody was treated with EndoS to remove the Fc N-glycan; 3G8 contains no N-glycosylation sequon in the Fab domains. EndoS was expressed in E. coli using a deposited plasmid and purified as described (AddGene). EndoS was incubated with 3G8 in 20 mm MOPS, 100 mm NaCl, pH 7.2, at a 1:50 (EndoS/3G8) molar ratio for 18 h at room temperature. The digestion was assessed by resolving the protein with SDS-PAGE and staining with Coomassie Blue. Glutathione beads were incubated with 3G8 and GST-EndoS to remove the GST-EndoS before coupling antibody to agarose beads.
Recombinant CD16a expression
The soluble extracellular fragment of CD16a (srCD16a) was prepared as a recombinant GFP-fusion protein and cleaved with tobacco etch virus protease as described previously using HEK293F cells (15). Open reading frames encoding the full-length recombinant CD16a protein (frCD16a) and the Fc ϵ receptor subunit γ (γ-chain) were cloned from cDNA obtained from NK cells and monocytes, respectively. Total RNA was isolated from 10 × 106 monocytes (prepared with negative selection, Stem Cell Tech) and 5 × 106 NK cells using the TRIzol reagent following the manufacturer's protocol (Thermo Fisher Scientific). A high capacity RNA to cDNA kit (Applied Biosystems) was used to reverse-transcribe 500 ng of RNA from each cell type and amplified with AccuPrime Pfx DNA polymerase (Life Technologies, Inc.) to generate full-length cDNA. Primers pairs for frCD16a were 5′-cagcggccgccCAGTGTGGCATCATGTGGCAG and 5′-ggatccaagcttTTTGTCTTGAGGGTCCTTTCT containing NotI and HindIII sites, respectively. Similarly, two primers 5′-gaattcCTCCAGCCCAAGATGATTCCAG and 5′-ggatcctcaCTACTGTGGTGGTTTCTCATGC with EcoRI and BamHI sites, respectively, were used to amplify genes for γ-chain. Primers were designed to include the native signal sequence form both genes. Both PCR fragments were cloned into pGEM-T Easy vectors following the manufacturer's guidelines (Promega). Full-length CD16a was subcloned into pGEN2 vector using the NotI and HindIII restriction sites. The γ-chain was subcloned into pGEef1Puro vector using the EcoRI and BamHI restriction sites. pGEN2-frCD16a and pGEef1Puro-γ-chain vector were cotransfected in a 1:2 mass ratio into the HEK293F cell line as described (57). HEK cells were transfected at a density of 3.0 × 106 cells ml−1 with a viability of >96%. Expressions usually proceeded for 5 days or until cell viability fell below 50%. Typical yields for srCD16a were 0.2 mg protein ml−1 culture medium. Cell-surface expression of CD16a in HEK293F cells was confirmed by flow cytometry using the anti-CD16 mIgG1 (3G8) antibody before freezing the cell pellets at −80 °C
CD16a immunoprecipitation
These methods were adapted from a previously published protocol to achieve a preparative scale purification (30). NK cells or HEK293F cells expressing full-length CD16a were lysed by resuspending the frozen pellet in ice-cold lysis buffer (100 mm Tris, 100 mm sodium chloride, 5 mm EDTA, 5 mm oxidized glutathione, 10 μm potassium ferricyanide, 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride, 10 mg/ml dodecyl maltoside, pH 8.0) with 200 μl added for every 1 × 107 cells by repeated pipetting, followed by a 20-min incubation on ice. Cell debris was removed by centrifugation at 10,000 × g for 10 min at 4 °C. Supernatant was incubated for 1 h at 4 °C on a rocker with 5 μl of protein G-Sepharose (Sigma) per 200 μl of lysate. Protein G-Sepharose was removed by centrifuging the lysate at 500 × g for 5 min. The recovered supernatant was sonicated in a bath sonicator for 1 min, followed by freezing at −80 °C, and slowly thawed on ice. 3G8-coupled agarose beads (20 μl) were added per 200 μl of NK cell lysate and incubated for 1 h at 4 °C on a rocking shaker. The resin was centrifuged at 500 × g for 5 min and washed twice with lysis buffer using 10 times the resin volume for each wash followed by two washes with 50 mm Tris, 100 mm sodium chloride, pH 8.0, and final two washes with 100 mm ammonium carbonate, pH 8.0. CD16 was eluted in 45:55:0.1 (HOH/acetonitrile/TFA) by centrifugation through a spin column to retain the resin.
PNGase F digestion of CD16a
PNGase F digestion of CD16a isolated from NK cells was performed per the manufacturer's protocol. NK cell CD16a bound to 3G8-agarose was boiled at 100 °C for 10 min in 1× denaturing buffer (0.5% SDS, 40 mm dithiothreitol) followed by addition of GlycoBuffer2 and 10% Nonidet P-40 to the reaction (New England Biolabs). After adding 1 μl of PNGase F, the sample was incubated at 37 °C for 4 h, and the reaction was stopped by heating at 95 °C for 5 min after the addition of 1 volume of 2× SDS-PAGE sample buffer containing 10% β-mercaptoethanol. A control reaction with CD16a was treated in an identical manner, except without the addition of PNGase F.
Western blotting
Eluted protein was resolved on an SDS-polyacrylamide gel (12% unless mentioned otherwise) and transferred onto a polyvinylidene difluoride membrane using the one-step electroblotting system (Thermo Fisher Scientific). The membrane was blocked with 5% dry milk in TBS Tween 20 (TBST) buffer for 1 h at room temperature. All incubations and washes were performed on an orbital shaker. Following blocking, the membrane was stained for 18 h at 4 °C with anti-hCD16 (Research and Diagnostic Systems, 0.1 μg/ml) in 5% milk in TBST buffer. The blot was washed four times with TBST buffer for 5 min and stained with secondary anti-goat HRP (Research and Diagnostic Systems, dilution 1:2000) in 5% milk in TBST buffer for 1 h at room temperature. After washing four times, the blot was imaged on a ChemiDoc XRS+ Image system (Bio-Rad) using the ECL Western blotting substrate (Thermo Fisher Scientific).
Peptide mass spectrometry
CD16 for peptide sequencing was isolated as described under “CD16a immunoprecipitation.” In the first instance, CD16a was excised from 12% SDS-PAGE at the same size as detected by the Western blotting detection system. The gel was subjected to standard in-gel trypsin digest on the Molecular Dynamics ProGest (Genomic Solutions) instrument. The sample was then dried down and resuspended in 1.25 μl of buffer B (0.1% formic acid in acetonitrile). In the second instance, peptides retained by the C18 Sep-Pak during the isolation of N-glycans from donor 3 (following treatment with PNGase F and trypsin) were eluted with isopropyl alcohol and lyophilized. Both samples were resuspended in 23.75 μl of buffer A (0.1% formic acid in water). Next, the sample was mixed, spun down, and put into a sample vial. The vial was then loaded onto a Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with a 1260 HPLC (Agilent) and C18 column and eluted as described previously (58). Peaks from MS1 and MS2 spectra were searched with the mammalian Mascot database server. Raw data were processed with Sequest HT (Version 1.17) and Mascot 2.2.07 (Matrix Science). The peptides identified were then analyzed using Proteome Discoverer Version 2.1 software (Thermo Fisher Scientific).
Procainamide labeling of PNGase F-released N-glycans
Following heat denaturation of CD16a for 5 min at 90 °C, 1 μl of sequencing grade trypsin (Promega; 1 mg/ml in 50 mm ammonium carbonate, pH 8.0) was added and incubated at 37 °C for 18 h after mixing. The trypsinized sample was again incubated at 90 °C for 5 min to denature the trypsin, followed by cooling for 5 min on ice. PNGase F (1 μl; glycerol-free; New England Biolabs) was then added to the sample and incubated at 37 °C for 18 h. A Hypersep C18 column (Thermo Fisher Scientific) was washed three times with 1 ml of methanol and then three times with 1 ml of 5% acetic acid. Free N-glycans were isolated by applying the sample, diluted in 5% acetic acid, over the C18 column and collecting the flow- through. Wash fractions (3× 1 ml) of 5% acetic acid were collected in the same tube, then frozen, and lyophilized. The lyophilized sample was then resuspended in 200 μl of double-distilled water by vortexing and transferred to a microcentrifuge tube, frozen, and lyophilized again. The sample was then resuspended in 10 μl of water. A stock solution containing 2 m sodium cyanoborohydride, 1 m procainamide, and 50% acetic acid (10 μl) was added to the sample, mixed, and incubated for 18 h at 37 °C. The sample was then lyophilized and resuspended into 10 μl of double-distilled water.
Hydrophilic interacting chromatography-mass spectrometry
The HPLC instrument used was an Agilent 1260 HPLC with an Acquity UPLC Glycoprotein BEH Amide, 300 Å, 1.7-μm HILIC column (Waters). The lyophilized procainamide-labeled glycan samples were resuspended in double-distilled water, and 9 μl was injected onto the column prewashed with 25% Solvent A (0.1% formic acid and 0.01% TFA in double-distilled water) and 75% Solvent B (0.1% formic acid and 0.01% TFA in acetonitrile) with a flow rate of 0.1 ml/min. The glycans were then eluted by a gradient at 0.1 ml/min flow rate unless otherwise indicated; 0.5–10.5 min (gradient from 25 to 35% A), 10.5–40.5 min (a gradient from to 35 to 50% A), 40.5–41.5 min (50 to 100% A), 41.5–43.5 min (100% A), 46.5–47 min (25% A, 75% B) at 0.05 ml/min and 47–60 min (25% A, 75% B) at 0.1 ml/min at 45 °C. Upon elution, the samples were coupled in-line to a Q ExactiveTM Hybrid Quadrupole-OrbitrapTM mass spectrometer (Thermo Fisher Scientific). The mass spectrometer (scan range 266.7–4000 m/z) subjected ions to higher-energy collisional dissociation (HCD) fragmentation (scan range of 200–2000 m/z) at a normalized collisional energy of 27 eV. In-between samples, identical chromatography and MS analyses were performed following an injection of water. These were scanned to ensure no carryover between samples. Sample spectra were then analyzed by converting the RAW spectra file to text format with MSConvert. The output was then scanned against a list of N-glycans calculated for singly, doubly, and triply charged procainamide-derivatized species to identify peaks with corresponding retention times and intensities. These matches were then validated manually by analyzing the RAW file in XCaliber (Thermo Fisher Scientific). Average mass error for each data set was calculated according to Equation 1.
| (1) |
IgG1 Fc binding assays by SPR
Expression, purification, and preparation of IgG1 Fc in the G0F glycoform and CD16a-binding analyses by surface plasmon resonance were performed as described previously (52). Dissociation constants were fitted to the equilibrium response units for each condition.
NMR spectroscopy
Isotope-labeled srCD16a expression and NMR experiments were conducted as described previously (39). Chemical shift perturbations (CSP) were calculated using Equation 2.
| (2) |
Author contributions
K. R. P., J. T. R., G. P. S., and A. W. B. conceptualization; K. R. P., J. T. R., and G. P. S., investigation; K. R. P., J. T. R., G. P. S., and A. W. B. methodology; K. R. P., J. T. R., G. P. S., and A. W. B. writing-original draft; K. R. P., J. T. R., G. P. S., and A. W. B. writing-review and editing; A. W. B. supervision; A. W. B. funding acquisition; A. W. B. project administration.
Supplementary Material
Acknowledgments
We thank Christine Hayes and the staff of the LifeServe Blood Center (Ames and Des Moines, IA) and the staff of the DeGowin Blood Center (University of Iowa Medical Center) for the Trima LRS filters; Dr. William Alley (Waters) for help with the HILIC separations; Prof. Kelley Moremen (University of Georgia) for the pGEef1 vector; Dr. Shawn Rigby of the Iowa State University Flow Cytometry Facility for flow cytometry analysis; and Joel Nott of the Iowa State University Protein Facility for mass spectrometric analyses.
This work was supported by National Institutes of Health Award R01 GM115489 from NIGMS and by funds from the Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S7 and supporting Spreadsheet.
- ADCC
- antibody-dependent cell mediated cytotoxicity
- NK
- natural killer
- FcγR
- Fc γ receptor
- ER
- endoplasmic reticulum
- LRS
- leukocyte reduction system
- PNGase F
- peptide:N-glycosidase F
- ESI-MS/MS
- electrospray ionization-tandem mass spectrometry
- HILIC
- hydrophilic interaction liquid chromatography
- SPR
- surface plasmon resonance
- HSQC
- heteronuclear single quantum coherence
- EndoS
- IgG-specific endoglycosidase.
References
- 1. Battella S., Cox M. C., Santoni A., and Palmieri G. (2016) Natural killer (NK) cells and anti-tumor therapeutic mAb: unexplored interactions. J. Leukocyte Biol. 99, 87–96 10.1189/jlb.5VMR0415-141R [DOI] [PubMed] [Google Scholar]
- 2. Caligiuri M. A. (2008) Human natural killer cells. Blood 112, 461–469 10.1182/blood-2007-09-077438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lanier L. L., Le A. M., Civin C. I., Loken M. R., and Phillips J. H. (1986) The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 136, 4480–4486 [PubMed] [Google Scholar]
- 4. Guillerey C., Huntington N. D., and Smyth M. J. (2016) Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 17, 1025–1036 10.1038/ni.3518 [DOI] [PubMed] [Google Scholar]
- 5. Dekkers G., Treffers L., Plomp R., Bentlage A. E., de Boer M., Koeleman C. A., Lissenberg-Thunnissen S. N., Visser R., Brouwer M., Mok J. Y., Matlung H., van den Berg T. K., van Esch W. J., Kuijpers T. W., Wouters D., et al. (2017) Decoding the human immunoglobulin G-glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front. Immunol. 8, 877 10.3389/fimmu.2017.00877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mellor J. D., Brown M. P., Irving H. R., Zalcberg J. R., and Dobrovic A. (2013) A critical review of the role of Fc γ receptor polymorphisms in the response to monoclonal antibodies in cancer. J. Hematol. Oncol. 6, 1 10.1186/1756-8722-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koene H. R., Kleijer M., Algra J., Roos D., von dem Borne A. E., and de Haas M. (1997) Fc γRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc γRIIIa, independently of the Fc γRIIIa-48L/R/H phenotype. Blood 90, 1109–1114 [PubMed] [Google Scholar]
- 8. Congy-Jolivet N., Bolzec A., Ternant D., Ohresser M., Watier H., and Thibault G. (2008) FcγRIIIa expression is not increased on natural killer cells expressing the FcγRIIIa-158V allotype. Cancer Res. 68, 976–980 10.1158/0008-5472.CAN-07-6523 [DOI] [PubMed] [Google Scholar]
- 9. Thomann M., Schlothauer T., Dashivets T., Malik S., Avenal C., Bulau P., Rüger P., and Reusch D. (2015) In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS ONE 10, e0134949 10.1371/journal.pone.0134949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deleted in proof.
- 11. Chan A. C., and Carter P. J. (2010) Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 10, 301–316 10.1038/nri2761 [DOI] [PubMed] [Google Scholar]
- 12. Weiner G. J. (2015) Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370 10.1038/nrc3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moremen K. W., Tiemeyer M., and Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 10.1038/nrm3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H. (eds) (2017) Essentials of Glycobiology. 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 15. Subedi G. P., Hanson Q. M., and Barb A. W. (2014) Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcγRIIIa binding. Structure 22, 1478–1488 10.1016/j.str.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Varki A. (2017) Biological roles of glycans. Glycobiology 27, 3–49 10.1093/glycob/cww086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edberg J. C., and Kimberly R. P. (1997) Cell type-specific glycoforms of Fcγ RIIIa (CD16): differential ligand binding. J. Immunol. 159, 3849–3857 [PubMed] [Google Scholar]
- 18. Zeck A., Pohlentz G., Schlothauer T., Peter-Katalinić J., and Regula J. T. (2011) Cell type-specific and site directed N-glycosylation pattern of FcγRIIIa. J. Proteome Res. 10, 3031–3039 10.1021/pr1012653 [DOI] [PubMed] [Google Scholar]
- 19. Cosgrave E. F., Struwe W. B., Hayes J. M., Harvey D. J., Wormald M. R., and Rudd P. M. (2013) N-Linked glycan structures of the human Fcγ receptors produced in NS0 cells. J. Proteome Res. 12, 3721–3737 10.1021/pr400344h [DOI] [PubMed] [Google Scholar]
- 20. Hayes J. M., Frostell A., Cosgrave E. F., Struwe W. B., Potter O., Davey G. P., Karlsson R., Anneren C., and Rudd P. M. (2014) Fc γ receptor glycosylation modulates the binding of IgG glycoforms: a requirement for stable antibody interactions. J. Proteome Res. 13, 5471–5485 10.1021/pr500414q [DOI] [PubMed] [Google Scholar]
- 21. Hayes J. M., Frostell A., Karlsson R., Müller S., Martín S. M., Pauers M., Reuss F., Cosgrave E. F., Anneren C., Davey G. P., and Rudd P. M. (2017) Identification of Fc γ receptor glycoforms that produce differential binding kinetics for rituximab. Mol. Cell. Proteomics 16, 1770–1788 10.1074/mcp.M117.066944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taniguchi T., Woodward A. M., Magnelli P., McColgan N. M., Lehoux S., Jacobo S. M., Mauris J., and Argüeso P. (2017) N-Glycosylation affects the stability and barrier function of the MUC16 mucin. J. Biol. Chem. 292, 11079–11090 10.1074/jbc.M116.770123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Higel F., Seidl A., Sörgel F., and Friess W. (2016) N-Glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 100, 94–100 10.1016/j.ejpb.2016.01.005 [DOI] [PubMed] [Google Scholar]
- 24. Nagae M., and Yamaguchi Y. (2012) Function and 3D structure of the N-glycans on glycoproteins. Int. J. Mol. Sci. 13, 8398–8429 10.3390/ijms13078398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Passlick B., Flieger D., and Ziegler-Heitbrock H. W. (1989) Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 74, 2527–2534 [PubMed] [Google Scholar]
- 26. Néron S., Thibault L., Dussault N., Côté G., Ducas E., Pineault N., and Roy A. (2007) Characterization of mononuclear cells remaining in the leukoreduction system chambers of apheresis instruments after routine platelet collection: a new source of viable human blood cells. Transfusion 47, 1042–1049 10.1111/j.1537-2995.2007.01233.x [DOI] [PubMed] [Google Scholar]
- 27. Lanier L. L., Ruitenberg J. J., and Phillips J. H. (1988) Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol. 141, 3478–3485 [PubMed] [Google Scholar]
- 28. Edberg J. C., Redecha P. B., Salmon J. E., and Kimberly R. P. (1989) Human Fc γ RIII (CD16). Isoforms with distinct allelic expression, extracellular domains, and membrane linkages on polymorphonuclear and natural killer cells. J. Immunol. 143, 1642–1649 [PubMed] [Google Scholar]
- 29. Ravetch J. V., and Perussia B. (1989) Alternative membrane forms of Fc γ RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J. Exp. Med. 170, 481–497 10.1084/jem.170.2.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edberg J. C., Barinsky M., Redecha P. B., Salmon J. E., and Kimberly R. P. (1990) Fc γ RIII expressed on cultured monocytes is a N-glycosylated transmembrane protein distinct from Fc γ RIII expressed on natural killer cells. J. Immunol. 144, 4729–4734 [PubMed] [Google Scholar]
- 31. Fleit H. B., Wright S. D., and Unkeless J. C. (1982) Human neutrophil Fc γ receptor distribution and structure. Proc. Natl. Acad. Sci. U.S.A. 79, 3275–3279 10.1073/pnas.79.10.3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blázquez-Moreno A., Park S., Im W., Call M. J., Call M. E., and Reyburn H. T. (2017) Transmembrane features governing Fc receptor CD16A assembly with CD16A signaling adaptor molecules. Proc. Natl. Acad. Sci. U.S.A. 114, E5645–E5654 10.1073/pnas.1706483114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takahashi N., Cohen-Solal J., Galinha A., Fridman W. H., Sautès-Fridman C., and Kato K. (2002) N-Glycosylation profile of recombinant human soluble Fcγ receptor III. Glycobiology 12, 507–515 10.1093/glycob/cwf063 [DOI] [PubMed] [Google Scholar]
- 34. Reeves P. J., Callewaert N., Contreras R., and Khorana H. G. (2002) Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U.S.A. 99, 13419–13424 10.1073/pnas.212519299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferrara C., Stuart F., Sondermann P., Brünker P., and Umaña P. (2006) The carbohydrate at FcγRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 281, 5032–5036 10.1074/jbc.M510171200 [DOI] [PubMed] [Google Scholar]
- 36. Ferrara C., Grau S., Jäger C., Sondermann P., Brünker P., Waldhauer I., Hennig M., Ruf A., Rufer A. C., Stihle M., Umaña P., and Benz J. (2011) Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. U.S.A. 108, 12669–12674 10.1073/pnas.1108455108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mizushima T., Yagi H., Takemoto E., Shibata-Koyama M., Isoda Y., Iida S., Masuda K., Satoh M., and Kato K. (2011) Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells 16, 1071–1080 10.1111/j.1365-2443.2011.01552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Subedi G. P., Falconer D. J., and Barb A. W. (2017) Carbohydrate-polypeptide contacts in the antibody receptor CD16A identified through solution NMR spectroscopy. Biochemistry 56, 3174–3177 10.1021/acs.biochem.7b00392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subedi G. P., and Barb A. W. (2015) The structural role of antibody N-glycosylation in receptor interactions. Structure 23, 1573–1583 10.1016/j.str.2015.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu Y., Chen J., Sethi A., Li Q. K., Chen L., Collins B., Gillet L. C., Wollscheid B., Zhang H., and Aebersold R. (2014) Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol. Cell. Proteomics 13, 1753–1768 10.1074/mcp.M114.038273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. West M. B., Segu Z. M., Feasley C. L., Kang P., Klouckova I., Li C., Novotny M. V., West C. M., Mechref Y., and Hanigan M. H. (2010) Analysis of site-specific glycosylation of renal and hepatic γ-glutamyl transpeptidase from normal human tissue. J. Biol. Chem. 285, 29511–29524 10.1074/jbc.M110.145938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Medzihradszky K. F., Kaasik K., and Chalkley R. J. (2015) Tissue-specific glycosylation at the glycopeptide level. Mol. Cell. Proteomics 14, 2103–2110 10.1074/mcp.M115.050393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sethi M. K., Kim H., Park C. K., Baker M. S., Paik Y. K., Packer N. H., Hancock W. S., Fanayan S., and Thaysen-Andersen M. (2015) In-depth N-glycome profiling of paired colorectal cancer and non-tumorigenic tissues reveals cancer-, stage- and EGFR-specific protein N-glycosylation. Glycobiology 25, 1064–1078 10.1093/glycob/cwv042 [DOI] [PubMed] [Google Scholar]
- 44. Loke I., Østergaard O., Heegaard N. H. H., Packer N. H., and Thaysen-Andersen M. (2017) Paucimannose-rich N-glycosylation of spatiotemporally regulated human neutrophil elastase modulates its immune functions. Mol. Cell. Proteomics 16, 1507–1527 10.1074/mcp.M116.066746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamamoto M., Yamamoto F., Luong T. T., Williams T., Kominato Y., and Yamamoto F. (2003) Expression profiling of 68 glycosyltransferase genes in 27 different human tissues by the systematic multiplex reverse transcription-polymerase chain reaction method revealed clustering of sexually related tissues in hierarchical clustering algorithm analysis. Electrophoresis 24, 2295–2307 10.1002/elps.200305459 [DOI] [PubMed] [Google Scholar]
- 46. Nairn A. V., York W. S., Harris K., Hall E. M., Pierce J. M., and Moremen K. W. (2008) Regulation of glycan structures in animal tissues: transcript profiling of glycan-related genes. J. Biol. Chem. 283, 17298–17313 10.1074/jbc.M801964200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaburagi T., Kizuka Y., Kitazume S., and Taniguchi N. (2017) The inhibitory role of α2,6-sialylation in adipogenesis. J. Biol. Chem. 292, 2278–2286 10.1074/jbc.M116.747667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Y. C., Stein J. W., Lynch C. L., Tran H. T., Lee C. Y., Coleman R., Hatch A., Antontsev V. G., Chy H. S., O'Brien C. M., Murthy S. K., Laslett A. L., Peterson S. E., and Loring J. F. (2015) Glycosyltransferase ST6GAL1 contributes to the regulation of pluripotency in human pluripotent stem cells. Sci. Rep. 5, 13317 10.1038/srep13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang H. H., and Stanley P. (2010) A testis-specific regulator of complex and hybrid N-glycan synthesis. J. Cell Biol. 190, 893–910 10.1083/jcb.201004102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang H. H., Hassinen A., Sundaram S., Spiess A. N., Kellokumpu S., and Stanley P. (2015) GnT1IP-L specifically inhibits MGAT1 in the Golgi via its luminal domain. Elife 4, 10.7554/eLife.08916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barb A. W. (2015) Intramolecular N-glycan/polypeptide interactions observed at multiple N-glycan remodeling steps through [13C,15]-N-acetylglucosamine labeling of immunoglobulin G1. Biochemistry 54, 313–322 10.1021/bi501380t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Subedi G. P., and Barb A. W. (2016) The immunoglobulin G1 N-glycan composition affects binding to each low affinity Fc γ receptor. MAbs 8, 1512–1524 10.1080/19420862.2016.1218586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dalbeth N., and Callan M. F. (2002) A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. 46, 1763–1772 10.1002/art.10410 [DOI] [PubMed] [Google Scholar]
- 54. Jönsen A., Gunnarsson I., Gullstrand B., Svenungsson E., Bengtsson A. A., Nived O., Lundberg I. E., Truedsson L., and Sturfelt G. (2007) Association between SLE nephritis and polymorphic variants of the CRP and FcγRIIIa genes. Rheumatology 46, 1417–1421 10.1093/rheumatology/kem167 [DOI] [PubMed] [Google Scholar]
- 55. Izumi Y., Ida H., Huang M., Iwanaga N., Tanaka F., Aratake K., Arima K., Tamai M., Kamachi M., Nakamura H., Origuchi T., Kawakami A., Anderson P., and Eguchi K. (2006) Characterization of peripheral natural killer cells in primary Sjogren's syndrome: impaired NK cell activity and low NK cell number. J. Lab. Clin. Med. 147, 242–249 10.1016/j.lab.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 56. Schepis D., Gunnarsson I., Eloranta M. L., Lampa J., Jacobson S. H., Kärre K., and Berg L. (2009) Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus. Immunology 126, 140–146 10.1111/j.1365-2567.2008.02887.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Subedi G. P., Johnson R. W., Moniz H. A., Moremen K. W., and Barb A. (2015) High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. J. Vis. Exp. 2015, e53568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marcella A. M., and Barb A. W. (2017) The R117A variant of the Escherichia coli transacylase FabD synthesizes novel acyl-(acyl carrier proteins). Appl. Microbiol. Biotechnol. 101, 8431–8441 10.1007/s00253-017-8586-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.